
Abstract
Genetic testing is routinely performed on infants with critical congenital heart disease (CHD). This project reviewed the effect of implementing a genetic testing protocol in this population. Charts of infants with critical CHD were reviewed for genetic testing and results across two time periods: the time before implementation of a genetic testing protocol (pre-protocol) and the time after implementation (post-protocol). The use of karyotype, 22q11.2 Deletion testing, and chromosomal microarray were compared across these two time periods. Records of 891 infants were reviewed. 562 (63%) had at least one of the target genetic tests completed. During the pre-protocol time period, 66% of patients who had genetic testing underwent multiple tests versus 24% during the post-protocol time period (p < 0.01). The rate of patients who underwent genetic testing increased from 60% in the pre-protocol time period to 77% in the post-protocol time period (p < 0.01). The rate of diagnosis of genetic conditions during the pre-protocol period was 26% versus 36% during the post-protocol period (p = 0.01). There was a reduction in cost to patients by $5105.59 per diagnosis during the post-protocol period. Patients with critical CHD in the post-protocol period were less likely to undergo multiple genetic tests and more likely to have a diagnosis of genetic disease. In addition there was a significant reduction in cost per diagnosis during the post-protocol time period. Genetic testing protocols for infants with critical CHD promoted more efficient use of genetic testing and increased the rate of diagnosis of genetic conditions in this population.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Patients with critical congenital heart disease (CHD) have a high rate of copy number variants, particularly 22q11.2 Deletion Syndrome, yet clinical use of chromosomal microarray (CMA) in patients with CHD is unpredictable and not standardized [1,2,3,4,5]. Previous work has demonstrated copy number variants demonstrated by CMA in 18–31% of patients with CHD [1, 3, 6,7,8]. These studies differ on inclusion of syndromic versus isolated CHD, critical CHD versus all CHD, as well as description of CHD. Clinically relevant recommendations for testing and evaluation are available, but have yet to be fully embraced [9]. Previous assessments of rate of genetic diagnosis following genetic evaluation in infants with critical CHD were 25% [10]. This quality improvement project was focused on increasing the consistency of genetic testing in infants with critical CHD and looking at the effects a genetic testing protocol had on genetic testing utilization and diagnosis rate.
Materials and Methods
Study Overview
In order to determine the rates of genetic testing in infants with critical CHD the genetic testing practices and yields for two time periods were examined. Baseline data were obtained through a project approved by the Institutional Review Board which resulted in the genetic testing protocol. Follow-up data were obtained as part of an Institutional Review Board exempt status quality improvement project determining the adherence to the protocol.
Patient Population
The Society for Thoracic Surgeons Database was queried for infants who had CHD requiring surgical intervention at less than a year of age. The first time period, or “pre-protocol” time period, includes infants who underwent cardiac surgery at less than one year of age on or after January 1, 2010 who were born on or before December 31, 2014. The second time period, or “post-protocol” time period, includes infants born between January 1, 2015 and June 30, 2016 who had cardiac surgery at less than a year of age before June 30, 2016. Infants born at less than 30 weeks gestation or who underwent patent ductus arteriosus repair were excluded.
Genetic Testing
All genetic testing recorded in the medical record was identified and was marked as normal or abnormal based on how it was clinically reported. Focus was on the three most commonly utilized genetic tests addressed by the protocol: G-Banded Karyotype, diagnostic testing for 22q11.2 deletion, and chromosomal microarray (CMA). The G-banded Karyotype has a resolution of identifying chromosomal abnormalities greater than five megabases. Karyotype testing can detect balanced translocations and structural changes not resulting in a change in copy number that could be missed by microarray and can detect low level chromosomal mosaicism. The method for diagnostic testing for 22q11.2 deletion included florescence in situ hybridization (FISH) for TUPLE1 and quantitative polymerase chain reaction (QPCR) for TBX1. FISH probe for TUPLE1 has a sensitivity of greater than 95% for 22q11.2 deletion syndrome and was available for the duration of the study. QPCR for TBX1 copy number became available to infants at Children’s Hospital of Wisconsin in August 2011 and has a sensitivity of greater than 95% for 22q11.2 deletion syndrome. In September 2012 this assay began to include copy number of CRKL to further enhance the sensitivity of detecting 22q11.2 deletions. The CMA utilized at Children’s Hospital of Wisconsin reports deletions larger than 200 kilobases and duplications larger than 500 kilobases. CMA is the most sensitive genetic test out of these options, and will pick up everything detectable by karyotype and 22q11.2 deletion testing with the exclusion of balanced translocations and low level chromosomal mosaicism which may only be detectable by karyotype.
