
Abstract
Aphids are known to be associated with a variety of symbiotic bacteria. To improve our knowledge of the bacterial diversity of polyphagous aphids, in the present study, we investigated the microbiota of the cosmopolitan agricultural pest Myzus persicae (Sulzer). Ninety-two aphid samples collected from different host plants in various regions of China were examined using high-throughput amplicon sequencing. We comprehensively characterized the symbiont diversity of M. persicae and assessed the variations in aphid-associated symbiont communities. We detected a higher diversity of symbionts than has been previously observed. M. persicae hosted the primary endosymbiont Buchnera aphidicola and seven secondary symbionts, among which Wolbachia was the most prevalent and Rickettsia, Arsenophonus, and Spiroplasma were reported for the first time. Ordination analyses and statistical tests revealed that the symbiont flora associated with M. persicae did not change with respect to host plant or geography, which may be due to frequent migrations between different aphid populations. These findings will advance our knowledge of the microbiota of polyphagous insects and will enrich our understanding of assembly of host-microbiome systems.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bacterial symbionts are widespread in insects, and their symbiotic associations range from obligate mutualism to facultative parasitism [1]. Xylem or phloem sap-feeding insect lineages typically host obligate symbionts that supply the nutrients necessary to supplement their unbalanced sap diets [2]. One typical example of obligate mutualism is the symbiosis between aphids and Buchnera aphidicola. Buchnera is the primary endosymbiont of aphids, resides in bacteriocytes, and can provide essential amino acids and vitamins for its hosts [3, 4]. Because of its strict vertical transmission, Buchnera has undergone long-term coevolution with its aphid hosts [5]. However, due to the metabolic losses caused by rapid genome deterioration in Buchnera [6], some aphid species of the subfamilies Lachninae, Chaitophorinae, and Aphidinae have established co-obligate associations with other symbiotic partners [7, 8].
In addition to the primary endosymbiont, aphids also carry a variety of secondary or facultative symbionts that are not required for aphid survival or reproduction but may confer fitness benefits. Secondary symbionts inhabit bacteriocytes, sheath cells, or hemocoel [9] and are maternally or horizontally transmitted [10]. Nine secondary symbionts have been widely reported in aphids, namely, Serratia symbiotica, Rickettsia, Wolbachia, Hamiltonella defensa, Regiella insecticola, Spiroplasma, Arsenophonus, Fukatsuia symbiotica, and Rickettsiella viridis. Secondary symbionts can provide various ecological benefits to aphids, including conferring resistance to parasitic wasps and fungal pathogens [11, 12], improving tolerance to thermal stress [13], manipulating aphid reproduction [14], affecting host plant utilization [15], and even altering aphid body color [16].
Individual aphid species generally exhibit symbiont variation at the population level. Many studies using diagnostic PCR have observed differences in symbiont composition and prevalence with respect to host plant, geography, and season in a given aphid species [17,18,19,20,21]. However, such PCR-based detection techniques are limited in that they depend on prior knowledge of symbiont diversity. In addition, it is important to elucidate the determinants of symbiont community structure based on a large number of samples that represent different environmental factors. In recent years, high-throughput amplicon sequencing has become an effective method for understanding insect microbial communities and their dynamics. Although high-throughput sequencing techniques have been used by many studies to investigate the bacterial communities of specific aphid groups [22, 23] and community changes within some aphid species [24, 25], the symbiont diversity of most aphid species remains unknown or poorly characterized.
The green peach aphid, Myzus persicae (Sulzer), is a cosmopolitan agricultural pest. M. persicae is heteroecious and holocyclic, alternating between its primary hosts (plants of the genera Amygdalus and Prunus) and more than 40 families of secondary host plants [26]. In the tropics and regions where the primary host is absent, M. persicae can also live parthenogenetically on secondary host plants all the year round. In addition, M. persicae is an important vector of many plant viruses. However, to date, few studies have investigated the bacterial diversity of M. persicae. In a survey study by Henry et al. [27], only three of 50 individual M. persicae were shown to be infected with H. defensa, R. insecticola, or S. symbiotica. Chen et al. [28] observed a high prevalence of Wolbachia in M. persicae from China and revealed geographic variation in its prevalence and infection pattern. Using high-throughput sequencing, Gallo-Franco et al. [29] examined the bacterial communities of M. persicae feeding on the pepper crop Capsicum annuum from two localities of southwestern Colombia and identified that Buchnera was the predominant symbiont, but they did not report any secondary symbionts.
To better understand the microbial profiles of M. persicae and to verify whether this species harbors a low diversity of symbionts as revealed by previous studies, using 16S rRNA amplicon Illumina sequencing, we characterized the bacterial diversity of M. persicae with a broad sampling from different host plants throughout China. The bacterial community dynamics over aphid populations was also evaluated.
Materials and Methods
Sampling and DNA Isolation
Ninety-two samples of M. persicae were collected from 16 families of plants in 30 geographic regions of China between 2002 and 2016 (Fig. 1, Table S1). Specimens preserved in 75% ethanol were used to make voucher specimens, and samples preserved in 100% ethanol at − 20 °C were used for DNA isolation. Species identification of the voucher slides was performed based on morphological characteristics. All samples were deposited in the National Zoological Museum of China, Institute of Zoology, Chinese Academy of Sciences, Beijing, China.

Collection regions and haplotype distribution of Myzus persicae. The haplotypes (H1–H16) are colored and labeled on the map. The abbreviations of geographic regions are provided in Table S2
The whole body of an adult aphid from a sample was used to extract total DNA, before which the aphid was rinsed in 70% ethanol for 5 min and then rinsed four times with sterilized water to eliminate the external microbial contaminants. DNA was isolated using the DNeasy Blood & Tissue Kit (QIAGEN, Germany). A sample of sterilized water was also included in the extraction procedure as a negative extraction control. All DNA extracts were stored at − 20 °C. To verify the aphid species identification, test the DNA quality, and exclude parasitized samples, COI barcodes were amplified using the universal primers LCO1490 and HCO2198 [30].
