
Abstract
Supravalvar aortic stenosis (SVAS) is a less common but clinically important form of left ventricular outflow tract obstruction, and commonly associated with Williams syndrome (WS). SVAS outside of WS may also occur sporadically or in a familial form, often with identifiable mutations in the elastin (ELN) gene. While risk of sudden cardiac death in patients with SVAS has been extensively described in the context of WS, less is known about risk in patients with isolated SVAS. We report a case of a nonsyndromic two-year-old boy with evolving manifestations of SVAS who developed sudden cardiac arrest and death during a sedated cardiac magnetic resonance imaging study. A strong family history of SVAS was present and targeted genetic testing identified an ELN gene mutation in the boy’s affected father and other paternal relatives. We review risk factors found in the literature for SCA in SVAS patients and utilize this case to raise awareness of the risk of cardiac events in these individuals even in the absence of WS or severe disease. This case also underscores the importance of genetic testing, including targeted panels specifically looking for ELN gene mutations, in all patients with SVAS even in the absence of phenotypic concerns for WS or other genetic syndromes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Congenital supravalvar aortic stenosis (SVAS) is relatively rare, accounting for less than 0.5% of all congenital heart defects, and is the least common form of congenital left ventricular outflow tract obstruction [1]. It is classically associated with Williams syndrome (WS), also known as Williams-Beuren syndrome [2]. However, isolated SVAS without phenotypic features of WS can also occur sporadically or as a familial inherited isolated mutation in the elastin (ELN) gene [3,4,5]. The increased risk of periprocedural sudden cardiac arrest in patients with WS and SVAS is well described [6,7,8,9,10,11]. However, less is known about the risk of acute cardiac events in nonsyndromic patients with SVAS, especially in the absence of severe disease [11, 12].
Case
This was a 2-year-old otherwise healthy and active toddler with no cardiorespiratory symptomatology referred for murmur evaluation. Prior history noted an echocardiogram performed in the postnatal period which showed mild peripheral pulmonary stenosis and no aortic stenosis. At 17 months of age, a newly appreciated murmur prompted referral with no antecedent cardiorespiratory symptoms or medical concerns. Family history revealed multiple family members with SVAS, including in the patient’s father, paternal uncle, and paternal grandfather, all of whom required surgical repair in childhood ranging from ages 3 to 20 years. Echocardiography at this time demonstrated normal aortic dimensions with no stenosis, mild right pulmonary artery stenosis (diameter Z-score − 2.4, peak Doppler gradient 29 mmHg), and normal left pulmonary artery size and flow (Z-score + 1.8, peak gradient 13 mmHg). A 6 month interval follow up echocardiogram (now 2.5 years of age) was technically challenging due to patient movement but demonstrated an improving right pulmonary artery gradient (peak of 20 mmHg). The echocardiogram also suggested evolving mild narrowing at the supravalvar aortic area with some size discrepancy between the sinotubular junction and the ascending aorta (sinotubular junction Z-score − 1.7, ascending aorta Z-score + 1.9). Peak Doppler velocity and gradient across this area was only borderline increased at 1.9 m/s and 15 mmHg, respectively. Biventricular size and systolic function were normal and there was no ventricular hypertrophy. EKG showed sinus rhythm with normal voltages for age and a normal corrected QT interval of 388 ms. The patient was clinically asymptomatic, nondysmorphic, and meeting normal growth and developmental parameters.
Referral was made for genetic evaluation given the significant family history and evolving cardiac manifestations. Previous genetic testing showed normal findings on prenatal karyotype and chromosome microarray. An extensive congenital heart disease panel (42 gene, Invitae) was ordered on the patient’s father in context of the extensive paternal history of SVAS, which demonstrated the presence of an isolated heterozygous pathogenic ELN gene mutation (c.2044G>T, p.Gly682*). While genetic testing results were pending, to better delineate the anatomy in this patient with a malignant family history for hemodynamically important SVAS and suboptimal echocardiography windows, cardiac magnetic resonance (CMR) was pursued.
