
Abstract
Kumbh Mela is one of the largest religious mass gathering events (MGE) involving bathing in rivers. The exponential rise in the number of devotees, from around 0.4 million in 1903 to 120 million in 2013, bathing in small specified sites can have a dramatic impact on the river ecosystem. Here, we present the spatiotemporal profiling of bacterial communities in Godavari River, Nashik, India, comprising five sites during the Kumbh Mela, held in 2015. Assessment of environmental parameters indicated deterioration of water quality. Targeted amplicon sequencing demonstrates approximately 37.5% loss in microbial diversity because of anthropogenic activity during MGE. A significant decrease in phyla viz. Actinobacteria, Chloroflexi, Proteobacteria, and Bacteroidetes was observed, while we noted substantial increase in prevalence of the phylum Firmicutes (94.6%) during MGE. qPCR estimations suggested nearly 130-fold increase in bacterial load during the event. Bayesian mixing model accounted the source of enormous incorporation of bacterial load of human origin. Further, metagenomic imputations depicted increase in virulence and antibiotic resistance genes during the MGE. These observations suggest the striking impact of the mass bathing on river ecosystem. The subsequent increase in infectious diseases and drug-resistant microbes pose a critical public health concern.
Similar content being viewed by others

Introduction
The Kumbh Mela is considered to be the world’s largest periodic religious mass gathering event (MGE) with as many as 120 million pilgrims recorded during the 2013 event at Allahabad, India [1,2,3,4]. It involves ritual bathing in the river on certain days, at one of four Indian cities in rotation every 3 years (Allahabad, Ujjain, Haridwar, and Nashik). The event is hosted by each city every 12 years based on the pre-determined zodiac combinations [5]. There has been an exponential rise in the number of pilgrims attending Kumbh Mela, from around 0.4 million in 1903 to 120 million in 2013. Such massive participation imposes substantial difficulties for the accommodation and flow of people. Thus, a temporary “pop-up city” is created, which is generally built in the flood plains of the rivers, that demands houses with proper electricity, sanitation, and healthcare services [3, 6]. Unfortunately, these efforts and their implications are largely undefined. However, lately, there has been a growing interest to document the concurrent events underlying the planning and management of world’s largest human gathering using multidisciplinary approach to minimize the impact of such events on public health. [4,5,6,7].
Kumbh Mela represents a periodic recreational use of water with religious paradigm, involving bathing in small specified sites that might drastically alter the biogeochemical balance of the river ecosystem [4, 8, 9]. Bacterial communities are sensitive to dynamic changes in their environments that could result in high spatiotemporal variations [10]. Bacteria are the key components of freshwater ecosystems and play a crucial role in primary production and nutrient cycling. However, the role of these bacterial communities in recreational waters is relatively unidentified. Hlavsa et al. [11] reported the relationship between various bacterial species such as Escherichia coli O157:H7, Legionella, Pseudomonas, and Shigella with waterborne illnesses in recreational freshwaters. Similarly, a recent study on recreational practices reported increased risk of waterborne infections during sea bathing [12]. Previous studies have also found dominance of unusual microorganisms under urban-impacted water bodies, which could thus elevate the risk to human health [13,14,15]. Therefore, perturbations of the microbial communities because of biotic and abiotic alterations under recreational usage could possibly have serious consequences on both the environment and public health. This could involve loss of autochthonous communities and/or elevated risk of gastrointestinal disorders and skin infections [4, 8, 16]. The magnitude of such waterborne infections is relative and might have long-term impact on human health.
The mechanisms underlying waterborne infections in response to the large recreational water use during MGE are largely unknown and have so far only been studied by conventional culture-based estimation of fecal indicator bacteria (FIB). The limitations associated with conventional techniques cannot be overlooked, and researchers have opted extensive investigation of bacterial communities using culture-independent methods, such as fluorescence in situ hybridization (FISH) [17, 18] and quantitative polymerase chain reaction (qPCR)-based detection [19]. Recent studies using next-generation sequencing (NGS) have already proved their efficiency in providing in-depth knowledge of microbial communities [20, 21]. Despite these technical advancements, understanding of complex microbial community dynamics in recreational freshwaters and its effects on ecological productivity and public health represents a major bottleneck. Thus, characterization of bacterial community structure under spatiotemporal variations would serve as a proxy to delineate the impact of MGE on freshwater microbial assemblage.
The current study attempts to elucidate alterations in bacterial community structure by deep sequencing and evaluates relationships among environmental factors, bacterial community composition, spread of infections, and drug resistance in the ecosystem. In this regard, we examined (1) the changes in bacterial communities of the Godavari River before and during the MGE and (2) the association between the environmental parameters and the river bacterial assemblage and (3) traced the source of invasive communities to the river ecosystem.
Materials and Methods
Sample Collection
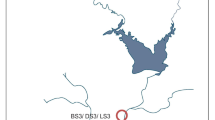
The Nashik Kumbh Mela event takes places on the bank of the Godavari River, the largest river of southern India which originates from “Brahmagiri” mountain ranges (located near Nashik, India) and travels approximately 1465 km southeast, before draining into the Bay of Bengal. The geographical location of sampling sites can be categorized by its altitudinal gradient. Thus, the earlier two sites located at higher altitude (mean 827.5 m a.s.l.) were named upstream (Gangadwar, Kushawart) while later three sites located at relatively lower altitude (mean 567 m a.s.l.) were named bathing sites (Anandvalli, Gharpure ghat, and Tapovan) (Fig. 1). The two upstream sites are situated in the mountain ranges and are relatively remote and in close vicinity of “Anjaneri” reserve forest which exhibits less civilization. Human activities at upstream sites may increase during the Kumbh Mela which is otherwise occasional. In contrast, the bathing sites are located within the Nashik city and are more prone to anthropogenic drivers during the MGE. Additionally, all the event sites have extended river banks which are built from concrete to accommodate the large number of pilgrims. Water samples were collected in triplicate from all the event sites immediately before (July 2015) and during (September 2015) the Kumbh Mela (hereafter, they will be referred to as before MGE and during MGE samples). Samples collected before the event are named as BS1, BS2, BS3, BS4, and BS5 while samples collected during the event are DS1, DS2, DS3, DS4, and DS5, and correspond to locations Gangadwar, Kushawart, Anandwalli, Gharpure ghat, and Tapovan, respectively.

