
Abstract
Large baleen and toothed whales play crucial ecological roles in oceans; nonetheless, very little is known about their intestinal microbiomes. Based on striking differences in natural history and thus in feeding behaviours, it can be expected that intestinal microbiomes of large baleen whales and toothed whales are different. To test this hypothesis, the phylogenetic composition of faecal microbiomes was investigated by a 16S rRNA gene amplicon sequence–based approach for Bacteria and Archaea. Faecal samples from free-ranging large whales collected off the Azores Archipelago (Portugal) were used, comprising 13 individual baleen whales (one sei, two blue and ten fin whales) and four sperm whales. The phylogenetic composition of the Bacteria faecal microbiomes of baleen and toothed whales showed no significant differences at the phylum level. However, significant differences were detected at the family and genus levels. Most abundant phyla were Firmicutes, Bacteroidetes, Proteobacteria, Tenericutes and Spirochaeta. Few highly abundant bacterial genera were identified as key taxa with a high contribution to differences among baleen and toothed whales microbiomes. Only few archaeal sequences were detected, primarily Methanomassiliicoccales representing potential methanogenic Archaea. This is the first study that directly compares the faecal bacterial and archaeal microbiomes of free-ranging baleen and toothed whales which represent the two parvorders of Cetacea which members are fully aquatic large mammals which were evolutionary split millions of years ago.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Baleen (parvorder Mysticeti) and toothed (parvorder Odontoceti) whales play essential roles in ocean ecosystem as apex predators, primary and secondary consumers and are, therefore, suitable as indicators of ocean health worldwide [1, 2]. Despite their ecological relevance, anthropogenic pressure has driven various large whales to endangered status, and even to near extinction by hunting, and overall degradation of marine environments [1,2,3,4].
Baleen and toothed whales comprise the two cetacean parvorders within the order Cetartiodactyla, suborder Cetacea, summing 89 extant species (14 and 75, respectively). Time-calibrated molecular phylogeny suggests that they split around 36 million years ago, and this divergence was in part driven by evolutionary innovations in feeding mechanisms [5,6,7]. Toothed whales evolved innovations combining echolocation and anatomical modifications, to target single prey items by raptorial biting or suction feeding, while baleen whales evolved keratinous plates (baleen) and modifications to the skull and mouth morphology, allowing bulk feeding on aggregations of small prey using filtration [6]. These specialisations resulted in a marked trophic separation between baleen (mean trophic level: 3.35) and toothed (mean trophic level: 4.23) whales, despite all being carnivorous [8, 9]. Taking these differences into account, one would expect striking differences in the two phyla also with respect to faecal microbiomes. This co-evolution of commensal/mutualistic microbes within marine vertebrate hosts may have had a strong contribution to their nutritional adaptation to specific diets [10]. Among a broad range of environmental factors and host genetics/phylogeny, diet composition (herbivore, omnivore or carnivore) is an essential factor in the acquisition and maintenance of a healthy intestinal microbiome, both in terrestrial and marine mammals [1, 11, 12]. Hence, largely different intestinal microbiomes can be expected in baleen and toothed whales.
The faecal microbiome as a proxy for vertebrate intestinal microbiome contains a high diversity of different microbes of multiple taxa and with different metabolic capacities including transient opportunistic microbes, food-borne communalists and often host-specific mutualistic microbes [10]. Especially the latter often play a critical role in the processing of food uptake but can also affect host metabolism, host development and immune system functioning [10]. In healthy individuals, intestinal microbiota are robust and resistant to perturbations and maintain their composition in physiological ranges in order to sustain homeostasis, symbiosis and development of proper innate and adaptive immunity [13].
Nonetheless, there are only a few studies focussing on faecal microbiomes of whales. Sanders et al. [14] performed a comprehensive study on the diversity and function of the faecal microbiome of baleen whales. In a phylogenetic diversity study based on 454 amplicon-generated 16S rRNA gene sequence libraries and generated shotgun metagenomes of selected samples, functions of the intestinal microbiome (total gene pool) of a sei whale (Balaenoptera borealis), seven right whales (Eubalaena glacialis) and four humpback whales (Megaptera novaeangliae) were studied. These samples were drawn from captured live whales or from faecal samples from the intestines of stranded carcasses. The faecal microbiomes of these baleen whale species were compared with those of captured belugas (Delphinapterus leucas) and several carnivorous or herbivores terrestrial mammals. The baleen whale microbiomes shared different functional traits either with faecal microbiomes of terrestrial carnivores, and on the other hand with those of terrestrial herbivores. Despite functional parallels of whale faecal microbiomes to those of terrestrial carnivores and herbivores, the phylogenetic composition of the faecal microbiome of the baleen whales sampled by Sanders et al. [14] was unique and clearly distinct to terrestrial species but also distinct to captured beluga whales (toothed whales).
There are only few other intestinal/faecal microbiome studies of other large marine mammals. Bik et al. [1] studied the faecal microbiomes (swab samples) compared to four other body sites of healthy dolphins and sea lions. They found that habitat, diet and the host phylogeny contributed to a specific composition of the intestinal microbiome. In another study of the faecal microbiome of bottlenose dolphins, Soverini et al. [15] compared their data to those of Sanders et al. [14] and concluded that the captive bottlenose dolphin intestinal microbiome was distinct from that of baleen whales, which they attributed to differences in the dietary niches. Furthermore, the authors indicated that the intestinal microbiome of dolphins showed a compositional similarity to intestinal microbiomes of carnivorous fishes and other marine piscivores, in contrast to earlier results on baleen whales [14].
Few other studies investigated the gut microbiome of baleen or toothed whales. Those studies included the investigation of the intestinal microbiome of stranded (dead) adult pygmy (Kogia breviceps) and dwarf (K. sima) sperm whales [16], southern right whale (Eubalaena australis) calves (baleen whales) [17], two free-ranging blue whales (B. musculus; baleen whales) [18] and different gut sections of 38 hunted bowhead whales (Balaena mysticetus, baleen whales) [19]. A direct comparison of toothed and baleen whale microbiomes studied in parallel was never performed.
Here we compared the faecal microbiomes of living, free-ranging, presumably healthy baleen whales of the family Balaenopteridae and of toothed whales (sperm whales Physeter macrocephalus), thereby analysing members of both whale phyla for the first time. Furthermore, sampling design was in full agreement with physiological defecation and did not induce any stress to these marine mammals or microbiome artefacts compared to stranded whales. For a comprehensive overview and unlike to most previous studies, our investigation also included assessment of Archaea as a separate prokaryotic group in 17 members of the Cetartiodactyla for the first time. The abundance and phylogenetic diversity of bacterial and archaeal members of faecal microbiomes were investigated by 16S rRNA gene target–based real-time PCR (qPCR) and 16S rRNA gene sequence–based Illumina amplicon sequencing using established Bacteria- and Archaea-specific primer systems.
Material and Methods
Sample Collection
Faecal samples of 13 baleen whales of the family Balaenopteridae, including one sei whale (Baleaenoptera borealis), two blue whales (B. musculus) and ten fin whales (B. physalus), and of four sperm whales (P. macrocephalus) were collected in 2011 and 2014 off the Azores Archipelago (Portugal; Supplementary Fig. S1; Supplementary Table S1). Samples were collected under research licences #51/2011/DRA and #20/2013/DRA granted by the Regional Government of the Azores. Collection methods have been described in Hermosilla et al. [20]. Briefly, samples were obtained during focal follows of individual whales considered healthy from visual inspection in the field. Animals were not disturbed. Upon defecation, samples were collected as quickly as possible using a mesh-dip net with a 400-μm mesh size. Approximately 10–50 ml of each faecal sample were transferred to sterile plastic Falkon vials and fixed with ethanol at 70% concentration and stored on ice. Upon arrival in the laboratory, samples were stored at 4 °C until being transferred to the Institute of Parasitology (Justus Liebig University, Giessen, Germany). For long-term storage, samples were stored at − 20 °C.
DNA Extraction from Whale Faecal Samples
For DNA extraction, cooled faecal samples in 70% ethanol were pelleted by centrifugation (13,780g for 20 min at 4 °C) and washed twice with 1x autoclaved phosphate-buffered saline (PBS; 130 mM NaCl, 7 mM Na2HPO4, 3 mM NaH3PO4 per litre; pH 7) to remove remaining ethanol. Approximately 150 mg faecal samples were extracted with the ZR Faecal DNA MiniPrep Kit (Zymo Research Europe GmbH, Freiburg, Germany) as described by the manufacturer. DNA was finally eluted with 100 μL Ultra Pure™ demineralised water (DNase, RNase Free; Invitrogen, Karlsruhe, Germany) from the final spin column and measured in a Nano drop (Peqlab, Erlangen, Germany). The DNA concentration was adjusted to a concentration of 10 ng μL−1 for molecular biological analysis. The quality of the DNA samples was checked by PCR amplification of 16S rRNA gene fragments using universal Bacteria 16S rRNA gene–targeting primers and subsequent denaturing gradient gel electrophoresis (DGGE) analysis according to Schellenberg et al. [21]. All samples gave good amplification products (Supplementary Fig. S2a) and specific bacterial community fingerprint patterns (Supplementary Fig. S2b) which had already indicated significant differences among bacterial communities of baleen and toothed whale samples (Supplementary Fig. S3).