The CMR was performed a few days after results became available. General anesthesia was administered by a pediatric cardiac anesthesiologist. Inhalation induction with peripheral intravenous line placement and endotracheal intubation occurred with no issues, with intubation utilized to permit suspension of respirations during CMR image acquisition. Cisatracurium was used for paralysis and anesthetic effect maintained using sevoflurane. There were no significant hemodynamic disturbances or electrocardiogram changes until the end of the examination, when the patient developed sudden ventricular fibrillation and acute cardiac arrest. CPR was initiated immediately and extensive resuscitative measures were performed including administration of various code medications and multiple attempts at defibrillation. ECMO was discussed but was not possible for small children in our institution at the time. Resuscitative efforts were not successful and the patient expired.
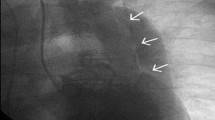
Available CMR images obtained were consistent with mild-to-moderate SVAS (Fig. 1), mild-to-moderate proximal right pulmonary artery hypoplasia, normal left pulmonary artery dimension, symmetric branch pulmonary artery flow distribution, no right ventricular hypertrophy, mild left ventricular hypertrophy, and normal biventricular size and systolic function. The family declined an autopsy.

CMR view of the LVOT. Systolic still frame from the patient’s CMR, LVOT CINE view in the coronal plane. Narrowing is seen at the aortic sinotubular junction (white arrow) with a turbulent dephasing jet which starts at this area (below the yellow star), consistent with supravalvar aortic stenosis. AAO ascending aorta, AOR aortic root, LV left ventricle, LVOT left ventricular outflow tract
Discussion
SVAS is characterized by narrowing of the supravalvar aortic area, and is typically associated with WS, a congenital multisystem disorder involving the cardiovascular, connective tissue, and central nervous systems caused by a large 1.5–1.8 Mb deletion on chromosome 7q11.23 incorporating up to 27 different genes including ELN [2, 13,14,15,16]. SVAS is the most common cardiac lesion in WS, estimated to occur in approximately 50% of patients, and can commonly coexist with supravalvar or peripheral branch pulmonary stenosis in these patients [17]. Nonsyndromic SVAS may occur outside of WS with identifiable genetic cause in the context of a familial autosomal-dominant inheritance of ELN gene mutation or as a sporadic mutation [18, 19]. Histologically, the ELN gene mutation results in reduced elastic tissue by formation of broken and disorganized elastin fibers, increased collagen content, and hypertrophied smooth muscle cells, all of which can lead to reduced elasticity and arterial narrowing [20, 21]. Hence, the term Elastin Arteriopathy (EA) has been coined to describe the spectrum of these associated conditions [4]. Not all patients with SVAS have an ELN mutation [22], and whether presence of an ELN mutation translates into worse disease or a higher risk profile is not fully clear and deserves further study. While there is evidence that some patients with isolated mutations in ELN may have less dysfunctional elastin compared to patients with WS who have deletion of the entire ELN gene [16, 22, 23], it appears that most ELN mutations result in functional haploinsufficiency and thus similar vascular histopathological findings to WS [22]. Nevertheless, significant variable expressivity, reduced penetrance, and varying degrees of clinical severity are seen in patients with EA, with some ranging from asymptomatic carriers never requiring cardiovascular interventions to others displaying early severe disease leading to death in early infancy [22, 24]. Even among cohorts with the same ELN mutation, significant intra- and inter-familial variation in phenotype and disease severity can exist [22].