Map showing transect of Godavari River, Nashik, India, hosting mass bathing event (the Kumbh Mela). Points indicate locations used for sampling during this study. BS and DS represent before and during MGE samples. Samples were collected from two upstream sites Gangadwar (BS1/DS1) and Kushawart (BS2/DS2) and three bathing sites Anandvalli (BS3/DS3), Gharpure ghat (BS4/DS4), and Tapovan (BS5/DS5)
Water samples were stored in sterile (gamma-irradiated) containers and transported immediately to the laboratory at 4 °C. Upon arrival, 100 ml of water samples from each site was filtered through 0.22-μ polyethersulfone filters (Merck Millipore, USA) and the filters were stored at − 80 °C for further processing. Physicochemical parameters such as pH (Pocket digital pH meter, Milwaukee pH 600) and temperature (Vintage USA E8000) were measured at the time of sampling. Simultaneously, each sample aliquot was processed to determine the turbidity (OD), total dissolved solid (TDS), total solids (TS), total suspended solids (TSS), dissolved oxygen (DO), biological oxygen demand (BOD), chemical oxygen demand (COD), fecal coliforms, and MPN of total coliforms at Department of Microbiology, KTHM College, Nashik, by following the protocols described by APHA (1998).
DNA Extraction and Amplicon Sequencing of Bacterial 16S rRNA Gene
DNA extraction was performed on the stored 0.22-μ polyethersulfone filter papers (Merck, Millipore, USA) using Power Water DNA extraction kit (Mobio, Carlsbad, CA, USA) according to the manufacturer’s instructions. The quality of DNA was assessed as described previously [22, 23] and stored at − 20 °C until further processing. The DNA was subject to amplification of V4 hypervariable region of 16S rRNA gene using forward primer 515F (5’-GTGCCAGCMGCCGCGGTAA-3′) and reverse primer 806R (5’-GGACTACHVGGGTWTCTAAT-3′) [24]. Template and library preparation was carried out according to the manufacturer’s protocol (Illumina, USA). 16S rRNA amplicons libraries were sequenced on Illumina MiSeq platform using paired end 2 × 250 bp v4 chemistry. The sequences obtained were submitted to the NCBI Sequence Read Archive (SRA) and can be accessed under Bio Project ID PRJNA383664.
Amplicon Sequence Analysis
The raw paired end reads obtained from Illumina MiSeq platform were assembled using FLASH tool (Fast Length Adjustment of Short reads) described by Magočand Salzberg [25]. The resultant raw paired reads were processed in MOTHUR v1.32 [26], to remove the adaptor sequences, ambiguous reads, and low-quality sequences with an average quality score below 30. The quality filtered amplicon reads from all the samples were pooled into a single FASTA file for further analysis using QIIME v1.8 (quantitative insights into microbial ecology) [27]. Closed reference-based OTU picking approach against the Greengenes reference database [28] was adapted to cluster reads into operational taxonomic units (OTUs) by using UCLUST algorithm with 97% similarity threshold [29]. Taxonomic assignments of OTUs were performed using RDP naïve Bayesian classifier with Greengenes reference database [30]. The reads were rarefied to the least number of reads per sample (i.e., 105,246 reads in sample BS4) before performing further analysis. Alpha diversity estimation such as OTU richness, Shannon, Chao1, and Good’s coverage associated with upstream and bathing sites was calculated using QIIME v1.8 based on sampling groups. The beta diversity analysis was performed using Bray-Curtis distances to microbial communities across the MGE.
Quantitative PCR Analysis
The total abundance of bacterial communities before and during the MGE was determined by absolute quantification of 16S rRNA gene copy number. The equimolar (50 ng/μl) template was subjected to qPCR amplification (in triplicate) with 341F (5’-CCTACGGGAGGCAGCAG-3′) and 518R (5’-ATTACCGCGGCTGCTGG-3′) in a 10 μl reaction of Power SYBR Green PCR Master Mix using 7300 Real time PCR system (Applied Biosystems Inc., USA). The qPCR assays involved initial denaturation at 95 °C for 10 min followed by 40 cycles with 95 °C for 15 s and 60 °C for 1 min. The standard curve was plotted by calculating the mean of tenfold dilutions of known concentration of PCR product which was later used to determine the 16S rRNA gene copy number [31]. The standard curve accounting for qPCR efficiency of > 90% and correlation coefficient of > 0.99 were considered as a threshold while calculating the copy number. We report all resultant data as copy number per liter of water sample.
Statistical Analysis
Statistical environment R (R Development Core Team, 2013), and R packages vegan, PhyloSeq, ggbiplot, ggplot2, corrplot, and pysch were used to carry out multivariate and statistical analysis at different taxonomic levels for the upstream and bathing samples 2013 [32]. Determinants of physicochemical factors from the water assessment were statistically validated and correlated with bacterial diversity by performing regression and principal component analysis to analyze the influence and the contribution of each environmental factor to the bacterial community structure. We also computed spearman correlation matrix at family level to dissect the complex microbial associations. Further, a function of PhyloSeq package was used to graphically visualize nonparametric alpha diversity parameter like OTU richness, Shannon, and Chao1, and further, it was used to perform nonmetric multidimensional scaling (NMDS) plots by calculating Bray-Curtis distances from the OTU table to illustrate differences between groups based on the overall bacterial community composition.