Quantification of Total Bacteria and Archaea 16S rRNA Gene Targets in Faecal Samples
Quantitative PCR (qPCR) analysis was performed with the SsoFast™ EvaGreen® Supermix (Bio-Rad, Feldkirchen, Germany) in a total volume of 10 μL including 1 μL template DNA, 1× SsoFast™ EvaGreen® Supermix, 0.2 μM of Bacteria (Univ-F, 5´-GTGSTGCAYGGYTGTCGTCA-3´, Univ-R, 5´-CCCCTCKGSAAAGCCTTCTTC-3´; [22]) and 0.5 μM of Archaea (ARC787F, 5´-ATTAGATACCCSBGTAGTCC-3´, ARC1059R, 5´-GCCATGCACCWCCTCT-3´; [23]) 16S rRNA gene targeting primer sets. A serial dilution of 16S rRNA gene fragments in defined concentrations (1 × 102 to 1 × 108 targets μL−1) was used as DNA standard for the qualification of bacterial and archaeal 16S rRNA gene targets. The standards were generated by the amplification of the 16S rRNA gene from reference strains, Citrobacter freundii ATCC 8090T (Bacteria) and Saccharolobus solfataricus P2 (Archaea) using universal primers as described by Cifuentes et al. [24]. The PCR products were purified (PCR purification kit; Qiagen) and quantified in a Tecan GENios FL fluorescence reader (Tecan Group Ltd., Männedorf, Switzerland) using PicoGreen (Quant-iT PicoGreen dsDNA reagent; Invitrogen, Germany) and a dilution series of Lambda DNA (Thermo Scientific, Germany) for quantification. The concentration of 16S rRNA gene targets μL−1 in the applied standards was calculated as described by Kolb et al. [25]. QPCRs were performed in a CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad) by the use of the following PCR program: 98 °C 2 min, followed by 35 cycles of 98 °C 20 s, 60 °C 20 s and 72 °C 20 s (detection), followed by melting curve analysis by heating from 65 to 95 °C (+0.5 °C/0.5 s cycle). Samples were run in technical triplicates and standards in technical duplicates. Primer dimers were not detected. Gene copy numbers and qPCR efficiencies were calculated using the Bio-Rad CFX Manager software (version 3.0). Efficiencies of qPCRs were 94.6% (R2 = 0.996) for the Bacteria and 77.6% (R2 = 0.992) for the Archaea primer system, respectively. Significant differences among samples were tested in SigmaPlot v12.5 (Systat Software, Erkrath, Germany) by one-way analysis of variance (ANOVA) using the Tukey test (post hoc test), the Shapiro Wilk normality test and the Brown Forsythe equal variance test. Concentrations of bacterial and archaeal 16S rRNA gene targets in baleen whales (all species summarised) compared to toothed whales were tested by T-tests assuming unequal variances. Tests were performed in Microsoft Excel (version 16.16.27).
Bacteria and Archaea 16S rRNA Gene Amplicon Sequencing Using an Illumina MiSeq Platform
The phylogenetic composition of bacterial and archaeal communities was analysed by 16S rRNA gene amplicon sequencing using universal 16S rRNA gene targeting primer systems, 341F (5´-CCTACGGGNGGCWGCAG-3´) and 785F (5´-GACTACHVGGGTATCTAAKCC-3´) for Bacteria and A340F (5´-CCCTAYGGGGYGCASCAG-3´) and A915R (5´-GTGCTCCCCCGCCAATTCCT-3´) for Archaea, respectively. Applied primer pairs were recommended by Klindworth et al. [26] for microbiome studies. PCR amplifications and Illumina 300 bp paired-end read sequencing using an Illumina MiSeq V3 system was performed by LGC Genomics (Berlin, Germany). The Illumina bcl2fastq 1.8.4 software (folder “RAW”) was used for dedublexing of all libraries. Reads were sorted by amplicon inline barcodes allowing one barcode mismatch. Reads with missing barcodes, one-sided barcodes or conflicting barcode pairs were discarded. Sequence adaptors were clipped in the following step and all reads with a length < 100 bp were discarded (adaptor clipping). Subsequently, primers (3 mismatches were allowed) were detected used for sequence orientation and clipped. Forward and reverse reads were combined using BBMerge 34.48 (http://bbmap.sourceforge.net/). The combined read pair data set was used for further analysis. Fastqc files of the combined reads were converted to fasta files using Galaxy (https://usegalaxy.org/) and submitted to the NGS analysis pipeline of the SILVA rRNA gene database project (SILVAngs 1.3; [27]). Each read was aligned using the SILVA Incremental Aligner (SINA; SINAv1.2.10 for ARB SVN [revision 21008]; [28]) against the SILVA SSU rRNA SEED and quality control [27]. Reads shorter than 50 aligned nucleotides and reads with more than 2% of ambiguities or 2% of homopolymers, respectively, were excluded from further processing. Putative contaminations, artefacts and reads with a low alignment quality (50 alignment identity, 40 alignment score reported by SINA) were excluded from downstream analyses. After these initial quality controls, identical reads were identified (dereplication), the unique reads (operational taxonomic units, OTUs) clustered on a per sample basis and the reference read of each OTU was classified based on the SILVA Taxonomy down to the genus level. Dereplication and clustering was done using cd-hit-est (version 3.1.2; http://www.bioinformatics.org/cd-hit;[29]) running in accurate mode, ignoring overhangs and applying identity criteria of 1.00 and 0.98, respectively. The classification was performed by a local nucleotide BLAST search against the non-redundant version of the SILVA SSU Ref dataset (release 128; http://www.arb-silva.de) using blastn (version 2.2.30+; http://blast.ncbi.nlm.nih.gov/Blast.cgi) with standard settings [30]. The classification of each OTU reference read was mapped onto all reads that were assigned to the respective OTUs and identical reads. Reads of all OTUs assigned to the same taxonomic paths were summarised for subsequent analysis. This yields quantitative information (number of individual reads per taxonomic path) within the limitations of PCR and sequencing technique biases as well as multiple rRNA operons. Reads without any BLAST hits or reads with weak BLAST hits, where the function “(% sequence identity + % alignment coverage)/2” did not exceed the value of 93, remained unclassified and were assigned to the meta group “No Relative” [31]. The applied method was first used in the publications of Klindworth et al. [26] and Ionescu et al. [32]. For relative abundance analysis of bacterial communities reads assigned as Archaea, mitochondria, chloroplasts, Eukarya or “No Relative” were excluded for the analysis of bacterial faecal microbiome. Bacteria-derived sequences were set to 100% for further analyses. Bacterial communities were analysed at three taxonomic path levels, phylum, family and genus. Due to the limited resolution of the 16S rRNA gene (genus level) subsequent analyses were not performed at the OTU level. Total sequence numbers per taxonomic paths and relative abundance patterns of the resulting bacterial community profiles (resolution: phyla, families, phylogenetic groups) were further used for detailed analysis in PAST4 (folk.uio.no/ohammer/past; [33]). Absolute sequence numbers per taxonomic path (genus level) were used for alpha diversity analysis including the Chao-1 index (estimating the number of present genera/phylogenetic groups), evenness and dominance values (describing the number and distribution of the phylogenetic groups within the microbiomes), and the Shannon diversity index (describing the overall taxa diversity), as well as rarefaction analysis (indicating taxa richness and diversity coverage). Relative abundance patterns of genera, families and phyla of baleen and toothed whale faecal microbiomes were compared by hierarchical clustering and non-metric multidimensional scaling (NMDS). Both analyses based on Bray-Curtis distances of the relative abundance patterns. For hierarchical clustering, the unweighted pair group method with arithmetic mean (UPGMA) method was used. One Way ANOSIM was performed to determine significant difference between community patterns of baleen and toothed whales. The analysis was based on Bray Curtis distances and 9999 permutations. Bonferroni-corrected p-values were used to indicate statistical significant differences among community patterns (p < 0.05). SIMPER analysis was used to calculate the contribution of each taxonomic group (given in % contribution) to the dissimilarity between each two groups, taxa that had the highest contribution to the differences among baleen and toothed whale microbiomes.
Results
Concentration of Bacterial and Archaeal 16S rRNA Gene Copies in Large Whale Faeces
The concentration of bacterial and archaeal 16S rRNA gene copies in whale faecal samples was in the range of 109 to 1011 and 106 to 109 copies per g fresh weight of faeces, respectively (Fig. 1; Supplementary Table S2). The concentration of bacterial 16S rRNA gene copies was always between one to four orders of magnitude higher than archaeal 16S RNA gene copies. No significant differences were obtained for the total bacterial and archaeal load in faecal samples among the whale families (t-tests; Bacteria: p = 0.18; Archaea; p = 0.74).