Patients with SVAS and WS are at increased risk for sudden cardiac death compared to the general population [5]. The preponderance of literature on cardiac arrest in SVAS patients is in those with WS, with particular emphasis on the periprocedural period with invasive procedures such as catheterizations, operations, or other noncardiac surgeries [5,6,7,8,9,10,11, 25,26,27,28,29]. The hemodynamic burdens from supravalvar aortic and/or pulmonary stenosis, higher incidence of coronary artery abnormalities, and repolarization abnormalities such as QTc prolongation, are some of the contributing factors. The risk of SCD in non-WS patients with SVAS, however, is less well defined and has likely historically been underestimated as data in non-WS SVAS patients are more sparse [11, 12]. We conjecture that past reports with similar EA phenotypes may have batched WS and non-WS patients into same cohorts. It is also possible that non-WS patients with isolated SVAS are less likely to require invasive procedures, or have a lower risk profile for sudden cardiac arrest, although this is unknown and warrants further investigation.. We are also unaware of any prior report of SCD in a non-WS patient during a non-invasive sedated diagnostic study, which is interesting given the common need for diagnostic evaluation often necessary in these patients [30]. While most procedures can be undertaken even in patients with WS without significant adverse events [9, 28, 29], the anesthesia and periprocedural period appears to confer a higher risk for cardiac compromise. Changes in hemodynamic conditions coupled with the arterial obstructive lesions in these patients can result in myocardial ischemia and subsequent sudden cardiac arrest. Sudden cardiac arrest in WS has even been reported during brief noninvasive procedures such as contrast enhanced CT [31].
Available data on risk for SCD in childhood SVAS has also primarily focused on WS patients. High anesthetic risk factors include age < 3 years, history of cardiovascular event or arrhythmia, more than moderate bilateral outflow tract obstruction severity, SVAS gradient > 40 mmHg with presence of left ventricular hypertrophy, known coronary artery involvement, and prolonged QTc interval [25, 26]. Data in children with isolated ELN mutations who do not have WS is limited. A recent single institution retrospective review examining risk factors for periprocedural complications in children with various types of EA did include a small cohort of nonsyndromic patients with SVAS (12% of all patients) [11]. In this review, cardiac arrest occurred in 5% of total procedures, all of whom were < 3 years old and had significant biventricular outflow tract obstruction (BVOTO), particularly when gradients across both sides of the heart approached > 40–50 mmHg [11]. Hemodynamic alterations during anesthesia are typically well tolerated in normal children but may be enough to precipitate cardiovascular collapse in children with EA and BVOTO. The presence of BVOTO and presumed resultant hypertrophy likely results in a tenuous myocardial oxygen supply versus demand relationship, which may be further strained in the setting of coronary artery anomalies [11, 32]. Unfortunately detecting the presence or characterization of coronary abnormalities is often not possible by echocardiography, especially in small children, and therefore commonly only noted by direct visual inspection in the operating room or on postmortem autopsy [33]. A high index of suspicion for coronary abnormalities should thus be maintained in all EA patients. However, sudden cardiac arrest can occur even in the absence of coronary issues, although this is usually in the context of severe outflow obstruction [11, 28, 31].
Importantly, while increased risk appears to be associated with coronary abnormalities or significant outflow tract obstruction (especially bilateral) [5, 11, 34], it can be clinically challenging to predict. With the exception of age (just under 3 years), our patient manifested no additional high risk features described for the WS SVAS population. However, the tragic outcome demonstrates the difficulty in predicting risk of cardiac decompensation in such patients, and that risk profile may also not be dependent on disease severity. While all attempts should be made to avoid non-essential sedated procedures in these patients, even if noninvasive in nature, many will nonetheless require diagnostic and interventional workup and treatment. It is strongly recommended that sedated procedures in children with EA, especially infants and toddlers, be performed at larger tertiary centers with highly trained professionals and expertise in caring for such patients, including ability for rapid ECMO cannulation. In places where cardiac CTA may be performed without or with minimal sedation even in small children, this can also be considered prior to sedated options. The risk–benefit ratio for any diagnostic test or anticipated sedation should be thoroughly considered, and frank discussions regarding the risks and benefits should occur with the family. Additionally, given the imaging-based disease progression noted even over a short 3 month period between the last echocardiogram and CMR, we suggest that every patient undergo a repeat EKG and echocardiogram for up to date assessment ideally within a month prior to a sedated procedure.