Source Tracker Analysis
Source Tracker is a computational model that uses a Bayesian approach to identify possible sources and proportions of bacterial contamination [33]. The model was applied to identify the potential source of microorganisms of human origin, into the river ecosystem during the MGE. The microbiome (human skin, stool and oral) data of Indian sub-population (unpublished study) was acquired and used as potential source of microbial communities and the data of MGE (before and during) was used as a sink.
Imputation of Microbial Functional Profiles
PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States) was used to categorize and compare the functional changes in the bacterial communities from the collection sites [34]. It involves the normalization of 16S rRNA gene copy number followed by prediction of the metagenome. The derived data-set was analyzed at KEGG (Kyoto Encyclopaedia of Genes and Genomes) level (L1 to L3). The LEfSe was performed to distinguish the functional elements of microbial communities before and during the MGE [35]. Furthermore, metagenomic contribution by each taxon was quantified by using normalized dataset.
Results
Assessment of Physico-chemical Parameters of the River Water
A significant increase in turbidity and optical density was observed in water samples during MGE (paired t test t = 4.41, p = 0.01). The elevated turbidity could be the consequence of rise in total dissolved solids, total suspended solids, and total solids (paired t test, p < 0.05) which reflects the introduction of inorganic and organic particles during MGE (supplementary Table 1). However, pH and temperature of the water (paired t test t = 0.10, p = 0.92 and t = 0.92, p = 0.4, respectively) do not differ significantly over the course of MGE.
The biological and chemical oxygen demand (paired t test t = 3.42, p = 0.02 and t = 3.67, p = 0.02, respectively) increased during the MGE probably due to higher release of organic particles. The notion is further supported by substantial reduction in the concentration of dissolved oxygen (DO) (paired t test t = 10.44, p = 0.0005) during the MGE. The substantial decrease in DO while increase BOD and COD could be linked with increased bacterial load in the river. The qPCR-based estimation of absolute 16S rRNA gene copy number also showed approximately 130-fold increased bacterial abundance during the MGE. In addition, MPN and estimation of fecal coliforms increased nearly 157- and 3-fold, respectively, during the MGE when compared with before MGE.
Bacterial Diversity
High-throughput sequencing yielded ~5.3 million raw reads which were processed for quality trimming and an average of 490,863 (accounts for ~91.2%) high-quality sequences per sample were retained for subsequent analysis. Clustering of the sequences with reference database resulted in 6529 OTUs. The average Good’s coverage for all the samples was found to be 99.31 ± 0.30% (mean ± SD) which indicates that the majority of bacterial diversity was captured.
The assessment of alpha diversity indices revealed significant differences at bathing sites before and during the MGE (Fig. 2) whereas no significant differences were noted at upstream sites. The estimates of bacterial species richness and observed OTUs were found to decrease at bathing sites during MGE. Significant variation across MGE was also noticed in non-parametric Shannon index (paired t test t = 3.04, p < 0.05) suggesting dominance of fewer taxa during MGE. Interestingly, among all samples, BS5 featured highest values for species richness (4207.89) and Shannon index (7.17) followed by BS4 and BS3 indicating higher species richness in bathing site samples before MGE. Species richness was found to decrease significantly in bathing site samples during MGE. Shannon evenness exhibits order of DS4 < DS5 < DS3 < BS3 < BS4 < BS5 which indicates diverse microbial community pattern between the samples from bathing site. (Table 1).

Elements of alpha diversity. Variations in alpha diversity indices among upstream and bathing sites of the event. Shapes denotes upstream (triangular) and bathing site (circle) while color code shows the two time points, i.e., before (red) and during (blue) the event
Linear regression analysis was applied to explore the relationship of species richness and environmental factors during MGE (Suppl. Fig. 1). The environmental factors such as optical density of water, TDS, TS, BOD, and COD estimates were higher during the event and showed inverse relationship with diversity (r = − 0.787, r = − 0.796, r = − 0.793, r = − 0.697, r = − 0.708, p< 0.05), except the dissolved oxygen (r = 0.70, p = 0.024). Therefore, it can be noted that physicochemical parameters render negative correlation with respect to event and further support the findings of the non-parametric alpha diversity estimation.
Bacterial Community Structure
A total of 25 bacterial phyla were detected of which the most abundant were Firmicutes, Proteobacteria, Bacteroidetes, Verrucomicrobia, Actinobacteria, Planctomycetes, and Cyanobacteria contributing to 98.97% of the total bacterial diversity before MGE whereas Firmicutes, Proteobacteria, and Bacteroidetes dominated during MGE and accounted for 98.49% of diversity (Fig. 3a). Bacteria belonging to phylum Firmicutes were ubiquitous and observed in higher abundance across all the bathing sties during MGE (DS4 98.88%, DS3 93.28%, and DS5 91.84%) as compared to the bathing sites before MGE (BS3 44.93%, BS5 40.95%, and BS4 36% respectively). In contrast, members of the Proteobacteria dominated bathing sites of before MGE samples (mean 33.67 ± 6.97%) over bathing sites of during MGE (mean 3.89 ± 3.79%). Likewise, Bacteroidetes were more abundant with mean of 9.79 ± 4.36% at the bathing sites before MGE and during MGE the mean of 0.87 ± 1.49% was noted (Suppl. Fig. 2). Bacterial taxa such as Chlorobi, Gemmatimonadetes, Armatimonadetes, and Fusobacteria and phyla like GN02 and OP3 were exclusively present before MGE. In contrast, Tenericutes, most of the members of candidate division like WS3, OD1, and SBR1093 were observed only during MGE.