Protocol Implementation
The genetic testing protocol was implemented on January 1, 2015. The protocol was introduced to the department of pediatrics and affected clinical divisions (cardiac intensive care unit, cardiology, cardiothoracic surgery, genetics, and neonatal intensive care unit) through departmental lectures and division meetings starting in April 2014. Our basic testing protocol is demonstrated in Table 1.
Diagnosis Rate
All genetic diagnoses listed in the medical record were noted (e.g., Hemophilia, Sickle Cell Disease). Genetic diagnoses associated with congenital heart disease (e.g., CHARGE Syndrome, Trisomy 21) were used to classify patients as being diagnosed with a genetic condition. For the specific protocol diagnosis rate the genetic diagnosis was only recorded if it was a direct result of the three target tests ordered. If a patient had a genetic diagnosis not made by the testing it was not recorded (e.g., a patient with Trisomy 21 underwent screening for 22q11.2 deletion only or if a patient with CHARGE Syndrome had a normal microarray) and this protocol diagnosis rate was utilized to determine the calculation of cost per diagnosis. All charts were reviewed for genetic diagnosis by a single author for consistency (GCG).
Value and Cost Analysis
In order to determine the effect of the protocol on cost to patients we assessed cost and compared pre-protocol to post-protocol periods. To determine cost baselines we queried three large genetic testing providers on the same day and averaged the quoted prices of the three providers and averaged them to create a cost for each test. The average costs at a single discrete time point were utilized to standardize for changes in cost over time. These were $895.17 for karyotype, $669.92 for 22q11.2 Deletion Testing, and $2615.33 for CMA. To determine the effect on value we used the cost data per patient and per diagnosis.
Data Analysis
Descriptive statistics identified the proportion of specific CHD group for each classification system represented within the sample and the frequency of abnormal genetic test results in proportion to tests completed. Two-tiered Fisher’s exact test was utilized to determine statistical significance of changes between groups.
Results
Population Description
Medical records of 891 infants were examined. There were 509 (57%) male infants and 382 (43%) female infants. Among these 891 infants 109 were diagnosed with Trisomy 21 and 36 were diagnosed with 22q11.2 Deletion Syndrome. Overall 562 (63%) had at least one of the three genetic tests completed. Population characteristics are summarized in Table 2 by time period.
Testing Practices
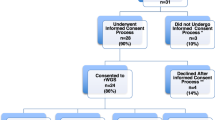
During the pre-protocol time period 66% of patients who had genetic testing underwent multiple tests. During the post-protocol time period 24% of patients who had genetic testing underwent multiple tests. This was found to be a significant decrease (p < 0.01) in utilization of multiple genetic tests. Testing patterns across the two time periods are demonstrated in Fig. 1.

Genetic testing patterns by Time Period. Pattern of using multiple genetic testing pre-protocol (left) and post-protocol (right) demonstrating reduction in frequency of undergoing multiple genetic tests at once
During the pre-protocol time period 60% of patients had genetic testing. During the post-protocol time period 77% of patient had genetic testing. This was found to be a significant increase (p < 0.01) of patients undergoing genetic testing in the post-protocol period.
Yield of Testing and Diagnosis Rate
In the pre-protocol time period the rate of abnormal karyotypes was 18%, the rate of abnormal 22q11.2 deletion testing was 9%, and the rate of abnormal microarray was 24%. In the post-protocol time period the rate of abnormal karyotype was 76%, the rate of abnormal 22q11.2 deletion testing was 26%, and the rate of abnormal microarray was 22%. The increase in yield of karyotype was found to be significant (p < 0.01) as was the increase in yield of 22q11.2 deletion testing (p = 0.03). There was no difference in yield of microarray between the pre-protocol and post-protocol time periods (p = 0.78). This is illustrated in Table 3. There were no results detected by karyotype or 22q11.2 deletion testing in this cohort that was not detectable with microarray in this patient population.