Genetic testing for patients with SVAS is an important consideration and likely has traditionally been underevaluated in nonsyndromic patients. First-line genetic testing methods typically include karyotype, FISH, or chromosomal microarray (CMA) [35]. CMA, in particular, will identify many of the genetic abnormalities associated with CHD, including WS. However, it generally lacks the sensitivity to detect isolated single gene mutations. Targeted gene panels are require, but are more widely available today and therefore promise to improve identification of ELN mutations in patients with SVAS. Newer, even broader molecular approaches such as Whole Exome Sequencing may also be useful when clinical features are subtle or non-specific [36]. Increased awareness across the scientific community for ELN mutations in patients with SVAS, even in the absence of concern for WS, is necessary to improve testing and identification of nonsyndromic patients. We recommend that all patients with SVAS (and/or peripheral pulmonary stenosis which does not resolve in the first couple years of life) undergo thorough genetic evaluation, including targeted testing for isolated ELN mutations. Improved identification of patients with EA would help optimize counseling, inform cascade family genetic testing, and prompt coordination of periprocedural multidisciplinary care, all of which can hopefully reduce adverse outcomes in this fragile and important patient population.
References
Kitchiner D, Jackson M, Malaiya N, Walsh K, Peart I, Arnold R (1994) Incidence and prognosis of obstruction of the left ventricular outflow tract in Liverpool (1960–91): a study of 313 patients. Br Heart J 71(6):588–595. https://doi.org/10.1136/hrt.71.6.588
Williams JC, Barratt-Boyes BG, Lowe JB (1961) Supravalvular aortic stenosis. Circulation 24:1311–1318. https://doi.org/10.1161/01.cir.24.6.1311
Schmidt MA, Ensing GJ, Michels VV, Carter GA, Hagler DJ, Feldt RH (1989) Autosomal dominant supravalvular aortic stenosis: large three-generation family. Am J Med Genet 32(3):384–389. https://doi.org/10.1002/ajmg.1320320324
Merla G, Brunetti-Pierri N, Piccolo P, Micale L, Loviglio MN (2012) Supravalvular aortic stenosis: elastin arteriopathy. Circ Cardiovasc Genet 5(6):692–696. https://doi.org/10.1161/circgenetics.112.962860
Burch TM, McGowan FX Jr, Kussman BD, Powell AJ, DiNardo JA (2008) Congenital supravalvular aortic stenosis and sudden death associated with anesthesia: what’s the mystery? Anesth Analg 107(6):1848–1854. https://doi.org/10.1213/ane.0b013e3181875a4d
Conway EE Jr, Noonan J, Marion RW, Steeg CN (1990) Myocardial infarction leading to sudden death in the Williams syndrome: report of three cases. J Pediatr 117(4):593–595. https://doi.org/10.1016/s0022-3476(05)80696-1
Bird LM, Billman GF, Lacro RV, Spicer RL, Jariwala LK, Hoyme HE, Zamora-Salinas R, Morris C, Viskochil D, Frikke MJ, Jones MC (1996) Sudden death in Williams syndrome: report of ten cases. J Pediatr 129(6):926–931. https://doi.org/10.1016/s0022-3476(96)70042-2
Wessel A, Gravenhorst V, Buchhorn R, Gosch A, Partsch CJ, Pankau R (2004) Risk of sudden death in the Williams–Beuren syndrome. Am J Med Genet A 127A(3):234–237. https://doi.org/10.1002/ajmg.a.30012
Olsen M, Fahy CJ, Costi DA, Kelly AJ, Burgoyne LL (2014) Anaesthesia-related haemodynamic complications in Williams syndrome patients: a review of one institution’s experience. Anaesth Intensive Care 42(5):619–624. https://doi.org/10.1177/0310057X1404200512
Bragg K, Fedel GM, DiProsperis A (2005) Cardiac arrest under anesthesia in a pediatric patient with Williams syndrome: a case report. AANA J 73(4):287–293