Concentrations of bacterial and archaeal 16S rRNA gene copies per g whale faeces. Analysis was performed by quantitative PCR using universal Bacteria and Archaea 16S rRNA gene targeting primer systems. Mean values and standard deviations of three technical replicates are given. FW: fresh weight
Phylogenetic Community Profiling of the Bacterial Whale Microbiome - Illumina Sequencing Results
A total of 817,474 high-quality combined sequences were obtained from a total of 2,094,076 raw reads (raw read pairs 1,047,038) obtained by the amplicon sequencing of the 17 whale samples (Supplementary Table S3). Only 645 sequences (0.08% of the combined reads) were rejected for not meeting the quality criteria. The average length of the remaining sequences was 409 nucleotides (minimal length 52, maximal length 480 nucleotides). Based on a 98% sequence similarity value, a total number of 90,379 OTUs was obtained from 470,405 clustered sequences including 256,045 replicates. Finally, 816,829 sequences were classified, among those, 801,411 sequences (98.1% of the total sequences) were classified as Bacteria (9368 to 72,925 per sample), 38 as mitochondria (0.01%), 65 as chloroplasts (0.01%), 13,284 as Archaea, Euryarchaeota (1.6%), 232 as Eukaryota (0.03%) and 1799 sequences (0.2%) were summarised under “No Relative” (Supplementary Table S3).
Eukaryotic sequences were found in 11 of the 13 samples from the three baleen whale species, but in none of the four sperm whales. The sequences obtained were all identified as Tetratrichomonas (Trichomonadea, Parabasalia, Metamonada, Excavata).
Relative Abundance of Different Bacterial Phyla in Large Whale Faecal Microbiomes
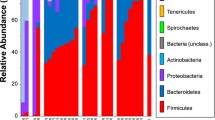
A total of 22 bacterial phyla were detected in the whale faecal samples (Fig. 2a; Supplementary Table S4), with nine phyla having a relative abundance above 1%. No distinct differences of the bacterial community patterns resolved at the level of phyla were visible by hierarchical clustering (Fig. 2a) and in the NMDS plot (Fig. 2b) between baleen and toothed whales. This was confirmed by One Way ANOSIM analysis which showed no significant differences in the phyla composition of baleen and toothed whale microbiomes (p = 0.2577).

Differences in the composition of the baleen and toothed whale faecal microbiomes studied at the level of bacterial phyla. a Hierarchical clustering (UPGMA clustering based on Bray Curtis distances) and relative abundance heatmap. b NMDS plot based on Bray Curtis distances. All analyses were performed in PAST4
Firmicutes was the predominant phylum in all cetacean faecal samples (53.9 to 91.2% of the sequenced 16S rRNA gene amplicons per sample; Fig. 2a; Supplementary Table S4). In 15 of the 17 samples, Bacteroidetes was the second most abundant phylum (0.9 to 32.4%). The two phyla together represented 80.1 to 96.7% of the bacterial 16S rRNA gene sequences obtained from the individual faecal samples. Changes in the relative abundance of Firmicutes and Bacteroidetes and the ratio among their relative abundance mainly affected the formation of different clusters in the hierarchical clustering based on relative abundance patterns of phyla (Fig. 2a, b).
The composition of the faecal microbiome at the level of bacterial phyla of the single sei whale (Bbo036) was clearly different to all other faecal microbiomes (Fig. 2). The microbiome of this sei whale sample showed a very high relative abundance of Firmicutes and a very low abundance of Bacteroidetes (79.2% and 0.9%; Fig. 2; Supplementary Table S4).
Beside the two dominating phyla, Proteobacteria followed by Spirochaetae and Tenericutes occurred at least in one of the samples with a relative abundance >6% and contributed with 8.1, 7.8 and 5.3%, respectively, to the differences among the individual faecal microbiomes (SIMPER analysis; Supplementary Tab. S4). Proteobacteria represented 0.2 to 4.9% of the bacterial 16S rRNA gene sequences in faecal microbiomes. Only in the sei whale sample (Bbo036) and in one fin whale sample (Bph053) the relative abundance of Proteobacteria was higher than that of Firmicutes, with 17.4% and 8.4%, respectively (Fig. 2a; Supplementary Tab. S4). The high abundance of Proteobacteria in those two samples was the main factor that led to the separation of the faecal microbiomes in the NMDS plot (Fig. 2b). The high abundance of Proteobacteria in the sei whale sample was outside the range of relative abundances of Proteobacteria obtained in the other microbiomes (Supplementary Table S4). The phylum Spirochaetae occurred in a relative abundance between 0.1 and 5.3%, and, except for one of the two blue whale samples (Bmu025), the relative abundance of Spirochaetae was with 8.7% above that range (Supplementary Table S4). The phylum Tenericutes occurred in a range of 0.1 to 1.8% and, except in two sperm whale samples (Pma179 and Pma190), the relative abundance was above the range (5.0% and 6.7%, respectively; Supplementary Table S4). The phyla Lentisphaerae, Actinobacteria, Cyanobacteria, Synergistetes and Kiritimatiellaeota occurred at least in one of the faecal microbiomes with a relative abundance of >1 to 2.5% and contributed with 1 to 2.7% to the differences among bacterial communities (Fig. 2a; Supplementary Table S4). Other detected but low abundant phyla (<1%) were Fusobacteria, Verrucomicrobia, Epsilonbacteraeota, Fibrobacteres, Elusimicrobia, Chloroflexi, Deinococcus-Thermus, Armatimonadetes, Planctomycetes, Deferribacteres and Marinimicrobia (SAR406 clade; Fig. 2a; Supplementary Table S4).
Family Level Resolution Showed Distinct Bacterial Faecal Microbiomes of Baleen and Toothed Whales
In total, 174 bacterial family level taxonomic paths were detected in the faecal microbiomes (Supplementary Table S5). Hierarchical clustering and the NMDS plots based on relative abundance patterns of family level taxonomic paths showed distinct non-overlapping patterns for baleen and toothed whale microbiomes (Fig. 3a). One Way ANOSIM confirmed that these differences were significant (p = 0.0041).

Differences in the composition of the baleen and toothed whale faecal microbiomes studied at the level of bacterial families (a) and genera (b). Analyses included hierarchical clustering based on Bray Curtis dissimilarity values and UPGMA clustering (left sides) and NMDS plots (right side) also using the Bray Curtis index for the generation of a dissimilarity matrix. Analysis was performed in PAST4
Different families of the orders Clostridiales (phylum Firmicutes) and Bacteroidiales (phylum Bacteroidetes) occurred with a high relative abundance either in baleen and toothed whale samples and had a high contribution to differences among the faecal microbiomes of the two groups of whales (SIMPER analysis; Supplementary Table S5; Supplementary Fig. 3). The families Clostridiaceae 1 (family taxon ID F-81) and Bacteroidaceae (F-27) occurred, respectively, with a 31.5- and 79-fold significant higher relative abundance in baleen than in toothed whales. In contrast, the Clostridiales vadinBB60 group (F-85) was present in considerable quantity in sperm whales (1.98–20.7%), but with only 0.03–1.41% in baleen whales. Carnobacteriaceae of the Lactobacillales (F-72; Firmicutes) were present in baleen whales with a relative abundance of 0.03 to 18.2% (mean 4.18 ± 5.7%), but not in sperm whales. The highest contribution to the differences of the bacterial microbiome composition (SIMPER analysis; Supplementary Table S5) was made with 22.11% by a Clostridiales 1 taxon (F-81). It occurred with a significantly higher relative abundance 82.75% [±26.3%]) in baleen than in toothed whales (0.87% [±0.89]). The second highest contributing family (14.35%) was Ruminococcaceae (F-95) of the order Clostridiales (Firmicutes). This family occurred with a mean relative abundance of 31.4% (±20.3%) in baleen and 28% (±13.3%) in toothed whales. An exception was the sei whale where the relative abundance of Ruminococcaceae was much lower (1.3%). Only few families occurred with a high relative abundance only in individual whale faeces microbiomes; Pasteurellaceae (F-156) and the Clostridiales family XIII (6.9%) occurred with a relative abundance of 15.9% and 6.9% only in the faecal sample of the sei whale and the Synthrophomonadaceae (F-96; Clostridia) and the Bacteroidetes BS11 group (F-28) with 11.0 and 7.9% only in one of the toothed whales (Pma185), respectively. The high relative abundance of F-96 and F-28 in the Pma185 faecal microbiome had also a high contribution to the differences of this toothed whale faecal microbiome compared to the other three toothed whales.