The breakdown of testing patterns and yield of testing by lesion is demonstrated in Table 4 which classifies cardiac lesions based on the National Birth Defect Prevention Study classification system [10]. Results of this breakdown are as anticipated. Trisomy 21 is more likely to be found in patients with atrioventricular septal defects making the karyotype more likely to be abnormal in patients with these lesions. Patients with 22q11.2 Deletion Syndrome are more likely to have conotruncal lesions and 22q11.2 deletion testing is more likely to be abnormal in this group. Of note, there was a higher yield for microarray testing in conotruncal lesions than 22q11.2 deletion testing alone, likely reflecting the increased clinical utility of microarray in detecting genetic disorders. There was no significant difference in yield of microarray across lesion types with a single exception. Patient with septal lesions who had microarray were significantly more likely to have an abnormal microarray (p = 0.0005). We suspect this is a reflection of clinical ascertainment bias given that only 29% patients with septal lesions had microarray testing completed.
The rate of diagnosis of genetic conditions associated with congenital heart disease during the pre-protocol period was 26%. The rate of diagnosis of genetic conditions associated with congenital heart disease during the post-protocol period was 36%. This increase in diagnosis rate was significant (p = 0.01). When patients with Trisomy 21 were excluded the rate of diagnosis of genetic conditions during the pre-protocol period was 16% and during the post-protocol period was 26%. This increase in diagnosis rate among patients without Trisomy 21 was also significant (p = 0.01).
Cost of Testing
Prior to the protocol initiation, fewer patients underwent genetic testing (60 vs. 77%), but of the patients who did undergo genetic testing, they underwent multiple tests more frequently (66 vs. 24%, p < 0.01). As result, the average cost per patient for genetic testing was higher prior to the initiation of the protocol, $2720.77 verse $2403.27. When we analyzed the cost per genetic diagnosis from testing ordered we found similar results with the average cost per genetic diagnosis being significant less after the protocol was initiated, $11,427.24 verse $6321.65. The cost of testing per time period is further demonstrated in Table 5.
Discussion
Our data demonstrate the utility of thoughtful genetic testing protocols in infants with critical CHD. The protocol significantly increased the rate of diagnosis of genetic conditions. A significant concern in implementing this protocol was that the yield of CMA would go down with expanded testing as appropriate individuals were undergoing testing and there was selection bias in the pre-protocol population. Our data demonstrate that the yield of microarray is consistent in this population and expanded testing resulted in an increased number of children identified to have chromosomal anomalies. Our diagnosis rate of genetic conditions associated with congenital heart disease is significantly higher than what has been published. The most recent assay of infants with critical congenital heart disease who underwent genetic assessment reported a diagnosis rate of 25% [11]. One potential influence on our data is that a dedicated clinical cardiovascular genetics program was started 6 months into the post-protocol period and the increased participation of geneticists in the care of this population. The diagnosis rate for this new program has been high compared to published rates at 39% (38/97) for infants with CHD assessed less than one year of age without Trisomy 21. This likely synergistically affected the diagnosis rate for this population of diagnoses not detected with CMA.
Importantly, our data suggest that the protocol reduced the overall cost of testing for patients. While the protocol resulted in more patients undergoing genetic testing, it reduced the number of patients undergoing multiple tests at once, which reduced the overall cost per patient by $317.50. Costs and charges are complex and can vary significantly between institutions. There was sincere concern that implementing this protocol could increase costs to patients and families. Our data suggest that a genetic testing protocol has the opposite effect. The cost per diagnosis was reduced significantly by $5105.59. This reflects that in addition to patients undergoing fewer redundant tests at once, more patients were undergoing more appropriate testing. More appropriate use of genetic testing is further evident by the statistically significant increase in yield for karyotype (18–76%) and 22q11.2 deletion testing (9–26%) after initiation of the protocol.
There are still clinical nuances to choosing genetic testing this protocol does not full address. Microarray testing has increased resolution to detect clinically significant chromosomal anomalies compared to karyotype, but important limitations of microarray include the inability to detect balanced translocations or determine the structural location of an anomaly as well as the inability to detect low level chromosomal mosaicism, which can be important when considering mosaic conditions like Turner Syndrome. Microarray is also not always the best test for the situation. For example, testing with karyotype Trisomy 21 is still appropriate as karyotype is the recommended standard of care to assess for translocations [12]. All of these genetic tests still have clinical roles, but our data suggest that a protocol helps patients with critical congenital heart disease undergo more appropriate clinical application of these tests.