16S rRNA Amplification and Sequencing
A nearly 420-bp V3–V4 region of 16S rRNA gene was amplified with the primers 338F (5′-ACTCCTACGGGAGGCAGCA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) [31]. Two polymerase chain reaction (PCR) procedures were performed, where the first reaction generated the primary 16S amplicons and the second converted the amplicons into libraries for sequencing. The first-step PCR mixture consisted of 10 μL 5× Q5 Reaction Buffer (New England Biolabs, USA), 0.4 μL Q5 High-Fidelity DNA Polymerase (New England Biolabs), 1 μL dNTPs, 10 μL 5× Q5 High GC Enhancer (New England Biolabs), 1.5 μL forward and reverse primers (10 μM), and 40–60 ng DNA. PCR amplification was performed using the following thermocycling program: a 5-min initial denaturation at 95 °C followed by 15 cycles of 1 min at 95 °C, 1 min at 50 °C, and 1 min at 72 °C, and a final elongation for 7 min at 72 °C. In the second-step PCR, the forward primer (5′-AATGATACGGCGACCACCGAGATCTACAC-NNNNNNNN-ACACTCTTTCCCTACACGACGCTCTTCCGATCT-3′) contained the Illumina i5 adapter, a unique 8-bp barcode sequence for each sample (indicated in N), and read 1 sequencing primer binding sites; the reverse primer (5′-CAAGCAGAAGACGGCATACGAGAT-NNNNNNNN-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3′) included the Illumina i7 adapter, a unique 8-bp barcode sequence for each sample (indicated in N), and read 2 sequencing primer binding sites. The reaction mixture contained 10 μL purified product from the first PCR, 20 μL 2× Phusion High-Fidelity PCR Master Mix (New England Biolabs) and 1 μL forward and reverse primers (10 μM). The reaction was heated for 30 s at 98 °C and then underwent 10 cycles of denaturation for 10 s at 98 °C, annealing for 30 s at 65 °C, and elongation for 30 s at 72 °C, with a final extension for 5 min at 72 °C. Samples of sterilized water were included in all PCRs as negative amplification controls. The PCR products were checked with 1.8% agarose gel electrophoresis. The positive samples were purified and mixed at a mass ratio of 1:1, after which the library was sequenced on an Illumina HiSeq 2500 platform, and 250-bp paired-end reads were generated.
Bioinformatics Analyses
Both operational taxonomic unit (OTU) picking and amplicon sequence variant (ASV) approaches were used in the present study. For OTU clustering, paired-end reads were first merged using FLASH v1.2.11 [32]. The raw reads with an average quality score below 20 in a 50-bp sliding window were trimmed using Trimmomatic v0.33 [33], and reads shorter than 300 bp were removed. Chimeras were filtered by UCHIME v8.1 [34]. The resulting high-quality clean reads were clustered using the QIIME UCLUST module [35], and reads with a similarity ≥ 97% were grouped into an OTU. ASVs were generated using the DADA2 pipeline [36]. The first eight nucleotides of each read were trimmed, and the forward and reverse reads were then truncated at 245 and 200 bp, respectively. We removed low-quality reads with default filtering parameters and merged paired-end reads. Chimeric sequences were filtered using the function removeBimeraDenovo. The OTU representative sequences and ASV sequences were aligned with the SILVA reference database v.128 for taxonomy annotation. Sequences identified as chloroplasts, mitochondria, or eukaryotes, and all OTUs/ASVs with an abundance less than 0.005% were filtered, unless they could be annotated [37]. The resulting OTU and ASV count matrices included 22,032–66,994 and 4919–32,368 sequences per sample, respectively. For both data sets, we rarefied all samples to the median number of sequences per sample prior to downstream analyses.
Statistical Analyses
Based on the OTU data set, Shannon and Simpson indices were calculated with the function diversity of the vegan package [38] in R v3.4.3 [39] to characterize the alpha diversity. Both indices are commonly used in estimating microbial diversity, with the Shannon index giving greater weight to richness, while the Simpson index stressing evenness [40]. The relative abundances of individual bacterial taxa were estimated by total sum scaling normalization with the function decostand in vegan. To assess variations in the symbiont and secondary symbiont communities with respect to distinct factors, we grouped all M. persicae samples according to host-plant family and geographic region (Table S2). First, variations in alpha diversity were examined. Because the alpha diversity data were not normally distributed (Shapiro–Wilk test: P < 0.05), two nonparametric methods Kruskal–Wallis test and Wilcoxon tests were performed to test for differences across all groups and between pairwise groups (n ≥ 3), respectively.
Then, we used the Bray–Curtis distances of relative abundance and presence/absence data to quantify the variations in symbiont and secondary symbiont communities among samples and examined how beta-diversity patterns were related to the host plant and geography. The Bray–Curtis dissimilarities were calculated based on both OTU and ASV data sets with the function vegdist of vegan. We logarithmically transformed the relative abundance data of the symbiont community (function decostand in vegan) to balance the bias caused by the most abundant Buchnera.