Genus Level Resolution Showed Distinct Bacterial Faecal Microbiomes of Baleen and Toothed Whales
The composition of the bacterial faecal microbiomes compared at the level of bacterial genera showed distinct differences for baleen and toothed whale microbiomes by hierarchical clustering and in the NMDS plot (Fig. 3b). One Way ANOSIM analysis confirmed that the differences for baleen and toothed whale bacterial community patterns were statistically significant (p = 0.0003).
Slight but non-significant differences were found between the faecal microbiomes of the different Balaenoptera species, although a statistical comparison was not possible because of the small sample sizes. One fin whale (BphF003) faecal microbiome differed from those of the remaining fin whales.
Distinct Abundant Genera Contributed to Baleen and Toothed Whale Faecal Microbiome Differences
In total 513 different genus level taxonomic paths have been identified. Among those, 58 occurred with a relative abundance of >1% in at least one faecal microbiome. Among those, the 21 most abundant contributed the most (>1%) differences among baleen and toothed whale faecal microbiomes as determined by SIMPER analysis (Supplementary Table S6; Fig. 4). Firmicutes (12 out of 21) followed by Bacteroidetes (6 out of 21) represented most of the abundant genera (Fig. 4a).

Impact of 21 most abundant genera (genus-like cluster) on differences among baleen and toothed whale faecal microbiomes. a Heatmap depicting relative abundances of the most abundant genera within the faecal microbiomes. Stars at the right side of the heatmap mark genera which showed significant differences in relative abundances in baleen and toothed whale faecal microbiomes (t-tests; p < =0.05). SIMPER values (calculated in PAST4) represent the contribution (in %) of individual phylogenetic groups on the differences of the baleen and toothed whale faecal microbiomes. A respective heatmap depicting all genera is given in Supplementary Table S6. b PCA plot depicting differences in the community profiles determined from the different whale species. The influence on individual genera on the community differences is depicted in form of biplots. Analysis was performed in PAST4 and based on relative abundance patterns of individual genera. Tax-IDs given at biplots are assigned to genera in a
Eight of the genera occurred with a statistically significant higher relative abundance in baleen whales (p < 0.05; pairwise t-tests; Fig. 4a). The most abundant genus with the highest contribution to community differences (SIMPER 17.0%; Fig. 4a) was the genus-like cluster Clostridium sensu stricto 1 (T-157) of the Clostridiaceae (F-81). Sequences assigned to this cluster occurred with a high relative abundance of up to 66% in the faecal samples of the different Balaenoptera species (Fig. 4). The taxon occurred with a very high relative abundance (29–56%; mean 53%) in six out of 13 Balaenoptera whales including one sei whale, two blue whales and three fin whales. The taxon mainly contributed to the distinction of those microbiomes from other microbiomes as shown by the box plot analysis in the PCA plot (Fig. 4b). The relative abundance of this taxon in the other fin whale faecal microbiomes was between 2.6 and 9.1% (mean 5.6%). The taxon was only detected with a very low relative abundance (0.1%) in one of the four sperm whale faecal microbiomes (Fig. 4a). A second Firmicutes genus (Granulicatella, T134) had the same occurrence pattern as T-157 and had with 2.6% a countable contribution to the differences of the baleen and toothed whale faecal microbiomes. The remaining six genera with a significantly higher relative abundance in baleen whales and high contribution to the microbiome differences were two uncultured Firmicutes genus-like cluster, Ruminococcaceae UCG-005 (T298) and Ruminiclostridium 9 (T292), two Bacteroidetes genera, Bacteroides (T-44) and Alloprevotella (T-68) and the genera Sutterella (T-439) of the Burkholderiaceae (Gammaproteobacteria) and Treponema 2 (T-49) of the Spirochaetaceae (Spirochaetes).
Several of the above-mentioned genera were less abundant in the faecal microbiome of the studied sei whale. The sei whale faecal microbiome contained, in contrast, a high relative abundance of three Pasteurellaceae genera, i.e. Actinobacillus (T470; 8.0%), Bisgaardia (T-472; 5.4%) and Avibacterium (T-471; 2.4%). The genera occurred only with a total of 0.8% in faecal samples of the other baleen whales and were not detected in the four sperm whales.
Conversely to the baleen whales, 12 genera occurred with a high relative abundance in toothed whale faecal microbiomes and had a high contribution to detected microbiome differences. The most abundant genera with high contribution to microbiome differences (SIMPER value >5%, Fig. 4a) were two Firmicutes genus-like cluster, Tyzzerella 3 (T-237; Lachnospiraceae) and the Clostridiales vadinBB60 group (T-169). Six additional Firmicutes genera, including three uncultured Ruminococcaceae genus-like cluster (T-305, T-301, T-312), the uncultured Christensenellaceae genus-like cluster R-7 (T-150) and the genus Synthrophomonas (T-315), three uncultured Bacteroidetes genus-like cluster assigned to the Rikenellaceae (T-83) and Bacteroidales (T-54, T-48) and one Tenericutes genus-like cluster (T-504; Izimaplasmatales of the Mollicutes), occurred with a higher relative abundance in toothed whale faecal microbiomes and contributed with 1.3 to 4.1% (SIMPER values; Fig. 4a) to the differences among the baleen and toothed whale faecal microbiomes.
Baleen and Toothed Whale Faecal Microbiomes Showed No Distinct Differences with Respect to Alpha Diversity Parameters
The alpha diversity values including the number of detected genera/genus-like cluster (Chao-1 index; Fig. 5a) and the distribution of those genera within the microbiomes (evenness, dominance values) and the overall diversity (Shannon diversity index; Fig. 5b–d) showed some variations among individual faecal microbiomes but without distinct patterns among baleen and toothed whales. Significant differences in genera distributions between baleen and toothed whale faecal microbiomes were only determined for the evenness (t-test; p < 0.05; Fig. 5d; Supplementary Table S7). Especially four of baleen whale faecal microbiomes were characterised by low evenness values combined with high dominance values indicating a high relative abundance of few bacterial genera (Bbo036, Bmu018, Bph044, Bph055; Fig. 5; Supplementary Table S7). Rarefaction curves for all faecal microbiomes indicated that the complete diversity at the genus level was not yet covered with the total number of analysed sequences per sample (Supplementary Fig. S4). However, the investigation based on the dataset of faecal microbiomes gave a clear overview of most abundant genera and indicated clear differences between faecal microbiomes of blue, sei, fin and sperm whales.

Comparison of baleen and toothed whale faecal microbiome at the genus level by alpha diversity parameters. a Richness (Chao-1). b Eveness. c Dominance. d Shannon index. Values obtained for baleen and toothed whales microbiomes were compared by t-tests. Significant differences (p < = 0.05) are marked with a star at top of the box-plots
Low Abundance of Archaeal 16S rRNA Gene Sequences in Large Whale Microbiomes
Because qPCR analysis indicated the presence of Archaea in all 17 large whale faecal samples, archaeal 16S rRNA gene amplicon libraries were generated as well as using a recommended archaeal-specific primer system [26]. In summary, only 70,038 combined reads could be subjected to phylogenetic analysis. Among these, 65,906 reads (94.1%) of the sequences were rejected because they did not match the quality criteria of the SILVAngs analysis pipeline. The average length of the finally aligned sequences was 173 nt (35–493 nt). A total number of 689 OTUs (1.0%) was obtained. In total, 456 sequences were clustered (0.65%) and 2987 reads counted as replicates (4.68%). Only 1875 sequences were classified (2.68%) and 2257 sequences were summarised under the criterion of “No Relative” (3.22%). From the 1875 classified sequences, only 19 sequences represented Archaea; the remaining sequences were identified as Bacteria, mainly of the Bacteroidetes and Firmicutes as well as some Spirochaetes, Alpha- and Gammaproteobacteria and Lentisphaerae (Fig. 6a; Supplementary Table S8)

Relative abundances and absolute numbers of archaeal 16S rRNA gene sequences detected by the 16S rRNA gene Illumina amplicon sequencing of baleen and toothed whale faecal microbiome using archaeal (a) and bacterial (b) specific primer systems
More archaeal sequences were determined with the Bacteria 16S rRNA gene targeting primer system (13,284 sequences; Fig. 6b). All detected Archaea sequences represented Euryarchaeota. The most abundant archaeal taxon in baleen whales was a genus level cluster of uncultured Methanomethylophilaceae (class Thermoplasmata; 42.9-100% of the archaeal sequences per samples). This taxon was less abundant in sperm whales (0–20% of archaeal sequences). Two additional genera of Thermoplasmata occurred in high relative abundances in the sei whale (Bbo036; Marine Group II; 57.1% of the archaeal sequences) and in one of the sperm whales (Pma185; Methanosarcina, 36.7%). Further archaeal sequences were assigned to the class Methanobacteria. These sequences were assigned to the genera Methanosphaera (class Methanobacteria) and Methanocorpusculum (class Methanomicrobia) and Methanosarcina (class Methanomicrobia).