Latham GJ, Ross FJ, Eisses MJ, Richards MJ, Geiduschek JM, Joffe DC (2016) Perioperative morbidity in children with elastin arteriopathy. Paediatr Anaesth 26(9):926–935. https://doi.org/10.1111/pan.12967
Groenewald CB, Latham GJ (2013) An unexpected cause of cardiac arrest during laparoscopy in an infant with supravalvar aortic stenosis. Paediatr Anaesth 23(1):91–93. https://doi.org/10.1111/pan.12069
Beuren AJ, Schulze C, Eberle P, Harmjanz D, Apitz J (1964) The syndrome of supravalvular aortic stenosis, peripheral pulmonary stenosis, mental retardation and similar facial appearance. Am J Cardiol 13:471–483. https://doi.org/10.1016/0002-9149(64)90154-7
Kaplan P, Wang PP, Francke U (2001) Williams (Williams Beuren) syndrome: a distinct neurobehavioral disorder. J Child Neurol 16(3):177–190. https://doi.org/10.1177/088307380101600305
Pober BR (2010) Williams–Beuren syndrome. N Engl J Med 362(3):239–252. https://doi.org/10.1056/NEJMra0903074
Urbán Z, Riazi S, Seidl TL, Katahira J, Smoot LB, Chitayat D, Boyd CD, Hinek A (2002) Connection between elastin haploinsufficiency and increased cell proliferation in patients with supravalvular aortic stenosis and Williams–Beuren syndrome. Am J Hum Genet 71(1):30–44. https://doi.org/10.1086/341035
Collins RT 2nd, Kaplan P, Somes GW, Rome JJ (2010) Long-term outcomes of patients with cardiovascular abnormalities and williams syndrome. Am J Cardiol 105(6):874–878. https://doi.org/10.1016/j.amjcard.2009.10.069
Li DY, Toland AE, Boak BB, Atkinson DL, Ensing GJ, Morris CA, Keating MT (1997) Elastin point mutations cause an obstructive vascular disease, supravalvular aortic stenosis. Hum Mol Genet 6(7):1021–1028. https://doi.org/10.1093/hmg/6.7.1021
Eisenberg R, Young D, Jacobson B, Boito A (1964) Familial supravalvar aortic stenosis. Am J Dis Child 108:341–347. https://doi.org/10.1001/archpedi.1964.02090010343002
Perou ML (1961) Congenital supravalvular aortic stenosis. A morphological study with attempt at classification. Arch Pathol 71:453–466
O’Connor WN, Davis JB Jr, Geissler R, Cottrill CM, Noonan JA, Todd EP (1985) Supravalvular aortic stenosis: Clinical and pathologic observations in six patients. Arch Pathol Lab Med 109(2):179–185
Metcalfe K, Rucka AK, Smoot L, Hofstadler G, Tuzler G, McKeown P, Siu V, Rauch A, Dean J, Dennis N, Ellis I, Reardon W, Cytrynbaum C, Osborne L, Yates JR, Read AP, Donnai D, Tassabehji M (2000) Elastin: mutational spectrum in supravalvular aortic stenosis. Eur J Hum Genet EJHG 8(12):955–963. https://doi.org/10.1038/sj.ejhg.5200564
Urbán Z, Zhang J, Davis EC, Maeda GK, Kumar A, Stalker H, Belmont JW, Boyd CD, Wallace MR (2001) Supravalvular aortic stenosis: genetic and molecular dissection of a complex mutation in the elastin gene. Hum Genet 109(5):512–520. https://doi.org/10.1007/s00439-001-0608-z
Chowdhury T, Reardon W (1999) Elastin mutation and cardiac disease. Pediatr Cardiol 20(2):103–107. https://doi.org/10.1007/s002469900415
Matisoff AJ, Olivieri L, Schwartz JM, Deutsch N (2015) Risk assessment and anesthetic management of patients with Williams syndrome: a comprehensive review. Paediatr Anaesth 25(12):1207–1215. https://doi.org/10.1111/pan.12775
Collins Ii RT, Collins MG, Schmitz ML, Hamrick JT (2017) Peri-procedural risk stratification and management of patients with Williams syndrome. Congenit Heart Dis 12(2):133–142. https://doi.org/10.1111/chd.12447
Horowitz PE, Akhtar S, Wulff JA, Al Fadley F, Al Halees Z (2002) Coronary artery disease and anesthesia-related death in children with Williams syndrome. J Cardiothorac Vasc Anesth 16(6):739–741. https://doi.org/10.1053/jcan.2002.128407