We used ordination analyses and statistical tests to detect whether there were changes in the symbiont and secondary symbiont community structures. We conducted unconstrained nonmetric multidimensional scaling (NMDS; function metaMDS of vegan; stress values < 0.05 were considered indicative of excellent representations) and constrained principal coordinate analysis (cPCoA; functions capscale and anova.cca of vegan) to visualize the Bray–Curtis dissimilarity. These two ordination methods are useful for summarizing community changes in relation to environmental factors. We performed analysis of similarities (ANOSIM) and permutational multivariate analysis of variance (PERMANOVA) with the Bray–Curtis dissimilarity to assess differences in the symbiont and secondary symbiont community compositions. The statistical tests were implemented using the functions anosim and adonis in vegan with 999 permutations to test for significance. ANOSIM and PERMANOVA are both widely used in microbiome studies, with the latter typically being more robust [41]. We then examined the relative abundance of each symbiont defined by OTU picking using the nonparametric Kruskal–Wallis test to further identify which symbiont contributed to the community variation. Fisher’s least significant difference (LSD) post hoc tests with a Bonferroni correction were performed for pairwise group comparisons of the relative abundances of specific symbionts. These two tests were applied using the function kruskal in agricolae [42].
Finally, we used Mantel tests with the Pearson correlation coefficient (function mantel in vegan) to examine whether the symbiont and secondary symbiont community changes were associated with spatial distance. The Mantel test determined the amount of correlation between the geographic distance matrix and the Bray–Curtis dissimilarity of the OTU data through a permutation procedure. The distances among sampling sites were calculated with Geographic Distance Matrix Generator v1.2.3 [43].
Results
Bacterial Community Profiling
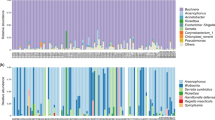
After filtering the noisy raw data, a total of 3,219,756 reads (34,997 reads per sample) were assigned to 1505 OTUs. The bacterial communities of M. persicae were dominated by the phylum Proteobacteria (average relative abundance across all samples: 96.83%), with other phyla accounting for less than 1% (Table S3). At the family level, the most abundant OTUs were associated with Enterobacteriaceae (91.37%). At the genus level, Buchnera was the most represented taxon (90.77%), followed by Rickettsia (1.59%), Acinetobacter (0.98%), and Duganella (0.56%) (Fig. 2, Table S3). The alpha diversity estimates of bacteria associated with all M. persicae samples ranged from 0.080 to 3.185 for the Shannon index and from 0.308 to 0.983 for the Simpson index (Table S4).

Bar plots of microbial communities associated with Myzus persicae across all samples. a Bacterial communities. b Secondary symbiont communities. Each bar shows the relative abundances of bacteria detected in each aphid sample
Core Symbiont Community
Eight common symbionts of aphids were detected in M. persicae, with B. aphidicola detected in each sample at an extremely high relative abundance (90.77%) (Fig. 2a, Table 1). Secondary symbionts were detected within 72 out of 92 samples (Fig. 2b). Single OTUs were identified for Rickettsia, S. symbiotica, R. insecticola, H. defensa, and Spiroplasma. Two and three OTUs of Wolbachia and Arsenophonus were detected, respectively, where each OTU was shared between different aphid samples, and multiple OTUs of Wolbachia or Arsenophonus were detected in individual aphids for a few samples. Wolbachia was the most prevalent secondary symbiont (infection frequency: 53/92), followed by Rickettsia (15/92), Arsenophonus (15/92), S. symbiotica (15/92), and R. insecticola (12/92) (Table 1). H. defensa and Spiroplasma were only detected in four and three samples, respectively.
The abundances of secondary symbionts were low relative to Buchnera, with average relative abundances across all samples far below 1%, except that of Rickettsia (1.59%) (Table 1). This result was attributed to the high abundance of Rickettsia in two aphid samples, M10499 (96.84%) and M22883 (45.89%), feeding on Crepidiastrum sonchifolium (Fig. 2a). This finding was further supported by three biological replicates (i.e., three aphid individuals from one colony) and two PCR replicates (56.14–78.76%) (Fig. S1). The relative abundance of Rickettsia across all samples remained 0.04% when these two samples were not counted. The alpha diversity estimates of symbiont communities within all M. persicae samples ranged from 0.023 to 0.717 for the Shannon index and from 0.498 to 0.995 for the Simpson index. After excluding the primary endosymbiont Buchnera, the alpha diversity estimates of secondary symbiont communities ranged from 0 to 1.468 for the Shannon index and from 0 to 1 for the Simpson index (Table S4).
Variations in Symbiont and Secondary Symbiont Communities
In general, the host plant and geography appeared to have no significant impacts on the symbiont flora of M. persicae. The Kruskal–Wallis and Wilcoxon tests based on the OTU data set revealed no variation in the alpha diversity of the symbiont community in relation to the host plant (Kruskal–Wallis: P = 0.520–0.540; Wilcoxon: P = 0.178–0.904). Differences in alpha diversity were also not significant between geographic regions (Kruskal–Wallis: P = 0.150–0.300; Wilcoxon: P = 0.104–0.907). Similarly, for the secondary symbiont community, no significant differences in the alpha diversity were observed with respect to the host plant [Kruskal–Wallis: P = 0.170–0.410; Wilcoxon: P = 0.141–0.978 (Solanaceae: a higher Shannon index than the mean value across all groups, P = 0.024)] or geography [Kruskal–Wallis: P = 0.140–0.520; Wilcoxon: P = 0.127–0.954 (Xinjiang4: a higher Shannon index than the mean value across all groups, P = 0.029)].