Bacterial community composition across the mass gathering. a Phylum level distribution of dominant bacterial groups between before (BS) and during (DS) the event samples. b Family-level distribution of dominant bacterial groups between before (BS) and during (DS) the event samples
Our investigation of spatiotemporal variation in the bacterial community composition showed differential abundance of OTUs (abundance ≥ 0.5%) corresponding to 15 bacterial families. It was observed that families such as Comamonadaceae, Flavobacteriaceae, Verrucomicrobiaceae, Chitinophagaceae, Moraxellaceae, Oxalobacteraceae, candidate family ACK-M1, Microbacteriaceae, Methylophilaceae, Cytophagaceae, and Weeksellaceae were prevalent before the MGE while Planococcaceae, Sphingomonadaceae, Prevotellaceae, and Xanthobacteraceae predominate during the MGE (Fig. 3b). Likewise, a comparison at genus level depicted 15 differentially abundant genera (abundance ≥ 0.5%) across MGE out of which Flavobacterium, Sediminibacterium, Prosthecobacter, candidate genus C39, Hydrogenophaga, Acinetobacter, Polynucleobacter, Hylemonella, Limnohabitans, Rhodoferax, and Cloacibacterium were significantly abundant before the MGE. In contrast, Planomicrobium, Novosphingobium, Prevotella, Akkermansia, Uliginosibacterium, and Dechloromonas were highly prevalent during MGE. Collectively, these results suggested significant differences in the microbial community composition across the MGE.
Determining the Impact of MGE on the Bacterial Community Composition
Beta diversity was analyzed to determine variations in bacterial community structure between two sampling points, i.e., before and during the MGE and to compare upstream and bathing sites before and during MGE. NMDS (Non-metric multidimensional scaling) plots based on Bray-Curtis dissimilarity metrics showed four distinct groups based on spatial (i.e., upstream and bathing sites) as well as temporal variation (i.e., before and during MGE) (Fig. 4). The inter-group variation between these groups was statistically validated. It was observed that spatial variation during the MGE was quite higher as compared to before the MGE.

Beta diversity analysis. Non-metric multidimensional scaling plot based on Bray-Curtis distance represents the spatiotemporal variation in the bacterial community structure. Spatial variation is denoted by shapes upstream (triangular) and bathing site (circle). Color code defines the temporal divergence, before (red) and during (blue) the event
The assessment of complex microbial association network and their correlation with physiochemical parameters revealed that bacterial families like Planococcaceae and Bacillaceae showed strong negative correlation with other bacterial families. Additionally, we found a positive correlation between the bacterial families Planococcaceae and Bacillaceae with the physiochemical parameters (OD, TS, TDS, TSS, BOD and COD, and MPN) during the MGE (Suppl. Fig. 3). In contrast, bacterial families like Comamonadaceae, Oxalobacteraceae, candidate family ACK−M1, Chthoniobacteraceae, Rhodobacteraceae, Gemmataceae, and Pirellulaceae showed a negative correlation with environmental factors such as OD, TS, TDS, TSS, BOD, and COD during the MGE. A positive correlation was noted between fecal coliform, MPN, and bacterial families viz. Lachnospiraceae, Ruminococcaceae, Campylobacteraceae, Clostridiaceae, and Methylocystaceae during the MGE. Similarly, principal component analysis of physiochemical parameters such as turbidity, solute concentration, and MPN was found to modulate bacterial communities during the MGE (p < 0.05). In contrast, microbial communities experience the influence of dissolved oxygen before the MGE (p < 0.05).
Tracking the Signature of Human Microbiota
The efforts of comparing human microbiota from healthy Indian subpopulation with MGE samples, by applying Bayesian mixing model (Source tracker), depicted increase in skin microbiota into the river upon mass bathing. We noted ~2.32-fold increase in skin-associated bacterial communities during the MGE (Fig. 5a). Similarly, the abundance of human fecal-associated bacterial communities showed a net change of ~2.89-fold which align with the results of MPN and fecal coliform estimations. However, we could not detect microbial communities associated with oral microbiota. Comprehensive assessment of microbial source information would serve to mitigate the extent of pollution and ensure the human health under higher fecal contamination.

a Pie chart depicts the signatures of human associated microbiota (skin, stool and oral) recovered during the mass bathing event. The color code denotes the source of bacterial communities associated with human-oral (green), skin (blue), and stool (red). Box plots represent functional acquaintance of bacterial communities across the gathering. The differential abundance of gene families associated with b infectious diseases and c drug resistance. Before samples (blue) and during samples (red)
Imputed Metagenomics
We noted significant differences in major metabolic functions of the river microbial assemblage before and during MGE (Suppl. Fig. 4). Predominantly, gene families associated with glycanbiosynthesis and metabolism, genetic information processing, biosynthesis of secondary metabolites, lipid metabolism, and amino acid metabolism were significantly higher in before MGE samples. In contrast, gene families such as cellular processing, motility, bacterial chemotaxis, xenobiotic degradation, and infectious diseases were significantly higher during the MGE. From the functional profiles of microbial communities, we targeted gene families related to infectious diseases and drug resistance to investigate the impact of MGE on public health. The analysis revealed elevated levels of genes related to infections, described as fold change for Helicobacter pylori infection (~0.469), Legionellosis (~0.58), pertussis (~0.157), Salmonella infection (~1.051), Staphylococcus aureus infection (~3.433), and tuberculosis (~0.092) during MGE (Fig. 5b). Similarly, increased drug resistant-related gene includes antifolate resistance (~0.25), beta-lactam resistance (~0.15), cationic antimicrobial peptide (~0.483), platinum drug resistance (~0.809), and vancomycin resistance (~0.346) during MGE (Fig. 5c). The above observations pose a critical challenge ensuring public health during the MGE which could otherwise lead to unfavorable outcomes of epidemics.
Discussion
In the current spatiotemporal investigation, we observed significant alterations in the bacterial communities during the world’s largest and unique mass bathing event. Our analysis depicted loss of diversity, with distinguished augmentation of bacterial taxa during the MGE. We further noted an increase in the drug resistance and waterborne infections which might pose serious threats to public health.