Pham PP, Moller JH, Hills C, Larson V, Pyles L (2009) Cardiac catheterization and operative outcomes from a multicenter consortium for children with williams syndrome. Pediatr Cardiol 30(1):9–14. https://doi.org/10.1007/s00246-008-9323-z
Brown ML, Nasr VG, Toohey R, DiNardo JA (2018) Williams syndrome and anesthesia for non-cardiac surgery: high risk can be mitigated with appropriate planning. Pediatr Cardiol 39(6):1123–1128. https://doi.org/10.1007/s00246-018-1864-1
Park JH, Kim HS, Jin GY, Joo CU, Ko JK (2008) Demonstration of peripheral pulmonary stenosis and supravalvular aortic stenosis by different cardiac imaging modalities in a patient with Williams syndrome-usefulness of noninvasive imaging studies. Int J Cardiol 128(3):e95-97. https://doi.org/10.1016/j.ijcard.2007.05.080
Gupta P, Tobias JD, Goyal S, Miller MD, Melendez E, Noviski N, De Moor MM, Mehta V (2010) Sudden cardiac death under anesthesia in pediatric patient with Williams syndrome: a case report and review of literature. Ann Card Anaesth 13(1):44–48. https://doi.org/10.4103/0971-9784.58834
van Son JA, Edwards WD, Danielson GK (1994) Pathology of coronary arteries, myocardium, and great arteries in supravalvular aortic stenosis: report of five cases with implications for surgical treatment. J Thorac Cardiovasc Surg 108(1):21–28
Stamm C, Kreutzer C, Zurakowski D, Nollert G, Friehs I, Mayer JE, Jonas RA, del Nido PJ (1999) Forty-one years of surgical experience with congenital supravalvular aortic stenosis. J Thorac Cardiovasc Surg 118(5):874–885. https://doi.org/10.1016/s0022-5223(99)70057-7
van Pelt NC, Wilson NJ, Lear G (2005) Severe coronary artery disease in the absence of supravalvular stenosis in a patient with Williams syndrome. Pediatr Cardiol 26(5):665–667. https://doi.org/10.1007/s00246-004-0845-8
Xia Y, Huang S, Wu Y, Yang Y, Chen S, Li P, Zhuang J (2019) Clinical application of chromosomal microarray analysis for the diagnosis of Williams–Beuren syndrome in Chinese Han patients. Mol Genet Genom Med 7(2):e00517. https://doi.org/10.1002/mgg3.517
Hou C, Zheng J, Liu W, Xie L, Sun X, Zhang Y, Xu M, Li Y, Xiao T (2021) Identification and characterization of a novel ELN mutation in congenital heart disease with pulmonary artery stenosis. Sci Rep 11(1):14154. https://doi.org/10.1038/s41598-021-93736-1
Acknowledgements
We thank the parents of our patient, “Charlie,” who provided consent for use of this case in the medical literature. Their interest was a driving force to help us increase awareness about ELN mutations in children and risk of cardiac compromise in all patients with elastin arteriopathy.
Funding
Open access funding provided by SCELC, Statewide California Electronic Library Consortium.
Author information
Authors and Affiliations
Contributions
Writing of main manuscript: DM, PS, RW, RG. Revision and editing of manuscript: DM, PS, KG, RW, RG. All authors reviewed the manuscript and approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical Approval
This study was approved by the local institutional review board. Parental/legal guardian informed consent was obtained.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Markush, D., Sanchez-Lara, P.A., Grand, K. et al. Sudden Cardiac Arrest During a Sedated Cardiac Magnetic Resonance Study in a Nonsyndromic Child with Evolving Supravalvar Aortic Stenosis Due to Familial ELN Mutation. Pediatr Cardiol 44, 946–950 (2023). https://doi.org/10.1007/s00246-022-03089-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00246-022-03089-3