In the NMDS and cPCoA analyses of the Bray–Curtis dissimilarity of the symbiont community, there was no recognizable clustering of samples representing either different host plant families or geographic regions at both the OTU and ASV levels (NMDS: stress = 0.073–0.141 for OTU, ASV data were insufficient for NMDS; cPCoA: P = 0.077–0.680 for OTU, P = 0.180–0.860 for ASV; Fig. S2), except for the ordinations of seven geographic groups with a sample size ≥ 3 (OTU) (cPCoA: 15.4% of variance, P = 0.035; Fig. 3a), three geographic groups on Brassicaceae (OTU/ASV) (cPCoA: 33.1% of variance, P = 0.006 for OTU, Fig. 3b; cPCoA: 47.2% of variance, P = 0.025 for ASV, Fig. 3e), two geographic groups on Asteraceae (OTU) (NMDS: stress = 0; Fig. 3c), and three host plant groups from Beijing (ASV) (cPCoA: 23.1% of variance, P = 0.012; Fig. 3d). In general, ANOSIM and PERMANOVA revealed no significant differences in the symbiont community in response to the host plant or geography (P > 0.05, Table 2). The symbiont compositions were only observed to differ among the three host plant groups from Beijing (ANOSIM: P < 0.05 for ASV; PERMANOVA: P < 0.05 for OTU and ASV; Table 2) and among the three geographic groups on the plants of Brassicaceae (PERMANOVA: P < 0.05 for OTU and ASV; Table 2), which was consistent with the cPCoA results. The Kruskal–Wallis tests conducted on the OTU data set further showed that Buchnera (P = 0.049), Rickettsia (P = 0.037), and Wolbachia (P = 0.027) were present in significantly different relative abundances among these groups. The Asteraceae-feeding aphid populations in Beijing harbored a significantly low abundance of Buchnera (LSD: P < 0.05; Fig. 3f) but a high abundance of Rickettsia (LSD: P < 0.05; Fig. 3g). Within the three geographic aphid groups occurring on Brassicaceae, the relative abundance of Wolbachia in Xinjiang4 was statistically higher than that in Ningxia (LSD: P < 0.05; Fig. 3h).

Variations observed in the symbiont communities associated with Myzus persicae. a–e Constrained principal coordinate analyses (cPCoA) (a, b, d, e) and nonmetric multidimensional scaling (NMDS) (c) of Bray–Curtis distances of symbiont communities at the OTU (a–c) and ASV (d, e) levels. a Seven geographic groups (n ≥ 3). b, e Three geographic groups on Brassicaceae (n ≥ 3). c Two geographic groups on Asteraceae (n ≥ 3). d Three host plant groups from Beijing (n ≥ 3). f–h Comparisons of relative abundances of Buchnera (f), Rickettsia (g), and Wolbachia (h) based on the OTU data. The different letters above boxes indicate significant differences (P < 0.05) following Fisher’s least significant difference (LSD) post hoc tests. Ast, Asteraceae; Bra, Brassicaceae; Ros, Rosaceae; Oth, other plant families; BJ, Beijing; NX, Ningxia; QH1, Qinghai1; SX1, Shaanxi1; XJ2, Xinjiang2; XJ4, Xinjiang4; XJ6, Xinjiang6
When investigating the beta-diversity pattern of the secondary symbiont community, only cPCoA analyses of the OTU data set were performed, as the OTU data were insufficient for NMDS, and the ASV data were insufficient for both ordination analyses. Constrained PCoA plots of the OTU data showed that the secondary symbiont compositions did not change significantly with the host plant or geography (P = 0.170–0.630; Fig. S3). ANOSIM and PERMANOVA performed on both OTU and ASV data sets suggested no impacts of host plant or geography on the secondary symbiont community structure (P > 0.05, Table 2).
Furthermore, at the OTU level, no correlation between geographic distances and Bray–Curtis dissimilarities of symbiont (r = − 0.014, P = 0.589) or secondary symbiont communities (r = 0.009, P = 0.432) was observed in the Mantel tests.
Discussion
In the present study, the microbiota of M. persicae was dominated by relatively few bacteria, which were primarily heritable symbionts. As in most other aphid species [22,23,24,25, 44], B. aphidicola was the predominant symbiont in M. persicae, being present in all samples and showing the highest average relative abundance. This is due to the fact that B. aphidicola is required for aphid development and reproduction [3]; it is strictly vertically transmitted and has undergone long-term parallel evolution with its aphid hosts [5]. Apart from four secondary symbionts that have been reported in previous studies (i.e., H. defensa, R. insecticola, S. symbiotica, and Wolbachia) [27, 28], Rickettsia, Arsenophonus, and Spiroplasma were also detected for the first time in the present study. Gallo-Franco et al. [29] did not report any secondary symbionts in their examined M. persicae samples, which may have been due to improper processing of the data. In their study, many OTUs were only annotated to bacterial families, and OTUs with relative abundances lower than 1% were ignored. However, secondary symbionts may occur at very low titers in their host aphids (e.g., Xu et al. [25]).
It is worth noting that two M. persicae samples on C. sonchifolium (Asteraceae) harbored a high abundance of Rickettsia. In most cases, the relative abundance of Rickettsia in C. sonchifolium-feeding aphid populations was greater than that of Buchnera (Figs. 2a and S1), which also led to the significantly higher relative abundance of Rickettsia in the Asteraceae-feeding populations in Beijing. Sakurai et al. [45] observed a significantly suppressed population density of Buchnera in the presence of Rickettsia in Acyrthosiphon pisum, which may also be the case for the two M. persicae samples occurring on C. sonchifolium assayed in the present study. Łukasik et al. [12] demonstrated that Rickettsia in the pea aphid could confer resistance against entomopathogens. In this study, the high abundance of Rickettsia was host-plant specific, suggesting that Rickettsia may have positive effects on the C. sonchifolium-feeding aphid populations. Further experiments are required to elucidate the potential role of Rickettsia in the adaptation of M. persicae to C. sonchifolium.