We evaluated the physicochemical parameters of river water which represent significant deterioration in the quality of water during the MGE and are previously reported to influence the bacterial diversity and their composition [36,37,38,39,40,41,42]. We assessed the turbidity, solute concentration, oxygen availability, and fecal contamination, which provided the primary leads about spatiotemporal differences in the bacterial communities. These results were not surprising given the numerous introductions of human waste being routinely discharge into the river over the course of MGE [4, 5]. Additionally, sediment re-suspension and release of the industrial and house hold waste have shown an influence on the bacterial community composition of freshwater ecosystems [18, 43, 44]. In particular, we observed a significantly higher particle load and salinity (TS and TSS), whereas reduced oxygen availability (BOD, COD, and DO) during the event [45,46,47,48]. However, the concrete constructions along the bank of Godavari River minimize the accumulation and re-suspension of particulate matter into the ecosystem which otherwise reported to associated with potential risk of pollution and human health [49,50,51,52,53]. Instead, we believe that sources of nutrients and contaminants to the river are due to the impaired waste disposal at bathing sites [4, 5]. This is further proved by the linear regression analysis indicating the negative association between physiochemical parameters and bacterial communities along the temporal variations. Similarly, results of spearman correlation analysis demonstrated loss of common freshwater bacterial families under the relatively high salt and oxygen-limiting conditions. In contrast, we found augmentation of human fecal signature communities due to the practice of open defecation by the pilgrims and abrupt sewage management at the MGE, which could pose potential risk of gastrointestinal diseases [4, 54].
Our results of 16S rRNA gene-based microbial profiling demonstrated drastic differences in the bacterial diversity of MGE-impacted water bodies. Alpha diversity estimates a significant reduction in the microbial diversity during the MGE (p < 0.05) [55]. The water bodies before the MGE harbored bacterial groups such as Proteobacteria, Actinobacteria, and Bacteroidetes [56, 57], while Firmicutes were predominant at the bathing sites. The members of phylum Proteobacteria such as Alphaproteobacteria and Betaproteobacteria are known as the major player in the primary production of freshwater ecosystem, while reduced light availability could lead to decrease in these predominant freshwater communities [37, 57, 58]. Similarly, the members of phylum Actinobacteria were found to be depleted which otherwise are reported as autochthonous communities with potentials to degrade variety of complex compounds [56]. Moreover, the abundance of family Planococcaceae belonging to phylum Firmicutes was also found to be higher during the MGE. The members of this family have been isolated from the variety of halophilic environments, explaining its distinguished augmentation under high solute conditions reported in the current study [59]. Absolute quantification of 16S rRNA gene copy number using qPCR showed the influence of MGE under temporal variation with ~130-fold increase in the bacterial load which substantiates the fact of bathers being the potential source of non-point pollution [60]. Moreover, previous epidemiological studies have shown the correlation between the concentrations of bacteria in bathing water to the bather’s health [61]. Interestingly, our results of spatiotemporal analysis also delineate extent of distinctness and diversity in bacterial communities before and during the MGE.
Looking at the number of pilgrims who attended the event, we attempted to differentiate the contribution of humans associated microbiota within the river which could deplete common freshwater bacterial diversity. The efforts of understanding the sources of human microbiota using a signature approach reflected ~2.32- and ~2.89-fold rise in human skin and fecal microbiota, respectively, during the gathering. It also provides information about the differential distribution of the human microbiota at individual sites of the event. Therefore, the risk associated with use of such contaminated water remains uncertain, as it is difficult to differentiate the impact of these microorganisms in correlation to the gastrointestinal illness, skin, and eye infections [62,63,64,65,66,67,68]. The influence of the MGE was also reflected in predicted metabolic functions of river bacterial communities where we observed a higher abundance of genes responsible for infectious diseases and drug resistance during the MGE. A previous study on the Ganga pilgrimages by targeting blaNDM-1 gene has also suggested substantial increase in antimicrobial resistance [69]. Resistance towards cefotaxime shown by the recently described Corynebacterium godavarianum isolated from the Godavari River during the Kumbh Mela 2015 supports the presence of high drug-resistant bacterial communities observed in the samples collected during the event [70]. These observations collectively indicate that numerous influxes of human microbiota during MGE contribute to alteration in microbial community structure and modulate metabolic functions.
Alternatively, previous studies have indicated the association of inadequate planning and management of participants at MGEs with spread of infectious diseases such as 1817–1824 Asiatic cholera pandemic during the Kumbh Mela, W135 meningococcal infections at Hajj and influenza outbreak, Australia [71,72,73,74,75]. Further, David and Roy suggested that rising population and better connectivity increase the participation at such MGEs which may substantiate the risk to public health. Therefore, MGE organizers have adapted the preparatory guidelines amended by local health commission in conjugation with WHO [76,77,78] which have proven to maintain the harmony of the event by supplementing adequate sanitation, randomize clinical inspections, and vaccinations etc.
We conclude that this study is the first report addressing impact of Kumbh Mela, the world’s largest MGE on river bacterial communities and its likely impact on human health. The significant changes in the bacterial diversity and composition with its potential impact on public health could be collectively attributed to the response of spatiotemporal variation of MGE. Also, our findings merit detailed investigation to evaluate long-term impact of such event on human health and river ecosystem.