Discussion
Specificity of the Research on the Intestinal Microbiomes of Free-Ranging Large Whales
Research on intestinal microbiomes of free-ranging, presumably healthy large whales is scarce. A key problem is the specificity of the sampling technique. Due to the challenges in sampling free-swimming cetaceans, currently only faecal samples can be used to get information on the gut microbiome of live animals in the wild. The available information on the microbiome composition for different regions of the gastrointestinal (GI) tract is limited to few studies [19, 34]. For example, Wan et al. [34] studied the stomach, foregut and hindgut in comparison to the faeces microbiomes of five East Asian finless porpoises immediately after their death due to by-catch. The study showed that the richness of the different gut microbiomes was even, but the phylogenetic composition was different between hindgut and faeces to stomach and foregut [34]. In conclusion, the faecal microbiome studied here is representative for the large intestine (i.e. colon, caecum, rectum); differences of microbiomes at more proximally located intestinal segments cannot be ruled out.
Another problem created by sampling floating faeces is that it cannot be excluded that bacterioplankton of the surrounding water may be present in the sequenced microbiome. No seawater analysis has been conducted here, but earlier studies have already shown that marine mammal microbiomes are different from those of, e.g., dietary fish and seawater [1]. This was also confirmed by Apprill et al. [35] for the skin microbiome of humpback whales (Megaptera novaeangliae).
Methanogenic Archaea in the Faecal Microbiomes of Large Whales
The present study is one of the first to specifically attempt to analyse also the Archaea composition besides that of Bacteria in the faecal microbiome of large whales. Archaeal sequences were found in all faecal samples by quantitative PCR using a common archaeal 16S rRNA gene targeting primer systems, but with a low concentration of approximately two log scores compared to Bacteria. The vast minority of these sequences may have been archaeal 16S rRNA gene sequence targets as determined by the archaeal 16S rRNA gene amplicon sequencing. The detected archaeal sequences were primarily assigned to the order Methanomassiliicoccales representing potential methanogenic Archaea. So far it is known that habitat-specific clades of Methanomassiliicoccales occur in the intestinal tracts of different animals [36] and the composition of Methanomassiliicoccales can vary with different diets as shown for example in a study of caprine intestine [37]. Other archaeal sequences detected here, Methanosphaera (class Methanobacteria) and Methanocorpusculum and Methanosarcina (both Methanomicrobia), also represented different methanogens which differed in their metabolic pathways to produce methane [38,39,40].
Abundance of mitochondriate Unicellular Protists
The Bacteria 16S rRNA gene amplicon approach gave also some information on mitochondriated intestinal flagellated unicellular protists with parasitical lifestyle: Tetratrichomonas (Trichomonadea, Parabasalia, Metamonada, Excavata). Trichomonads in general are found in the intestine and urogenital tract as common parasites of many vertebrate and invertebrate species [41,42,43]. In previous studies on occurrence of gastrointestinal parasites of sperm [3], blue [44,45,46], sei and fin whales [44, 45], the genus Tetratrichomonas or any other closely related genera were not detected as whale parasites [44].
Comparison of Bacterial Microbiome Data Available for Large Whales and Other Marine Mammals
A high abundance of Bacteroidetes and Firmicutes has been found in other whale microbiomes and also in other terrestrial mammals [14]. However, Bacteroidetes were generally more abundant than Firmicutes. Studies of the faecal microbiomes of humans and mice and several other terrestrial mammals showed a relative abundance of 60–80% Bacteroidetes and 20–40% Firmicutes [12]. Dominant roles for these two phyla were found in faecal microbiomes of different baleen whale species [14], two live blue whales [18], stranded pygmy and dwarf sperm whales [16], one sperm whale [47] and southern right whale calves [17]. The faecal samples of the two blue whales analysed by Guass et al. [18] were dominated by Firmicutes (relative abundance >98%); in the study of Sanders et al. [14] some faecal microbiomes were also dominated by Firmicutes while others showed a higher abundance of Bacteroidetes followed by Firmicutes. In contrast to our study, the taxonomic composition of the Odontoceti faecal microbiomes investigated by Sanders et al. [14] strongly varied among the samples and was enriched in Proteobacteria and Fusobacteria. In the present study, the microbiome composition was quite stable among the four sperm whale samples (analysed at the level of genera) and Proteobacteria were detected only in low abundances (0.4 to 2.6%). Fusobacteria were only detected in a single sperm whale sample and in very low abundance (0.004%). These contrasting results may be due to differences in intestinal microbiome related to feeding ecology within the Odontoceti. All of the Odontoceti samples in our study belong to sperm whales, while in Sanders et al. [14], Odontoceti samples originated from three different dolphin species. In temperate latitudes, such as in the Azores, sperm whales are chiefly teuthophagous, although they may occasionally consume small amounts of fish [48, 49]. Dolphins, on the other hand, have a more generalist diet. For example, the Atlantic white-sided dolphin (Lagenorhynchus acutus), the bottlenose dolphin (Tursiops truncatus) and the beluga whale that were investigated by Sanders et al. [14] feed on coastal and pelagic fish and cephalopods (and other invertebrates in the case of the beluga), and their diet can vary seasonally and geographically [50,51,52]. Furthermore, aside from one Atlantic white-sided dolphin, samples from Odontoceti in the study of Sanders et al. [14] were all from captive dolphins; thus, the results may not be completely representative of wild populations.
On the other hand, our study is in agreement with the findings of Sanders et al. [14] regarding a relatively high abundance of Spirochaetae in the faecal microbiome of baleen whales. This was pointed out as a clear difference to the low abundance of Spirochaetae in faecal microbiome of terrestrial mammals (Sanders et al. [14]). In line with our results, Sanders et al. [14] also detected a very low proportion of Proteobacteria in baleen whale samples. Within this phylum, Pasteurellaceae were exclusively found in the sei whale in significant numbers (15.9%), and this group was also detected with a relative abundance of 4 ± 1.9% in a 16S rRNA gene–based Illumina amplicon sequencing study of the faecal microbiome of adult dolphins [15]. The ecological niche of Bisgaardia spp. is the oropharynx of seals and sea elephants, but the other two members of Pasteurellaceae, Actinobacillus and Avibacterium, also contain species colonising terrestrial mammals and birds, respectively.
Although cetaceans evolved from herbivorous land-living artiodactyls and still share a number of “ruminant-like” anatomical homologies to this group, large baleen whale microbiota composition parallels that of both terrestrial herbivores and carnivores, especially with respect to functional capacity (chitin degradation) and taxonomic level [14]. Sperm whale microbiomes determined in this study differed in several aspects from that of baleen whales. Specifically, differences were found for the relative abundance of different Firmicutes and Bacteroidetes taxa which were either present in the baleen or toothed whale faecal samples. Among those were several taxa of uncultured Ruminococcaceae and uncultured clostridia, which may have specific nutritional functions in the intestinal microbiota of sperm and baleen whales. This suggests that their marine diet compositions have significantly influenced co-evolution of commensal intestinal microbes in these two whale families.
The sei whale faecal microbiome analysed in our study showed strong differences to the other two baleen whale species, which can be explained by different diet and spatial distribution. The chief difference that was found was the high abundance of Pasteurellaceae (Proteobacteria). Another sei whale microbiome, inferred from a sample collected in 2011 off the Canadian Atlantic coast, revealed a much lower abundance of Proteobacteria, which may indicate spatiotemporal or individual differences and warrant further investigation into these aspects [14, 16].
Tenericutes and Proteobacteria were, together with Bacteroidetes, determined by 454 pyrosequencing among the most abundant phyla in the foregut of dolphins [1]. In contrast, Firmicutes was among the most abundant phyla detected in the rectal samples of dolphins together with Proteobacteria and Fusobacteria while Bacteroidetes occurred only in a very low abundance in those samples (<1%; [1]). Tenericutes and Proteobacteria were also quite abundant in faecal microbiomes in this study, while Fusobacteria occurred in a very low relative abundance in all whale faecal samples (<0.8%). Another study of adult dolphins determined Firmicutes and Proteobacteria as the most dominating phyla in the faecal microbiome [15]. In that study, Actinobacteria, Bacteroidetes, Fusobacteria and Tenericutes were determined together (each of them in a relative abundance of 3 to 5%) and thus were accounted as subdominant phyla in respective faecal samples. Studies of faecal microbiomes of wild young South American and subarctic fur seals as well as Australian sea lions also revealed that Firmicutes represented the most abundant phylum with more than 80% in wild marine mammals [53, 54]. In contrast to our study, the relative abundance of Bacteroidetes was in general very low. Fusobacteria occurred with a very low relative abundance. The distribution of Actinobacteria and Proteobacteria varied between the four seal species and was higher than that found in the whale samples. Besides our faecal whale microbiome study, a skin microbiome study (epimicrobiome study) of 19 North Pacific humpback whales revealed also a high proportion of Bacteroidetes [35].