Wolbachia was the most prevalent secondary symbiont of M. persicae, a symbiont that is widespread in arthropods [46] but was previously considered to be rare in aphids [47]. Augustinos et al. [48] detected Wolbachia in 8.7% of European aphids and speculated that its occurrence in aphids was underestimated. Some studies have shown that Wolbachia is much more prevalent in aphids than previously believed [23, 44, 49, 50]. The high occurrence of Wolbachia indicates that it may play important roles in aphids. Wolbachia is well known for manipulating the reproduction of its arthropod hosts [51], although its exact role in aphids remains ambiguous. Wolbachia was suggested to be correlated with the prevalence of asexual lineages in some aphid species [49, 52], but this possibility has not been confirmed. De Clerck et al. [53] proposed that Wolbachia is involved in the nutrient production in Pentalonia nigronervosa, although this theory is controversial [54]. Therefore, the effects of Wolbachia in aphids should be investigated further.
S. symbiotica, R. insecticola, and H. defensa were reported to be present in M. persicae with very low prevalence (each symbiont: 1/50) in a study of Henry et al. [27] and were also detected in our samples at low infection frequencies. The relative abundances of S. symbiotica and R. insecticola were extremely low (< 0.02% across infected samples). H. defensa was only detected in four of 92 samples; however, it showed an average relative abundance of 3.45% across infected samples (Table 1). Because H. defensa can confer resistance to parasitoid attack [11], relatively high abundance of H. defensa in infected samples may indicate its importance as a defensive symbiont of aphids. In experimental populations of the pea aphid, the dynamics of H. defensa infection were observed to be related to the pressure from parasitoid wasps [55]. Therefore, low parasitism pressure may account for the low incidence of H. defensa observed in our study.
Arsenophonus and Spiroplasma were also reported here for the first time in the present study. Arsenophonus was harbored by 15 samples at low abundance. Arsenophonus has been identified in many insect groups [56] and in recent years has been shown to be a widespread secondary symbiont in aphids [23, 25, 57] that can enhance aphid performance on specific host plants [58]. However, Arsenophonus was not detected in previous studies of M. persicae [29, 57]. Although Arsenophonus may have truly been absent from these previously examined samples, the possibility that its absence was caused by false negatives in their screenings (diagnostic PCRs and improper data analyses) cannot be discarded. Among all seven secondary symbionts associated with M. persicae, Spiroplasma showed the lowest infection frequency and relative abundance across all samples. Its relative abundance across infected samples was also extremely low (0.006%). To date, Spiroplasma has rarely been detected in aphids [25, 59]. In A. pisum, Spiroplasma was shown to be abundant [60], and it can reduce male production [14] and provide resistance against natural enemies [12, 61]. However, in other aphids, its occurrence may have been overlooked probably due to its low titer. High-throughput sequencing can be helpful for Spiroplasma detection (e.g., Xu et al. [25] and the present study), and quantitative real-time PCR and fluorescence in situ hybridization (FISH) are certainly needed for its further precise detection.
The ordination analyses and statistical tests based on both the OTU and ASV data sets suggested that host plant and geography contributed little to the community structures of symbionts and secondary symbionts in M. persicae. Many studies on polyphagous aphids have revealed a crucial role of host plants in shaping symbiont composition [20, 25, 27]. However, in our present study, the secondary symbiont community did not change with the host plant, and symbiont community variation with respect to the host plant was only observed among the three Beijing groups on different plants (Fig. 3d, Table 2). As mentioned above, the high abundance of Rickettsia in C. sonchifolium-feeding populations contributed to these differences. Some studies have also shown geographical variation in aphid symbiont flora [17, 19, 44]. However, we observed no correlation between the spatial distances of sampling sites and the symbiont or secondary symbiont community structures. We also detected no significant community differences over geographical space, except in the three Brassicaceae-feeding symbiont community groups from different regions, which was supported by both ordination plots and statistical tests at the OTU and ASV levels (Fig. 3b, e, Table 2). Previous studies on the polyphagous Aphis gossypii have reported that geography has an effect on the symbiont communities of aphid populations that colonize a limited number of host plants [19, 25, 44]. However, spatial variation in the two geographic groups occurring on Asteraceae was only observed in the NMDS plot (Fig. 3c), which may be due to the two C. sonchifolium-feeding samples from Beijing, and this variation was not corroborated by statistical tests (Table 2). Therefore, it is also possible that the symbiont variation in Brassicaceae-feeding groups was caused by factors other than geography.
The results of the present study revealed no significant impacts of host plant or geography on the symbiont flora associated with M. persicae. Aphids and their bacterial partners are involved in a dynamic ecosystem. The prevalence and relative abundance of specific symbionts may be related to other ecological factors and selection pressures from the varying environment, such as ant attendance [27] and exposure to natural enemies [55].
M. persicae is a heteroecious holocyclic species, with anholocyclic populations occurring in warmer climates [26]. This species moves seasonally between primary and secondary host plants and migrates among diverse secondary hosts. We investigated the population genetic structure of M. persicae using the COI gene (detailed methods are provided in the Supplementary Methods). Among all 16 haplotypes identified, H1 was the most common haplotype and was shared by different populations across most sampling regions (Fig. 1). Furthermore, no significant isolation by distance (IBD) relationship was detected between population genetic differentiation and geographic distance (r = 0.142, P = 0.067). The widespread sharing of haplotype H1 and the absence of an IBD pattern suggests that M. persicae in China have undergone repeated migrations between populations. Because the host plants of M. persicae are primarily crops and ornamental plants, human activities may have greatly facilitated its dispersal. It appears that the absence of distinct patterns of symbiont community is normal within such a large panmictic population of M. persicae. In addition, secondary symbionts can be horizontally transmitted between aphids during sexual reproduction [62] as well as via the host plant [63] or parasitoids [64]. Same phylotype (OTU) of secondary symbionts was shared by different populations of M. persicae, suggesting that horizontal transmission may have occurred. Frequent migrations between different populations, coupled with intraspecific horizontal transmission of secondary symbionts, may have caused the absence of distinct symbiont community patterns in M. persicae.