References
Abubakar I, Gautret P, Brunette GW, Blumberg L, Johnson D, Poumerol G, Memish ZA, Barbeschi M, Khan AS (2012) Global perspectives for prevention of infectious diseases associated with mass gatherings. Lancet Infect Dis 12(1):66–74
Banks AL (1961) Religious fairs and festivals in INDIA. Lancet:162–163
Koch K (2013) Inside India’s pop-up city. Harvard Gazette http://news.harvard.edu/gazette/story/2013/01/inside-indias-pop-up-city/ Accessed 04 June 2017
Vortmann M, Balsari S, Holman SR, Greenough PG (2015) Water, sanitation, and hygiene at the world’s largest mass gathering. Curr Infect Dis Rep 17(2):461. https://doi.org/10.1007/s11908-015-0461-1
David S, Roy N (2016) Public health perspectives from the biggest human mass gathering on earth: KumbhMela, India. Int J Infect Dis 47:42–45
Baranwal A, Anand A, Singh R, Deka M, Paul A, Borgohain S, Roy N (2015) Managing the earth’s biggest mass gathering event and wash conditions: Maha kumbh mela (India). PLoS Curr doi: https://doi.org/10.1371/currents.dis.e8b3053f40e774e7e3fdbe1bb50a130d
World Health Organization. Communicable disease alert and response for mass gatherings. Technical workshop. Geneva, Switzerland, April 29–30, 2008. Geneva: WHO. 2008 http://www.who.int/csr/resources/publications/WHO_HSE_EPR_2008_8c.pdf Accessed 04 June 2017
Imperial Gazette of India. Volume 13 (1903) http://dsal.uchicago.edu/reference/gazetteer/pager.html?objectid=DS405.1.I34_V13_058.gif. Accessed 04 June 2017
Labbate M, Seymour JR, Lauro F, Brown MV (2016) Editorial: anthropogenic impacts on the microbial ecology and function of aquatic environments. Front Microbiol 7:1044. https://doi.org/10.3389/fmicb.2016.01044
Lee CS, Kim M, Lee C, Yu Z, Lee J (2016) The microbiota of recreational freshwaters and the implications for environmental and public health. Front Microbiol 7:1826. https://doi.org/10.3389/fmicb.2016.01826
Hlavsa MC, Roberts VA, Anderson AR, Hill VR, Kahler AM, Orr M, Garrison LE, Hicks LA, Newton A, Hilborn ED, Wade TJ, Beach MJ, Yoder JS; CDC (2011) Surveillance for waterborne disease outbreaks and other health events associated with recreational water—United States, 2007-2008. MMWR Surveill Summ 60(12):1–32
Paíga P, Delerue-Matos C (2017) Anthropogenic contamination of Portuguese coastal waters during the bathing season: assessment using caffeine as a chemical marker. Mar Pollut Bull 120(1–2):355–363
Paerl HW, Dyble J, Moisander PH, Noble RT, Piehler MF, Pinckney JL, Steppe TF, Twomey L, Valdes LM (2003) Microbial indicators of aquatic ecosystem change: current applications to eutrophication studies. FEMS Microbiol Ecol 46(3):233–246
Newton RJ, Bootsma MJ, Morrison HG, Sogin ML, McLellan SL (2013) A microbial signature approach to identify fecal pollution in the waters off an urbanized coast of Lake Michigan. Microb Ecol 65(4):1011–1023
King GM (2014) Urban microbiomes and urban ecology: how do microbes in the built environment affect human sustainability in cities. J Microbiol 52(9):721–728. https://doi.org/10.1007/s12275-014-4364-x
Vitousek PM, Mooney HA, Lubchenco J, Melillo JM (1997) Human domination of Earth’ s ecosystems. Science 277(5325):494–499
Sekar R, Pernthaler A, Pernthaler J, Warnecke F, Posch T, Amann R (2003) An improved protocol for quantification of freshwater Actinobacteria by fluorescence in situ hybridization. Appl Environ Microbiol 69(5):2928–2935
Lindström ES, Kamst-Van Agterveld MP, Zwart G (2005) Distribution of typical freshwater bacterial groups is associated with pH, temperature, and lake water retention time. Appl Environ Microbiol 71(12):8201–8206
Hu C, Rea C, Yu Z, Lee J (2016) Relative importance of Microcystis abundance and diversity in determining microcystin dynamics in Lake Erie coastal wetland and downstream beach water. J Appl Microbiol 120(1):138–151
Tan B, Ng C, Nshimyimana JP, Loh LL, Gin KY, Thompson JR (2015) Next-generation sequencing (NGS) for assessment of microbial water quality: current progress, challenges, and future opportunities. Front Microbiol 6:1027. https://doi.org/10.3389/fmicb.2015.01027
Marti R, Ribun S, Aubin JB, Colinon C, Petit S, Marjolet L, Gourmelon M, Schmitt L, Breil P, Cottet M, Cournoyer B (2017) Human-driven microbiological contamination of benthic and hyporheic sediments of an intermittent peri-urban river assessed from MST and 16S rRNA genetic structure analyses. Front Microbiol 8:19. https://doi.org/10.3389/fmicb.2017.00019
Kumbhare SV, Dhotre DP, Dhar SK, Jani K, Apte DA, Shouche YS, Sharma A (2015) Insights into diversity and imputed metabolic potential of bacterial communities in the continental shelf of Agatti Island. PLoS One 10(6):e0129864
Sharma A, Paul D, Dhotre D, Jani K, Pandey A, Shouche YS (2017) Deep sequencing analysis of bacterial community structure of Soldhar hot spring, India. Microbiology 86(1):136–142
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R (2011) Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A 108(Suppl 1):4516–4522
Magoč T, Salzberg SL (2011) FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27(21):2957–2963
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75(23):7537–7541
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7(5):335–336
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL (2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72(7):5069–5072
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26(19):2460–2461
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73(16):5261–5267