The alpha diversity of the faecal bacterial microbiomes of the baleen and toothed whales showed no statistically significant differences except for four of the baleen whales’ faecal microbiomes, which were characterised by low evenness combined with high dominance values indicating a high relative abundance of few bacterial genera (Bbo036, Bmu018, Bph044, Bph055). Erwin et al. [16] also determined no significant differences in alpha diversity for intestinal microbial communities of kogiid hosts. Diversity and faecal microbiome composition of pygmy and dwarf sperm whales were recently found to be significantly more diverse (mean 416–432 OTUs) compared to other toothed whales [14, 16]. In contrast to rectal microbiomes of wild and captive dolphins, porpoises and beluga whales that contained 10–179 OTUs on average [1, 34, 47, 55] with 122 a higher number than in this study was noted in a single sperm whale faecal microbiome [47]. A mean number of 64–87 OTUs was found in the present study. Higher values of 163–364 OTUs on average were also reported in baleen whales [47] compared to 56–77 OTUs from this study.
Interestingly, the low level of diversity in intestinal and faecal microbiomes was also found in this study contrasts with other body sites of cetaceans, where a rich diversity of microbial communities has been documented. Bik et al. [1] detected twice the phylum level and four times the OTU level diversity in oral microbiomes of bottlenose dolphins compared to their intestinal microbiomes.
Detection of Potential Pathogens in the Bacterial Whale Microbiomes
Although the whales from this study were apparently healthy, the dataset was checked for known and potential bacterial pathogens in cetaceans and other marine mammals, in accordance with the One Health principle [56]. In these, members of genera Staphylococcus, Streptococcus, Erysipelothrix, Clostridium, Mycobacterium, Chlamydia, Brucella, Leptospira, Aeromonas, Pseudomonas, Plesiomonas, Edwardsiella, Salmonella, Campylobacter and Helicobacter were considered animal pathogens, sometimes also associated with human infections [17, 55, 57,58,59,60,61,62,63,64,65,66]. From the latter, very similar bacteria to the rat bite fever organism, Streptobacillus moniliformis, were recently described as Oceanivirga in fish, dolphins, sea lions and different cetacean species [1, 67,68,69]. In this regard, it needs to be explicitly mentioned that amplicon studies targeting partial 16S rRNA gene sequences alone cannot resolve taxa reliably at species level [21]. Sequences, assigned to the genera Chlamydia, Brucella, Leptospira, Aeromonas, Pseudomonas, Plesiomonas, Edwardsiella, Salmonella and Oceanivirga (Leptotrichiaceae) were not detected. Sequences of the following genera were detected in our dataset, although the resolution was not high enough to unequivocally identify certain taxa to species level (in brackets putative pathogens and possibly zoonotic microorganisms): Staphylococcus (St. aureus), Streptococcus (S. phocae, S. iniae, S. suis, S. pyogenes, S. canis, S. zooepidemicus), Erysipelothrix (E. rhusiopathiae), Clostridium (C. perfringens), Mycobacterium (M. pinnipedii, M. abscessus), Campylobacter (C. fetus) and Helicobacter (H. pylori, H. cetorum).
Conclusions
Albeit remarkable differences exist between large baleen and toothed whale families specifically with respect to feeding ecology, there was a high degree of homology in the microbiomes in both groups. In this study, differences were detected especially at the lower taxonomic levels (genera/taxonomic groups), that can help to address different pathways in food digestion. However, even the scarce number of similar studies has shown that striking differences remain within and between species, as well as between different biomes, questioning a strict host-microbe specificity and suggesting important influences of season, diet and even social contacts, as described earlier [55, 70]. Our results warrant further investigation on the role of these effects in shaping cetacean microbiomes. Highly abundant phyla in most faecal samples were Firmicutes, followed by Bacteroidetes, Proteobacteria, Tenericutes and Spirochaeta occurred in individual samples with a relative abundance of up to 17.4%. Lower abundances were found for Lentisphaerae, Cyanobacteria, Actinobacteria, Synergistetes, Verrucomicrobia and Fusobacteria in some of the samples. Archaea were found in the minority of samples and with a low number of different taxa, all known as methanogens. A relatively small number of potentially pathogenic and even zoonotic species were detected. Further studies are needed to better understand these differences, especially taking into account the poor conservation status of many cetacean species and populations worldwide. So far, most of the data available are descriptive and little is known about functions of the different taxa in the intestinal microbiota of marine mammals including whales.
Data Availability
Raw data of paired-end sequence read of bacterial and archaeal 16S rRNA gene amplicon sequences are available at the Sequence Read Archive (SRA) under BioSample accessions number SRR11586677- SRR11586693 within the BioProject PRJNA627228.
Code Availability
All details to applied software including version numbers are given in the text.
Change history
24 March 2021
The original version of this paper was updated to correct the ORCID of Angel Franco.
References
Bik EM, Costello EK, Switzer AD, Callahan BJ, Holmes SP, Wells RS, Carlin KP, Jensen ED, Venn-Watson S, Relman DA (2016) Marine mammals harbor unique microbiotas shaped by and yet distinct from the sea. Nat Commun 7:10516. https://doi.org/10.1038/ncomms10516
Hunt KE, Moore MJ, Rolland RM, Kellar NM, Hall AJ, Kershaw J, Raverty SA, Davis CE, Yeates LC, Fauquier DA, Rowles TK, Kraus SD (2013) Overcoming the challenges of studying conservation physiology in large whales: a review of available methods. Conserv Physiol 1(1):cot006. https://doi.org/10.1093/conphys/cot006
Hermosilla C, Hirzmann J, Silva LMR, Brotons JM, Cerda M, Prenger-Berninghoff E, Ewers C, Taubert A (2018) Occurrence of anthropozoonotic parasitic infections and faecal microbes in free-ranging sperm whales (Physeter macrocephalus) from the Mediterranean sea. Parasitol Res 117(8):2531–2541. https://doi.org/10.1007/s00436-018-5942-3
Hermosilla C, Hirzmann J, Silva LMR, Scheufen S, Prenger-Berninghoff E, Ewers C, Häussermann et al (2018) Gastrointestinal parasites and bacteria in free-living South American sea lions (Otaria flavescens) in Chilean Comau Fjord and new host record of a Diphyllobothrium scoticum-like cestode. Front Mar Sci 5. https://doi.org/10.3389/fmars.2018.00459
Zurano JP, Magalhaes FM, Asato AE, Silva G, Bidau CJ, Mesquita DO, Costa GC (2019) Cetartiodactyla: Updating a time-calibrated molecular phylogeny. Mol Phylogenet Evol 133:256–262. https://doi.org/10.1016/j.ympev.2018.12.015
Marshall CD, Pyenson ND (2019) Feeding in aquatic mammals: an evolutionary and functional approach. In: Bels V, Whishaw IQ (eds) Feeding in vertebrates: evolution, morphology, behavior, biomechanics. Springer International Publishing, Cham, pp 743–785
Goldbogen JA, Madsen PT (2018) The evolution of foraging capacity and gigantism in cetaceans. J Exp Biol 221(Pt 11):jeb166033. https://doi.org/10.1242/jeb.166033
Pauly D, Trites AW, Capuli E, Christensen V (1998) Diet composition and trophic levels of marine mammals. ICES J Mar Sci 55:467–481
Trites AW (2001) Marine mammal trophic levels and interactions. In: Steele JH (ed) Encyclopedia of ocean sciences. Academic Press, Oxford, pp 1628–1633
Moran NA (2006) Symbiosis. Curr Biol 16(20):R866–R871. https://doi.org/10.1016/j.cub.2006.09.019
Spor A, Koren O, Ley R (2011) Unravelling the effects of the environment and host genotype on the gut microbiome. Nat Rev Microbiol 9(4):279–290. https://doi.org/10.1038/nrmicro2540
Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R, Gordon JI (2008) Evolution of mammals and their gut microbes. Science (New York, NY) 320(5883):1647–1651. https://doi.org/10.1126/science.1155725
Ohno H (2015) Impact of commensal microbiota on the host pathophysiology: focusing on immunity and inflammation. Semin Immunopathol 37(1):1–3. https://doi.org/10.1007/s00281-014-0472-2
Sanders JG, Beichman AC, Roman J, Scott JJ, Emerson D, McCarthy JJ, Girguis PR (2015) Baleen whales host a unique gut microbiome with similarities to both carnivores and herbivores. Nat Commun 6:8285. https://doi.org/10.1038/ncomms9285
Soverini M, Quercia S, Biancani B, Furlati S, Turroni S, Biagi E, Consolandi C, Peano C, Severgnini M, Rampelli S, Brigidi P, Candela M (2016) The bottlenose dolphin (Tursiops truncatus) faecal microbiota. FEMS Microbiol Ecol 92(4):fiw055. https://doi.org/10.1093/femsec/fiw055
Erwin PM, Rhodes RG, Kiser KB, Keenan-Bateman TF, McLellan WA, Pabst DA (2017) High diversity and unique composition of gut microbiomes in pygmy (Kogia breviceps) and dwarf (K. sima) sperm whales. Sci Rep 7(1):7205. https://doi.org/10.1038/s41598-017-07425-z
Maron CF, Kohl KD, Chirife A, Di Martino M, Fons MP, Navarro MA, Beingesser J, McAloose D, Uzal FA, Dearing MD, Rowntree VJ, Uhart M (2019) Symbiotic microbes and potential pathogens in the intestine of dead southern right whale (Eubalaena australis) calves. Anaerobe 57:107–114. https://doi.org/10.1016/j.anaerobe.2019.04.003
Guass O, Haapanen LM, Dowd SE, Sirovic A, McLaughlin RW (2016) Analysis of the microbial diversity in faecal material of the endangered blue whale, Balaenoptera musculus. Antonie Van Leeuwenhoek 109(7):1063–1069. https://doi.org/10.1007/s10482-016-0698-1
Miller CA, Holm HC, Horstmann L, George JC, Fredricks HF, Van Mooy BAS, Apprill A (2020) Coordinated transformation of the gut microbiome and lipidome of bowhead whales provides novel insights into digestion. ISME J 14(3):688–701. https://doi.org/10.1038/s41396-019-0549-y
Hermosilla C, Silva LM, Navarro M, Taubert A (2016) Anthropozoonotic endoparasites in free-ranging “urban” South American sea lions (Otaria flavescens). J Vet Med 2016:7507145–7507147. https://doi.org/10.1155/2016/7507145
Schellenberg J, Reichert J, Hardt M, Klingelhöfer I, Morlock G, Schubert P, Bižić M, Grossart H-P, Kämpfer P, Wilke T, Glaeser SP (2020) The bacterial microbiome of the long-term aquarium cultured high-microbial abundance sponge Haliclona cnidata – sustained bioactivity despite community shifts under detrimental conditions. Front Mar Sci 7. https://doi.org/10.3389/fmars.2020.00266
Maeda H, Fujimoto C, Haruki Y, Maeda T, Kokeguchi S, Petelin M, Arai H, Tanimoto I, Nishimura F, Takashiba S (2003) Quantitative real-time PCR using TaqMan and SYBR Green for Actinobacillus actinomycetemcomitans, Porphyromonas gingivalis, Prevotella intermedia, tetQ gene and total bacteria. FEMS Immunol Med Microbiol 39(1):81–86. https://doi.org/10.1016/S0928-8244(03)00224-4
Yu Y, Lee C, Kim J, Hwang S (2005) Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction. Biotechnol Bioeng 89(6):670–679. https://doi.org/10.1002/bit.20347
Cifuentes Y, Glaeser SP, Mvie J, Bartz JO, Muller A, Gutzeit HO, Vilcinskas A, Kämpfer P (2020) The gut and feed residue microbiota changing during the rearing of Hermetia illucens larvae. Antonie Van Leeuwenhoek 113:1323–1344. https://doi.org/10.1007/s10482-020-01443-0
Kolb S, Knief C, Stubner S, Conrad R (2003) Quantitative detection of methanotrophs in soil by novel pmoA-targeted real-time PCR assays. Appl Environ Microbiol 69(5):2423–2429. https://doi.org/10.1128/aem.69.5.2423-2429.2003
Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glockner FO (2013) Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res 41(1):e1. https://doi.org/10.1093/nar/gks808
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glockner FO (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41(Database issue):D590–D596. https://doi.org/10.1093/nar/gks1219
Pruesse E, Peplies J, Glockner FO (2012) SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28(14):1823–1829. https://doi.org/10.1093/bioinformatics/bts252
Li W, Godzik A (2006) Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22(13):1658–1659. https://doi.org/10.1093/bioinformatics/btl158
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL (2009) BLAST+: architecture and applications. BMC Bioinformatics 10:421. https://doi.org/10.1186/1471-2105-10-421
Ondov BD, Bergman NH, Phillippy AM (2011) Interactive metagenomic visualization in a Web browser. BMC Bioinformatics 12:385. https://doi.org/10.1186/1471-2105-12-385
Ionescu D, Siebert C, Polerecky L, Munwes YY, Lott C, Hausler S, Bizic-Ionescu M, Quast C, Peplies J, Glockner FO, Ramette A, Rodiger T, Dittmar T, Oren A, Geyer S, Stark HJ, Sauter M, Licha T, Laronne JB, de Beer D (2012) Microbial and chemical characterization of underwater fresh water springs in the Dead Sea. PLoS One 7(6):e38319. https://doi.org/10.1371/journal.pone.0038319
Hammer Ø, Harper DAT, Ryan PD (2001) PAST: Paleontological statistics software package for education and data analysis. Palaeontol Electron 4(1):1–9
Wan XL, McLaughlin RW, Zheng JS, Hao YJ, Fan F, Tian RM, Wang D (2018) Microbial communities in different regions of the gastrointestinal tract in East Asian finless porpoises (Neophocaena asiaeorientalis sunameri). Sci Rep 8(1):14142. https://doi.org/10.1038/s41598-018-32512-0
Apprill A, Mooney TA, Lyman E, Stimpert AK, Rappe MS (2011) Humpback whales harbour a combination of specific and variable skin bacteria. Environ Microbiol Rep 3(2):223–232. https://doi.org/10.1111/j.1758-2229.2010.00213.x
Söllinger A, Urich T (2019) Methylotrophic methanogens everywhere - physiology and ecology of novel players in global methane cycling. Biochem Soc Trans 47(6):1895–1907. https://doi.org/10.1042/BST20180565
Jin W, Cheng Y, Zhu W (2017) The community structure of Methanomassiliicoccales in the rumen of Chinese goats and its response to a high-grain diet. J Anim Sci Biotechnol 8:47. https://doi.org/10.1186/s40104-017-0178-0
Boone DR, Whitman WB, Koga Y (2001) Order III. Methanosarcinales ord. nov. In: Boone DR, Castenholz RW, Garrity GM (eds) Bergey’s manual of systematic bacteriology, The Archaea and the deeply branching and phototrophic Bacteria, vol 12nd edn. Springer, New York, p 268
Zellner G, Stackebrandt E, Messner P, Tindall BJ, Conway de Macario E, Kneifel H, Sleytr UB, Winter J (1989) Methanocorpusculaceae fam. nov., represented by Methanocorpusculum parvum, Methanocorpusculum sinense spec. nov. and Methanocorpusculum bavaricum spec. nov. Arch Microbiol 151(5):381–390. https://doi.org/10.1007/bf00416595
Miller TL, Wolin MJ (1985) Methanosphaera stadtmaniae gen. nov., sp. nov.: a species that forms methane by reducing methanol with hydrogen. Arch Microbiol 141(2):116–122. https://doi.org/10.1007/bf00423270
Brugerolle G, Müller M (2000) Amitochondriate flagellates. In: Leadbeater BSC, Green JC (eds) The Flagellates. Taylor and Francis, London, pp 166–189
Amin A, Bilic I, Liebhart D, Hess M (2014) Trichomonads in birds--a review. Parasitology 141(6):733–747. https://doi.org/10.1017/s0031182013002096
Maritz JM, Land KM, Carlton JM, Hirt RP (2014) What is the importance of zoonotic trichomonads for human health? Trends Parasitol 30(7):333–341. https://doi.org/10.1016/j.pt.2014.05.005
Hermosilla C, Silva LM, Kleinertz S, Prieto R, Silva MA, Taubert A (2016) Endoparasite survey of free-swimming baleen whales (Balaenoptera musculus, B. physalus, B. borealis) and sperm whales (Physeter macrocephalus) using non/minimally invasive methods. Parasitol Res 115(2):889–896. https://doi.org/10.1007/s00436-015-4835-y
Flores-Cascante L, Gomez-Gutierrez J, Gomez Del Prado-Rosas MDC, Gendron D (2019) Helminth load in feces of free-ranging blue and fin whales from the gulf of California. Acta Parasitol 64(3):625–637. https://doi.org/10.2478/s11686-019-00069-1