Conclusions
Characterizing symbiont communities is fundamental to understanding host-symbiont systems. In the present study, based on a broad sampling, we provide the first comprehensive survey of symbiont diversity within M. persicae, a representative polyphagous aphid species. Heritable bacterial symbionts comprised the major components of the M. persicae microbiome. The symbiont compositions were not significantly altered across natural aphid populations. Specific biological characteristics of M. persicae and human activities may together have contributed to the absence of distinct patterns in symbiont community. Thus, the results of our study highlight the importance of the host and environment in microbiome assemblage.
References
Kikuchi Y (2009) Endosymbiotic bacteria in insects: their diversity and culturability. Microbes Environ 3:195–204. https://doi.org/10.1264/jsme2.me09140s
Baumann P (2005) Biology of bacteriocyte-associated endosymbionts of plant sap-sucking insects. Annu Rev Microbiol 59:155–189. https://doi.org/10.1146/annurev.micro.59.030804.121041
Douglas AE (1998) Nutritional interactions in insect-microbial symbioses: aphids and their symbiotic bacteria Buchnera. AnnuRev Entomol 43:17–37. https://doi.org/10.1146/annurev.ento.43.1.17
Nakabachi A, Ishikawa H (1999) Provision of riboflavin to the host aphid, Acyrthosiphon pisum, by endosymbiotic bacteria, Buchnera. J Insect Physiol 45:1–6. https://doi.org/10.1016/S0022-1910(98)00104-8
Moran NA, Munson MA, Baumann P, Ishikawa H (1993) A molecular clock in endosymbiotic bacteria is calibrated using the insect hosts. Proc R Soc Lond B Biol Sci 253:167–171. https://doi.org/10.1098/rspb.1993.0098
Pérez-Brocal V, Gil R, Ramos S et al (2006) A small microbial genome: the end of a long symbiotic relationship? Science 314:312–313. https://doi.org/10.1126/science.1130441
Manzano-Marín A, Coeur d’acier A, Clamens AL, Orvain C, Cruaud C, Barbe V, Jousselin E (2020) Serial horizontal transfer of vitamin-biosynthetic genes enables the establishment of new nutritional symbionts in aphids’ di-symbiotic systems. ISME J 14:259–273. https://doi.org/10.1038/s41396-019-0533-6
Monnin D, Jackson R, Kiers ET, Bunker M, Ellers J, Henry LM (2020) Parallel evolution in the integration of a co-obligate aphid symbiosis. Curr Biol 30:1949–1957. https://doi.org/10.1016/j.cub.2020.03.011
Fukatsu T, Nikoh N, Kawai R, Koga R (2000) The secondary endosymbiotic bacterium of the pea aphid Acyrthosiphon pisum (Insecta: Homoptera). Appl Environ Microbiol 66:2748–2758. https://doi.org/10.1128/aem.66.7.2748-2758.2000
Sandström JP, Russell JA, White JP, Moran NA (2001) Independent origins and horizontal transfer of bacterial symbionts of aphids. Mol Ecol 10:217–228. https://doi.org/10.1046/j.1365-294X.2001.01189.x
Oliver KM, Russell JA, Moran NA, Hunter MS (2003) Facultative bacterial symbionts in aphids confer resistance to parasitic wasps. Proc Natl Acad Sci U S A 100:1803–1807. https://doi.org/10.1073/pnas.0335320100
Łukasik P, van Asch M, Guo H, Ferrari J, Godfray HCJ (2013) Unrelated facultative endosymbionts protect aphids against a fungal pathogen. Ecol Lett 16:214–218. https://doi.org/10.1111/ele.12031
Montllor CB, Maxmen A, Purcell AH (2002) Facultative bacterial endosymbionts benefit pea aphids Acyrthosiphon pisum under heat stress. Ecol Entomol 27:189–195. https://doi.org/10.1046/j.1365-2311.2002.00393.x
Simon JC, Boutin S, Tsuchida T, Koga R, le Gallic JF, Frantz A, Outreman Y, Fukatsu T (2011) Facultative symbiont infections affect aphid reproduction. PLoS One 6:e21831. https://doi.org/10.1371/journal.pone.0021831
Tsuchida T, Koga R, Fukatsu T (2004) Host plant specialization governed by facultative symbiont. Science 303:1989. https://doi.org/10.1126/science.1094611
Tsuchida T, Koga R, Horikawa M, Tsunoda T, Maoka T, Matsumoto S, Simon JC, Fukatsu T (2010) Symbiotic bacterium modifies aphid body color. Science 330:1102–1104. https://doi.org/10.1126/science.1195463
Tsuchida T, Koga R, Shibao H, Matsumoto T, Fukatsu T (2002) Diversity and geographic distribution of secondary endosymbiotic bacteria in natural populations of the pea aphid, Acyrthosiphon pisum. Mol Ecol 11:2123–2135. https://doi.org/10.1046/j.1365-294X.2002.01606.x
Simon JC, Carré S, Boutin M, Prunier-Leterme N, Sabater-Muñoz B, Latorre A, Bournoville R (2003) Host-based divergence in populations of the pea aphid: insights from nuclear markers and the prevalence of facultative symbionts. Proc R Soc Lond B Biol Sci 270:1703–1712. https://doi.org/10.1098/rspb.2003.2430
Najar-Rodríguez AJ, McGraw EA, Mensah RK, Pittman GW, Walter GH (2009) The microbial flora of Aphis gossypii: patterns across host plants and geographical space. J Invertebr Pathol 100:123–126. https://doi.org/10.1016/j.jip.2008.10.005
Ferrari J, West JA, Via S, Godfray HCJ (2012) Population genetic structure and secondary symbionts in host-associated populations of the pea aphid complex. Evolution 66:375–390. https://doi.org/10.1111/j.1558-5646.2011.01436.x