Bhute S, Pande P, Shetty SA, Shelar R, Mane S, Kumbhare SV, Gawali A, Makhani H, Navandar M, Dhotre D, Lubree H, Agarwal D, Patil R, Ozarkar S, Ghaskadbi S, Yajnik C, Juvekar S, Makharia GK, Shouche YS (2016) Molecular characterization and meta-analysis of gut microbial communities illustrate enrichment of prevotella and megasphaera in Indian subjects. Front Microbiol 7:660. https://doi.org/10.3389/fmicb.2016.00660
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O'Hara RB, Simpson GL, Solymos P, Stevens MHH, Szoecs E, Wagner H (2013) Community ecology package (vegan). https://cranr-projectorg/web/packages/vegan/indexhtml Accessed 04 June 2017
Knights D, Kuczynski J, Charlson ES, Zaneveld J, Mozer MC, Collman RG, Bushman FD, Knight R, Kelley ST (2011) Bayesian community-wide culture-independent microbial source tracking. Nat Methods 8(9):761–763
Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, Beiko RG, Huttenhower C (2013) Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31(9):814–821
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C (2011) Metagenomic biomarker discovery and explanation. Genome Biol 12(6):R60. https://doi.org/10.1186/gb-2011-12-6-r60
Jani K, Ghattargi V, Pawar S, Inamdar M, Shouche Y, Sharma A (2017) Anthropogenic activities induce depletion in microbial communities at urban sites of the river Ganges. Curr Microbiol. https://doi.org/10.1007/s00284-017-1352-5
Newton RJ, McLellan SL (2015) A unique assemblage of cosmopolitan freshwater bacteria and higher community diversity differentiate an urbanized estuary from oligotrophic Lake Michigan. Front Microbiol 6:1028. https://doi.org/10.3389/fmicb.2015.01028
Booth DB, Jackson CR (1997) Urbanization of aquatic systems: degradation thresholds, stormwater detection, and the limits of mitigation. J Am Water Resour Assoc 33(5):1077–1090
Carpenter SR, Caraco NF, Correll DL, Howarth RW, Sharpley AN, Smith VH (1998) Nonpoint pollution of surface waters with phosphorus and nitrogen. Ecol Appl 8:559–568
Hale RL, Turnbull L, Earl SR, Childers DL, Grimm NB (2014) Stormwater infrastructure controls runoff and dissolved material export from arid urban watersheds. Ecosystems 18(1):62–75
Kaushal SS, Belt KT (2012) The urban watershed continuum: evolving spatial and temporal dimensions. Urban Ecosyst 15(2):409–435
Wollheim WM, Pellerin BA, Vörösmarty CJ, Hopkinson CS (2005) N retention in urbanizing headwater catchments. Ecosystems 8(8):871–884
Allgaier M, Grossart HP (2006) Seasonal dynamics and phylogenetic diversity of free-living and particle-associated bacterial communities in four lakes in northeastern Germany. Aquat Microb Ecol 45:115–128. https://doi.org/10.3354/ame045115
Newton RJ, Jones SE, Eiler A, McMahon KD, Bertilsson S (2011a) A guide to the natural history of freshwater lake bacteria. Microbiol Mol Biol Rev 75(1):14–49
Horner-Devine MC, Leibold MA, Smith VH, Bohannan BJM (2003) Bacterial diversity patterns along a gradient of primary productivity. Ecol Lett 6(7):613–622
Jankowski K, Schindler DE, Horner-Devine MC (2014) Resource availability and spatial heterogeneity control bacterial community response to nutrient enrichment in lakes. PLoS One 9(1):e86991
Yannarell CC, Triplett EW (2005) Geographic and environmental sources of variation in lake bacterial community composition. Appl Environ Microbiol 71(1):227–239
Spietz RL, Williams CM, Rocap G, Horner-Devine MC (2015) A dissolved oxygen threshold for shifts in bacterial community structure in a seasonally hypoxic estuary. PLoS One 10(8):e0135731. https://doi.org/10.1371/journal.pone.0135731
Amalfitano S, Corno G, Eckert E, Fazi S, Ninio S, Callieri C, Grossart HP, Eckert W (2017) Tracing particulate matter and associated microorganisms in freshwaters. Hydrobiologia 800:145–154
Foppen JWA, Schijven JF (2006) Evaluation of data from the literature on the transport and survival of Escherichia coli and thermotolerant coliforms in aquifers under saturated conditions. Water Res 40:401–426
Mahler BJ, Personn JC, Lods GF, Drogue C (2000) Transport of free and particulate associated bacteria in karst. J Hydrol 238:179–193
Bai S, Lung WS (2005) Modeling sediment impact on the transport of fecal bacteria. Water Res 39:5232–5240
Fries JS, Noble RT, Characklis GW (2006) Attachment of fecal indicator bacteria to particles in the Neuse River estuary, N.C. J Environ Eng Am Soc Civil Eng 132:1338–1345
Fisher JC, Levican A, Figueras MJ, McLellan SL (2014) Population dynamics and ecology of Arcobacter in sewage. Front Microbiol 5:525. https://doi.org/10.3389/fmicb.2014.00525
Gibbons SM, Jones E, Bearquiver A, Blackwolf F, Roundstone W, Scott N, Hooker J, Madsen R, Coleman ML, Gilbert JA (2014) Human and environmental impacts on river sediment microbial communities. PLoS One 9(5):e97435
Warnecke F, Amann R, Pernthaler J (2004) Actinobacterial 16S rRNA genes from freshwater habitats cluster in four distinct lineages. Environ Microbiol 6(3):242–253
Van der Gucht K, Vandekerckhove T, Vloemans N, Cousin S, Muylaert K, Sabbe K, Gillis M, Declerk S, De Meester L, Vyverman W (2005) Characterization of bacterial communities in four fresh water lakes differing in nutrient load and food web structure. FEMS Microbiol Ecol 2005;53(2):205–220
Rose KC, Hamilton DP, Williamson CE, McBride CG, Fischer JM, Olson MH, Saros JE, Allan MG, Cabrol N (2014) Light attenuation characteristics of glacially-fed lakes. J Geophys Res Biogeosci 119:1446–1457