de Vos A, Faux CE, Marthick J, Dickinson J, Jarman SN (2018) New determination of prey and parasite species for northern Indian ocean blue whales. Front Mar Sci 5:104. https://doi.org/10.3389/fmars.2018.00104
Li C, Tan X, Bai J, Xu Q, Liu S, Guo W, Yu C, Fan G, Lu Y, Zhang H, Yang H, Chen J, Liu X (2019) A survey of the sperm whale (Physeter catodon) commensal microbiome. PeerJ 7:e7257. https://doi.org/10.7717/peerj.7257
Whitehead H (2003) Sperm whales: social evolution in the ocean. University of Chicago Press, Chicago
Clarke MR, Martins HR, Pascoe P (1993) The diet of sperm whales (Physeter macrocephalus Linnaeus 1758) off the Azores. Philos Trans R Soc Lond Ser B Biol Sci 339(1287):67–82. https://doi.org/10.1098/rstb.1993.0005
Quakenbush LT, Suydam RS, Bryan AL, Lowry LF, Frost KJ, Mahoney BA (2015) Diet of beluga whales, Delphinapterus leucas, in Alaska from stomach contents, March-November. Mar Fish Rev 77:70–84
Wells RS, Scott MD (2009) Common bottlenose dolphin: Tursiops truncatus. In: Perrin WF, Würsig B, Thewissen JGM (eds) Encyclopedia of marine mammals, vol 2nd. Academic Press, London, pp 249–255
Cipriano F (2009) Atlantic white sided dolphin: Lagenorhynchus acutus. In: Perrin WF, Würsig B, Thewissen JGM (eds) Encyclopedia of marine mammals. Academic Press, London, pp 57–58
Medeiros AW, Giongo A, Valdez FP, Blaese de Amorin D, Tavares M, d’Azevedo PA, Franco AC, Frazzon J, Frazzon AP (2016) Characterization of the faecal bacterial community of wild young South American (Arctocephalus australis) and subantarctic fur seals (Arctocephalus tropicalis). FEMS Microbiol Ecol 92(3). https://doi.org/10.1093/femsec/fiw029
Lavery TJ, Roudnew B, Seymour J, Mitchell JG, Jeffries T (2012) High nutrient transport and cycling potential revealed in the microbial metagenome of Australian sea lion (Neophoca cinerea) faeces. PLoS One 7(5):e36478. https://doi.org/10.1371/journal.pone.0036478
Wan X, Ruan R, McLaughlin RW, Hao Y, Zheng J, Wang D (2016) Fecal bacterial composition of the endangered Yangtze finless porpoises living under captive and semi-natural conditions. Curr Microbiol 72(3):306–314. https://doi.org/10.1007/s00284-015-0954-z
Atlas RM (2013) One Health: Its origins and future. In: Mackenzie JS, Jeggo M, Daszak P, Richt JA (eds) One Health: the human-animal-environment interfaces in emerging infectious diseases. The concept and examples of a One Health approach. Springer, Berlin, Heidelberg, pp 1–13
Venn-Watson S, Daniels R, Smith C (2012) Thirty year retrospective evaluation of pneumonia in a bottlenose dolphin Tursiops truncatus population. Dis Aquat Org 99(3):237–242. https://doi.org/10.3354/dao02471
Valderrama Vasquez CA, Macgregor SK, Rowcliffe JM, Jepson PD (2008) Occurrence of a monophasic strain of Salmonella group B isolated from cetaceans in England and Wales between 1990 and 2002. Environ Microbiol 10(9):2462–2468. https://doi.org/10.1111/j.1462-2920.2008.01651.x
Taurisano ND, Butler BP, Stone D, Hariharan H, Fields PJ, Ferguson HW, Haulena M, Cotrell P, Nielsen O, Raverty S (2018) Streptococcus phocae in marine mammals of northeastern Pacific and arctic Canada: a retrospective analysis of 85 postmortem investigations. J Wildl Dis 54(1):101–111. https://doi.org/10.7589/2016-09-208
Santoro M, Iaccarino D, Di Nocera F, Degli Uberti B, Lucibelli MG, Borriello G, De Luca G, D’Amore M, Cerrone A, Galiero G (2019) Molecular detection of Chlamydia abortus in a stranded Mediterranean striped dolphin Stenella coeruleoalba. Dis Aquat Org 132(3):203–208. https://doi.org/10.3354/dao03320
Sanchez-Sarmiento AM, Carvalho VL, Meirelles ACO, Gravena W, Marigo J, Sacristan C, Costa-Silva S, Groch KR, Silva ND, Ferreira Neto JS, Catao-Dias JL (2018) Survey of Brucella spp. and Leptospira spp. antibodies in cetaceans and manatees of the Amazon basin and Atlantic Ocean, Brazil. Dis Aquat Org 132(1):1–11. https://doi.org/10.3354/dao03312
Roe WD, Lenting B, Kokosinska A, Hunter S, Duignan PJ, Gartrell B, Rogers L, Collins DM, de Lisle GW, Gedye K, Price-Carter M (2019) Pathology and molecular epidemiology of Mycobacterium pinnipedii tuberculosis in native New Zealand marine mammals. PLoS One 14(2):e0212363. https://doi.org/10.1371/journal.pone.0212363
Lee K, Kim HK, Park SK, Sohn H, Cho Y, Choi YM, Jeong DG, Kim JH (2018) First report of the occurrence and whole-genome characterization of Edwardsiella tarda in the false killer whale (Pseudorca crassidens). J Vet Med Sci 80(6):1041–1046. https://doi.org/10.1292/jvms.17-0590
Kersulyte D, Rossi M, Berg DE (2013) Sequence divergence and conservation in genomes of Helicobacter cetorum strains from a dolphin and a whale. PLoS One 8(12):e83177. https://doi.org/10.1371/journal.pone.0083177
Clayton LA, Stamper MA, Whitaker BR, Hadfield CA, Simons B, Mankowski JL (2012) Mycobacterium abscessus pneumonia in an Atlantic bottlenose dolphin (Tursiops truncatus). J Zoo Wildl Med 43(4):961–965. https://doi.org/10.1638/2012-0110r.1
Bonar CJ, Wagner RA (2003) A third report of “golf ball disease” in an Amazon river dolphin (Inia geoffrensis) associated with Streptococcus iniae. J Zoo Wildl Med 34(3):296–301. https://doi.org/10.1638/1042-7260(2003)034[0296:Atrogb]2.0.Co;2
Eisenberg T, Kämpfer P, Ewers C, Semmler T, Glaeser SP, Collins E, Ruttledge M, Palmer R (2016) Oceanivirga salmonicida gen. nov., sp. nov., a member of the Leptotrichiaceae isolated from Atlantic salmon (Salmo salar). Int J Syst Evol Microbiol 66(6):2429–2437. https://doi.org/10.1099/ijsem.0.001050
Palmer R, Fleming GTA, Glaeser S, Semmler T, Flamm A, Ewers C, Kämpfer P, Budich O, Berrow S, O’Brien J, Siebert U, Collins E, Ruttledge M, Eisenberg T (2020) Marine mammals are natural hosts of Oceanivirga salmonicida, a bacterial pathogen of Atlantic salmon. Dis Aquat Organ. 139:161–174. https://doi.org/10.3354/dao03478
Volokhov DV, Blom J, Amselle M, Delmonte P, Gao Y, Shen Z, Zhang S, Gulland FM, Chizhikov VE, Eisenberg T (2020) Oceanivirga miroungae sp. nov. isolated from oral cavity of northern elephant seal (Mirounga angustirostris). Int J Syst Evol Microbiol 70(5):3037–3048. https://doi.org/10.1099/ijsem.0.004127
Vendl C, Slavich E, Nelson T, Acevedo-Whitehouse K, Montgomery K, Ferrari B, Thomas T, Rogers T (2020) Does sociality drive diversity and composition of airway microbiota in cetaceans? Environ Microbiol Rep 12:324–333. https://doi.org/10.1111/1758-2229.12835
Acknowledgements
We kindly acknowledge Judy Kolberg who performed DNA extraction and real-time PCR analysis under supervision of SG at the Institute of Applied Microbiology of the Justus-Liebig University Giessen, Germany.
Funding
Open Access funding enabled and organized by Projekt DEAL. RP is supported by FCT (SFRH/BPD/108007/2015) and MAS through SUMMER (H2020-EU.3.2.3.1, GA 817806). We also acknowledge funds provided by FCT to MARE and Okeanos through strategic projects UID/MAR/04292/2019, UIDB/05634/2020, respectively. Data collection in the Azores was funded by FRCT, FCT, FEDER, COMPETE, QREN ESF through TRACE-PTDC/MAR/74071/2006 and MAPCET-M2.1.2/F/012/2011.
Author information
Authors and Affiliations
Contributions
SG, CH, AT, TE designed the microbiological study; RP, MAS collected the whale faecal samples; AF performed the quality check of DNA samples by DGGE; SG performed data analyses; SG, TE interpreted the data and wrote the manuscript; PK, CH, AT financially supported the study; all authors critically checked and contributed to the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Conflict of Interest
The authors declare no competing interests.
Supplementary Information
ESM 1
(DOCX 28824 kb).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Glaeser, S.P., Silva, L.M.R., Prieto, R. et al. A Preliminary Comparison on Faecal Microbiomes of Free-Ranging Large Baleen (Balaenoptera musculus, B. physalus, B. borealis) and Toothed (Physeter macrocephalus) Whales. Microb Ecol 83, 18–33 (2022). https://doi.org/10.1007/s00248-021-01729-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-021-01729-4