Liu XD, Lei HX, Chen FF (2019) Infection pattern and negative effects of a facultative endosymbiont on its insect host are environment-dependent. Sci Rep 9:4013. https://doi.org/10.1038/s41598-019-40607-5
Jousselin E, Clamens AL, Galan M, Bernard M, Maman S, Gschloessl B, Duport G, Meseguer AS, Calevro F, Coeur d'acier A (2016) Assessment of a 16S rRNA amplicon Illumina sequencing procedure for studying the microbiome of a symbiont-rich aphid genus. Mol Ecol Resour 16:628–640. https://doi.org/10.1111/1755-0998.12478
Xu TT, Chen J, Jiang LY, Qiao GX (2020) Diversity of bacteria associated with Hormaphidinae aphids (Hemiptera: Aphididae). Insect Sci. https://doi.org/10.1111/1744-7917.12746
Russell JA, Weldon S, Smith AH, Kim KL, Hu Y, Łukasik P, Doll S, Anastopoulos I, Novin M, Oliver KM (2013) Uncovering symbiont-driven genetic diversity across North American pea aphids. Mol Ecol 22:2045–2059. https://doi.org/10.1111/mec.12211
Xu S, Jiang L, Qiao G, Chen J (2020) The bacterial flora associated with the polyphagous aphid Aphis gossypii Glover (Hemiptera: Aphididae) is strongly affected by host plants. Microb Ecol 79:971–984. https://doi.org/10.1007/s00248-019-01435-2
Blackman RL, Eastop VF (2020) Aphids on the world’s plants. http://www.aphidsonworldsplants.info/. Accessed 10 Jan 2020
Henry LM, Maiden MCJ, Ferrari J, Godfray HCJ (2015) Insect life history and the evolution of bacterial mutualism. Ecol Lett 18:516–525. https://doi.org/10.1111/ele.12425
Chen R, Su X, Chen J, Jiang L, Qiao GX (2019) Wolbachia infection in two species: novel views on the colonization ability of Wolbachia in aphids. Environ Entomol 48:1388–1393. https://doi.org/10.1093/ee/nvz122
Gallo-Franco JJ, Duque-Gamboa DN, Toro-Perea N (2019) Bacterial communities of Aphis gossypii and Myzus persicae (Hemiptera: Aphididae) from pepper crops (Capsicum sp.). Sci Rep 9:5766. https://doi.org/10.1038/s41598-019-42232-8
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R (1994) DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol 3:294–299
Yu Y, Lee C, Kim J, Hwang S (2005) Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction. Biotechnol Bioeng 89:670–679. https://doi.org/10.1002/bit.20347
Magoč T, Salzberg SL (2011) FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27:2957–2963. https://doi.org/10.1093/bioinformatics/btr507
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200. https://doi.org/10.1093/bioinformatics/btr381
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. https://doi.org/10.1093/bioinformatics/btq461
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP (2016) DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. https://doi.org/10.1038/nmeth.3869
Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R, Mills DA, Caporaso JG (2013) Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods 10:57–59. https://doi.org/10.1038/nmeth.2276
Oksanen J, Blanchet FG, Friendly M et al (2018) Package ‘vegan’, community ecology package. Version 2.5-2. https://github.com/vegandevs/vegan. Accessed 24 Aug 2019
R Core Team (2017) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/. Accessed 2 Aug 2019
Nagendra H (2002) Opposite trends in response for the Shannon and Simpson indices of landscape diversity. Appl Geogr 22:175–186. https://doi.org/10.1016/S0143-6228(02)00002-4
Anderson MJ, Walsh DCI (2013) PERMANOVA, ANOSIM, and the Mantel test in the face of heterogeneous dispersions: What null hypothesis are you testing? Ecol Monogr 83:557–574. https://doi.org/10.1890/12-2010.1
De Mendiburu F (2014) Agricolae: statistical procedures for agricultural research. R package version 1.3-3. https://CRAN.R-project.org/package=agricolae. Accessed 24 Aug 2019
Ersts PJ (2020) Geographic Distance Matrix Generator v1.2.3. American Museum of Natural History, Center for Biodiversity and Conservation http://biodiversityinformatics.amnh.org/open_source/gdmg. Accessed 13 Jan 2020
Jones RT, Bressan A, Greenwell AM, Fierer N (2011) Bacterial communities of two parthenogenetic aphid species cocolonizing two host plants across the Hawaiian Islands. Appl Environ Microbiol 77:8345–8349. https://doi.org/10.1128/AEM.05974-11
Sakurai M, Koga R, Tsuchida T, Meng XY, Fukatsu T (2005) Rickettsia symbiont in the pea aphid Acyrthosiphon pisum: novel cellular tropism, effect on host fitness, and interaction with the essential symbiont Buchnera. Appl Environ Microbiol 71:4069–4075. https://doi.org/10.1128/AEM.71.7.4069-4075.2005
Jeyaprakash A, Hoy MA (2000) Long PCR improves Wolbachia DNA amplification: wsp sequences found in 76% of sixty-three arthropod species. Insect Mol Biol 9:393–405. https://doi.org/10.1046/j.1365-2583.2000.00203.x
Oliver KM, Degnan PH, Burke GR, Moran NA (2010) Facultative symbionts in aphids and the horizontal transfer of ecologically important traits. Annu Rev Entomol 55:247–266. https://doi.org/10.1146/annurev-ento-112408-085305
