
Abstract
A 38-year-old woman was discovered to have a systolic murmur for an unrelated complaint. Transesophageal echocardiography showed no atrial or ventricular septal defects, but multiple large collateral vessels in inter-ventricular septum. The origin of left coronary artery was not seen at the expected site on the aortic root. The 64-multislice computed tomography confirmed the diagnosis of an anomalous origin of the left coronary artery from the pulmonary artery. Left coronary artery was revascularized with a saphenous vein graft with an uneventful recovery.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
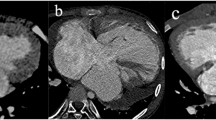
During physical examination, a 38-year-old woman was found to have an asymptomatic systolic murmur. The electrocardiogram was normal, and the chest radiograph showed mild cardiomegaly. Transthoracic echocardiography showed a moderately decreased systolic function (ejection fraction = 35%), no mitral regurgitation, and multiple muscular ventricular septal defects (VSDs), i.e., a possible “Swiss cheese” VSD. In contrast, the transesophageal echocardiogram showed no atrial or ventricular septal defects; instead, there were multiple large collateral vessels in the interventricular septum. Because the origin of the left coronary artery (LCA) was not visualized, and to better clarify the anatomy, a contrast-enhanced 64-multislice computed tomogragraphy (MSCT) was performed. The tomogram showed a dilated and tortuous right coronary artery arising from the right sinus of Valsalva and giving off extensive collateral vessels coursing over the right ventricular wall, the interventricular septum, and the apex to the LCA that originated from the proximal main pulmonary artery (Fig. 1a–c). Therefore, the diagnosis of anomalous origin of the LCA from the pulmonary artery (ALCAPA) was confirmed. Technically, transfer of the LCA from the PA was not feasible; thus, the LCA coronary origin was oversewn and a saphenous vein graft from the aorta to the LCA was performed. The patient’s postoperative course was uneventful.

(a) A 64-MSCT shows severe dilatation of right coronary artery. (b) Several collateral arteries connecting left and right coronary arteries are shown. (c) ALCAPA is shown
ALCAPA, also known as Bland–White–Garland syndrome, is a rare congenital anomaly and occurs in 1 in 300,000 live births (0.25% to 0.5%) [2, 4]. Few patients survive past childhood without surgical repair, and without surgical intervention nearly 90% die within the first year of life [3]. Improved imaging technologies now provide accurate diagnosis and provide opportunities for treatment planning. Two-dimensional Doppler echocardiography is considered the major diagnostic tool for ALCAPA; however, it has limitations, as illustrated by the present case [1]. The first echocardiographies in our patient did not show the anomaly. This may have been caused by an incomplete echocardiography examination due to the lack of awareness of doctors of this syndrome leading to multiple collateral arteries in the interventricular septum be misdiagnosed as multiple VSDs.
In conclusion, although ALCAPA is rare and usually an isolated anomaly, a high index of suspicion is required for diagnosis. MSCT is a useful modality for diagnosing congenital coronary anomalies. In addition, the surgeon should be aware of the possibility of misdiagnosing the collateral arteries in the septum by multiple VSDs and should not commit to repair the VSDs before a modality of high sensitivity for structural and vascular anomalies, such as 64-MSCT, makes the entity certain.
References
Drighil A, Chraibi S, Bennis A (2006) Adult type anomalous origin of the left coronary artery from the pulmonary artery: When should we be aware? Int J Cardiol 113:E119–E121
Lange R, Vogt M, Hörer J, Cleuziou J, Menzel A, Holper K et al (2007) Long-term results of repair of anomalous origin of the left coronary artery from the pulmonary artery. Ann Thorac Surg 83:1463–1471
Wesselhoeft H, Fawcett JS, Johnson AL (1968) Anomalous origin of the left coronary artery from the pulmonary trunk. Its clinical spectrum, pathology, and pathophysiology, based on a review of 140 cases with seven further cases. Circulation 38:403–425
Wollenek G, Domanig E, Salzer-Muhar U, Havel M, Wimmer M, Wolner E (1993) Anomalous origin of the left coronary artery: A review of surgical management in 13 patients. J Cardiovasc Surg (Torino) 34:399–405
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Jebelli, M., Kernstine, K., Mandegar, M.H. et al. The 64-Multislice Computed Tomogram Averts Misdiagnosis of an Anomalous Origin of the Left Main Coronary Artery. Pediatr Cardiol 30, 1184–1185 (2009). https://doi.org/10.1007/s00246-009-9508-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00246-009-9508-0