Jung YT, Kang SJ, Oh TK, Yoon JH, Kim BH (2009) Planomicrobium flavidum sp.nov., isolated from a marine solar saltern, and transfer of Planococcus stackebrandtii Mayilraj et al.2005tothegenus Planomicrobium as Planomicrobium stackebrandtii comb. nov. Int J Syst Evol Microbiol 59:2929–2933
Fewtrell L, Kay D (2015) Recreational water and infection: a review of recent findings. Curr Environ Health Rep 2(1):85–94
Stevenson AH (1953) Studies of bathing water quality and health. Am J Public Health Nations Health 43(5 Pt 1):529–538
Field KG, Samadpour M (2007) Fecal source tracking, the indicator paradigm, and managing water quality. Water Res 41(16):3517–3538
Guidelines for safe recreational water environments Volume 1: Coastal and Fresh Waters. WHO 2003. http://www.who.int/water_sanitation_health/publications/srwe1/en/Accessed 04 June 2017
McLellan SL, Eren AM (2014) Discovering new indicators of fecal pollution. Trends Microbiol 22(12):697–706
Wade TJ, Calderon RL, Sams E, Beach M, Brenner KP, Williams AH, Dufour AP (2006) Rapidly measured indicators of recreational water quality are predictive of swimming-associated gastrointestinal illness. Environ Health Perspect 114(1):24–28
Wade TJ, Calderon RL, Brenner KP, Sams E, Beach M, Haugland R, Wymer L, Dufour AP (2008) High sensitivity of children to swimming-associated gastrointestinal illness: results using a rapid assay of recreational water quality. Epidemiology 19(3):375–383
Fleisher JM, Fleming LE, Solo-Gabriele HM, Kish JK, Sinigalliano CD, Plano L, Elmir SM, Wang JD, Withum K, Shibata T, Gidley ML, Abdelzaher A, He G, Ortega C, Zhu X, Wright M, Hollenbeck J, Backer LC (2010) The BEACHES study: health effects and exposures from non-point source microbial contaminants in subtropical recreational marine waters. Int J Epidemiol 39:1291–1298
Papastergiou P, Mouchtouri V, Pinaka O, Katsiaflaka A, Rachiotis G, Hadjichristodoulou C (2012) Elevated bathing-associated disease risks despite certified water quality: a cohort study. Int J Environ Res Public Health 9(5):1548–1565
Ahammad ZS, Sreekrishnan TR, Hands CL, Knapp CW, Graham DW (2014) Increased waterborne blaNDM-1 resistance gene abundances associated with seasonal human pilgrimages to the upper Ganges River. Environ Sci Technol 48(5):3014–3020
Jani K, Khare K, Senik S, Karodi P, Vemuluri VR, Bandal J, Shouche Y, Rale V, Sharma (2017) A Corynebacterium godavarianum sp. nov., isolated from Godavari River. India Int J Syst Evol Microbiol. https://doi.org/10.1099/ijsem.0.002491
Hays JN (2005) Epidemics and pandemics: their impacts on human history. Santa Barbara, ABC-CLIO,CA, pp 214–219
Bryceson AD (1977) Cholera, the flickering flame. Proc R Soc Med 70(5):363–365
Aguilera JF, Perrocheau A, Meffre C, Hahné S, the W135 Working Group (2002) Outbreak of serogroup W135 meningococcal disease after the Hajj pilgrimage, Europe, 2000. Emerg Infect Dis 8(8):761–767
Blyth CC, Foo H, van Hal SJ, Hurt AC, Barr IG, McPhie K, Armstrong PK, Rawlinson WD, Sheppeard V, Conaty S, Staff M, Dwyer DE, World Youth Day (2008) Influenza study group (2010) influenza outbreaks during world youth day 2008 mass gathering. Emerg Infect Dis 16(5):809–815
Memish ZA, Stephens GM, Steffen R, Ahmed QA (2012) Emergence of medicine for mass gatherings: lessons from the hajj. Lancet Infect Dis Lancet Infect Dis 12(1):56–65
Memish ZA, Zumla A, Alhakeem RF, Assiri A, Turkestani A, Al Harby KD, Alyemni M, Dhafar K, Gautret P, Barbeschi M, McCloskey B, Heymann D, Al Rabeeah AA, Al-Tawfiq JA (2014) Hajj: infectious disease surveillance and control. Lancet 383(9934):2073–2082
Nsoesie EO, Kluberg SA, Mekaru SR, Majumder MS, Khan K, Hay SI, Brownstein JS (2015) New digital technologies for the surveillance of infectious diseases at mass gathering events. Clin Microbiol Infect 21(2):134–140
Memish ZA, Alrabeeah AA (2011) Jeddah declaration on mass gatherings health. Lancet Infect Dis. https://doi.org/10.1016/S1473-3099(11)70066-4
Acknowledgements
We would like to acknowledge director NCCS for providing infrastructure. We are grateful to Dr. Vilas Sinkar for providing valuable comments and critical review of the manuscript. Mr. Vishal Nawale is highly acknowledged for his help in sampling.
Funding
This work was supported by the “Department of Biotechnology (DBT), Government of India” (by Grant no. BT/PR/0054/NDB/52/94/2007), under the project “Establishment of Microbial Culture Collection.”
Author information
Authors and Affiliations
Contributions
K.J., J.B., Y.S. and A.S. were involved in the study design; J.B. was involved in the sample collection; K.J., J.B., Y.S. M.S. and A.S. were involved in the experimental work; K.J., D.D., Y.S., and A.S. were involved in the bioinformatics and statistical analysis; K.J., D.D., Y.S., V. R. and A.S interpretation of the data; K.J., D.D., Y.S., V. R. and A.S. were involved in the drafting of the manuscript; K.J., D.D., Y.S., V. R. and A.S were involved in the critical revision of the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests.
Electronic supplementary material
ESM 1
(DOCX 1338 kb).
Rights and permissions
About this article

{kind=link}
Cite this article
Jani, K., Dhotre, D., Bandal, J. et al. World’s Largest Mass Bathing Event Influences the Bacterial Communities of Godavari, a Holy River of India. Microb Ecol 76, 706–718 (2018). https://doi.org/10.1007/s00248-018-1169-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-018-1169-1