Augustinos AA, Santos-Garcia D, Dionyssopoulou E, Moreira M, Papapanagiotou A, Scarvelakis M, Doudoumis V, Ramos S, Aguiar AF, Borges PAV, Khadem M, Latorre A, Tsiamis G, Bourtzis K (2011) Detection and characterization of Wolbachia infections in natural populations of aphids: Is the hidden diversity fully unraveled? PLoS One 6:e28695. https://doi.org/10.1371/journal.pone.0028695
De Clerck C, Tsuchida T, Massart S, Lepoivre P, Francis F, Jijakli MH (2014) Combination of genomic and proteomic approaches to characterize the symbiotic population of the banana aphid (Hemiptera: Aphididae). Environ Entomol 43:29–36. https://doi.org/10.1603/EN13107
Wang Z, Su XM, Wen J, Jiang LY, Qiao GX (2014) Widespread infection and diverse infection patterns of Wolbachia in Chinese aphids. Insect Sci 21:313–325. https://doi.org/10.1111/1744-7917.12102
Werren JH, Baldo L, Clark ME (2008) Wolbachia: master manipulators of invertebrate biology. Nat Rev Microbiol 6:741–751. https://doi.org/10.1038/nrmicro1969
Gómez-Valero L, Soriano-Navarro M, Pérez-Brocal V, Heddi A, Moya A, García-Verdugo JM, Latorre A (2004) Coexistence of Wolbachia with Buchnera aphidicola and a secondary symbiont in the aphid Cinara cedri. J Bacteriol 186:6626–6633. https://doi.org/10.1128/JB.186.19.6626-6633.2004
De Clerck C, Fujiwara A, Joncour P et al (2015) A metagenomic approach from aphid’s hemolymph sheds light on the potential roles of co-existing endosymbionts. Microbiome 3:63. https://doi.org/10.1186/s40168-015-0130-5
Manzano-Marín A (2019) No evidence for Wolbachia as a nutritional co-obligate endosymbiont in the aphid Pentalonia nigronervosa. bioRxiv. https://doi.org/10.1101/609511
Oliver KM, Campos J, Moran NA, Hunter MS (2008) Population dynamics of defensive symbionts in aphids. Proc R Soc Lond B Biol Sci 275:293–299. https://doi.org/10.1098/rspb.2007.1192
Nováková E, Hypša V, Moran NA (2009) Arsenophonus, an emerging clade of intracellular symbionts with a broad host distribution. BMC Microbiol 9:143. https://doi.org/10.1186/1471-2180-9-143
Jousselin E, Coeur d’acier A, Vanlerberghe-Masutti F, Duron O (2013) Evolution and diversity of Arsenophonus endosymbionts in aphids. Mol Ecol 22:260–270. https://doi.org/10.1111/mec.12092
Wagner SM, Martinez AJ, Ruan YM, Kim KL, Lenhart PA, Dehnel AC, Oliver KM, White JA (2015) Facultative endosymbionts mediate dietary breadth in a polyphagous herbivore. Funct Ecol 29:1402–1410. https://doi.org/10.1111/1365-2435.12459
Guidolin AS, Cônsoli FL (2018) Diversity of the most commonly reported facultative symbionts in two closely-related aphids with different host ranges. Neotrop Entomol 47:440–446. https://doi.org/10.1007/s13744-017-0532-0
Gauthier JP, Outreman Y, Mieuzet L, Simon JC (2015) Bacterial communities associated with host-adapted populations of pea aphids revealed by deep sequencing of 16S ribosomal DNA. PLoS One 10:e0120664. https://doi.org/10.1371/journal.pone.0120664
Frago E, Mala M, Weldegergis BT, Yang C, McLean A, Godfray HCJ, Gols R, Dicke M (2017) Symbionts protect aphids from parasitic wasps by attenuating herbivore-induced plant volatiles. Nat Commun 8:1860. https://doi.org/10.1038/s41467-017-01935-0
Moran NA, Dunbar HE (2006) Sexual acquisition of beneficial symbionts in aphids. Proc Natl Acad Sci U S A 103:12803–12806. https://doi.org/10.1073/pnas.0605772103
Pons I, Renoz F, Noël C, Hance T (2019) Circulation of the cultivable symbiont Serratia symbiotica in aphids is mediated by plants. Front Microbiol 10:764. https://doi.org/10.3389/fmicb.2019.00764
Gehrer L, Vorburger C (2012) Parasitoids as vectors of facultative bacterial endosymbionts in aphids. Biol Lett 8:613–615. https://doi.org/10.1098/rsbl.2012.0144
Acknowledgments
Thanks to all collectors for sample collections. Thanks to Fendi Yang for making voucher slides.
Funding
The work was supported by the National Key R & D Program of China (No. 2016YFE0203100), the National Natural Science Foundation of China (Nos. 31620103916, 31772492), the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDA19050303), and the Youth Innovation Promotion Association of Chinese Academy of Sciences (No. 2020087).
Author information
Authors and Affiliations
Contributions
J.C. and G.Q. designed the project. G.Q. and L.J. identified voucher specimens. S.X. conducted molecular experiments and all analyses. J.C. assisted in data analyses. S.X. and J.C. wrote the manuscript and all authors contributed to revisions.
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Data Accessibility
All COI sequences were deposited in GenBank under accession numbers MT127004–MT127095. The raw data obtained from Illumina sequencing were deposited in the NCBI Sequence Read Archive (SRA) database under BioProject accession number PRJNA602138.
Electronic supplementary material
ESM 1
(DOCX 6620 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, S., Jiang, L., Qiao, G. et al. Diversity of bacterial symbionts associated with Myzus persicae (Sulzer) (Hemiptera: Aphididae: Aphidinae) revealed by 16S rRNA Illumina sequencing. Microb Ecol 81, 784–794 (2021). https://doi.org/10.1007/s00248-020-01622-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-020-01622-6