Our study does have limitations. We are a tertiary referral center, and as a result it is possible that additional testing following discharge was performed at an outside institution that was not available at the time of chart review. As all charts were reviewed for testing by a single author (GCG) with a consistent, standard method it is likely limitation would be uniform throughout each time period. In addition, cost and value calculations are highly variable and continue to evolve over time, making retrospective reviews challenging.
Despite these limitations, we feel that our data support a standardized genetic testing protocol for infants with critical cardiac disease can significantly impact the rate of identification of chromosomal anomalies and is cost effective in this complex, high risk population. We recommend institutions consider implementing genetic testing protocols for this population, ideally in conjunction with a medical geneticist or genetic counselor.
References
Bachman KK, DeWard SJ, Chrysostomou C, Munoz R, Madan-Khetarpal S (2013) Array CGH as a first-tier test for neonates with congenital heart disease. Cardiol Young 25:115–122
Baker K, Sanchez-de-Toledo J, Munoz R, Orr R, Kiray S, Shiderly D, Clemens M, Wearden P, Morell VO, Chrysostomou C (2012) Critical congenital heart disease- utility of routine screening for chromosomal and other extracardiac malformations. Congenit Heart Dis 7:145–150
Connor JA, Hinton RB, Miller EM, Sund KL, Ruschman JG, Ware SM (2014) Genetic testing practices in infants with congenital heart disease. Congenit Heart Dis 9(2):158–167
Geddes GC, Butterly M, Sajan I (2015) FISH for 22q11.2 deletion not cost-effective for infants with congenital heart disease with microarray. Pediatr Cardiol 36:531–536
Hartman RJ, Rasmussen SA, Botto LD, Riehle-Colarusso T, Martin CL, Cragen JD, Shin M, Correa A (2011) The contribution of chromosomal anomalies to congenital heart defect: a population-based study. Pediatr Cardiol 32:1147–1157
Breckpot J, Thienpont B, Peeters H, deRavel T, Singer A, Rayyan M, Allegaert K, Vanhole C, Eyskens B, Vermeesch JR, Gewillig M, Devriendt K (2010) Array comparative genomic hybridization as a diagnostic tool for syndromic heart defects. J Pediatr 156:810–817
Goldmuntz E, Paluru P, Glessner J, Hakonarson H, Biegel JA, White PS, Gai X, Shaikh TH (2011) Microdeletions and microduplications in patients with congenital heart disease and multiple congenital anomalies. Congenit Heart Dis 6:592–602
Thienpont B, Mertens L, de Ravel T, Eyskens B, Boshoff D, Maas N, Fryns JP, Gewillig M, Vermeesch JR, Devriendt K (2007) Submicroscopic chromosomal imbalances detected by array-CGH are a frequent cause of congenital heart defects in selected patients. Eur Heart J 28:2778–2784
Cowan JR, Ware SM (2015) Genetics and genetic testing in congenital heart disease. Clin Perinatol 42(2):373–393
Botto LD, Lin AE, Riehle-Colarusso T, Malik S, Correa A, NBDPS (2007) Seeking causes: classifying and evaluating congenital heart defects in etiologic studies. Birth Defects Res A 79:714–727
Ahrens-Nicklas RC, Khan S, Garbarini J, Woyciechowski S, D’Alessandro L, Zackai EH, Deardorff MA, Goldmuntz E (2016) Utility of genetic evaluation in infants with congenital heart defects admitted to the cardiac intensive care unit. Am J Med Genet Part A 9999A:1–8
Bull MJ, American Academy of Pediatrics Committee on Genetics (2011) Clinical report- health supervision for children with down syndrome. Pediatrics 128(2):393–406
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare they have no conflicts of interest.
Ethical Approval
There were no procedures performed on human subjects. This project was in accordance with the ethical standards of the institutional review board and in accordance with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. For this type of study formal consent is not required.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Geddes, G.C., Basel, D., Frommelt, P. et al. Genetic Testing Protocol Reduces Costs and Increases Rate of Genetic Diagnosis in Infants with Congenital Heart Disease. Pediatr Cardiol 38, 1465–1470 (2017). https://doi.org/10.1007/s00246-017-1685-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00246-017-1685-7