
Abstract
The subfamily Pothoideae belongs to the ecologically important plant family Araceae. Here, we report the chloroplast genomes of two species of the subfamily Pothoideae: Anthurium huixtlense (size: 163,116 bp) and Pothos scandens (size: 164,719 bp). The chloroplast genome of P. scandens showed unique contraction and expansion of inverted repeats (IRs), thereby increasing the size of the large single-copy region (LSC: 102,956 bp) and decreasing the size of the small single-copy region (SSC: 6779 bp). This led to duplication of many single-copy genes due to transfer to IR regions from the small single-copy (SSC) region, whereas some duplicate genes became single copy due to transfer to large single-copy regions. The rate of evolution of protein-coding genes was affected by the contraction and expansion of IRs; we found higher mutation rates for genes that exist in single-copy regions as compared to those in IRs. We found a 2.3-fold increase of oligonucleotide repeats in P. scandens when compared with A. huixtlense, whereas amino acid frequency and codon usage revealed similarities. The ratio of transition to transversion mutations was 2.26 in P. scandens and 2.12 in A. huixtlense. Transversion mutations mostly translated in non-synonymous substitutions. The phylogenetic inference of the limited species showed the monophyly of the Araceae subfamilies. Our study provides insight into the molecular evolution of chloroplast genomes in the subfamily Pothoideae and family Araceae.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The plant family Araceae belongs to the order Alismatales. Araceae is a large and ancient monocot family that dates back to the Early Cretaceous period (Nauheimer et al. 2012). This family consists of 144 genera and 3645 species (Boyce and Croat 2018). It is the most diverse monocotyledon family in terms of morphology, encompassing the smallest known angiosperms (Wolffia Schkeid. species) as well as some of the plants having the largest vegetative and reproductive structures [Amorphophallus titanum (Becc) Becc. ex Arcang] (Gunawardena and Dengler 2006). Ecologically, species of Araceae are highly versatile, ranging from submerged, emergent or free-floating aquatics to epiphytic, climbing and terrestrial plants (Cabrera et al. 2008). Species of Araceae have been subdivided into eight subfamilies, distributed in tropical and temperate regions (Cabrera et al. 2008; Cusimano et al. 2011; Nauheimer et al. 2012). Pothoideae is the second largest subfamily, with approximately 1010 described species and approximately 2072 estimated species (Boyce and Croat 2018). The subfamily is divided into two tribes: Tribe Potheae which includes the genera Pothos L., Pedicellarum M.Hotta, and Pothoidium Schott, and the monogeneric Tribe Anthurieae comprised of species of Anthurium Schott (Mayo et al. 1997; Cabrera et al. 2008; Cusimano et al. 2011; Chartier et al. 2014). Members of Pothoideae are recognized by fine reticulate venation, complete lack of laticifers, usually aerial stems, apically geniculate petioles, bisexual perigoniate flowers, and a simple spathe not enclosing the spadix (Mayo et al. 1997). Pothoideae contains many climbing and hemiepiphytic species that live in humid tropical forests. The largest genera are Pothos and Anthurium, with approximately 57 and 950 described, and approximately 70 and 2000 estimated species, respectively (Boyce and Croat 2018). The other two genera, Pothoidium and Pedicellarum, are monospecific. Pothos is distributed in south and Southeast Asia, Australia, the Malagasy region, and the Malay Archipelago, while Anthurium is a strictly Neotropical genus that ranges from southern Mexico to southern Brazil, extending into the West Indies (Mayo et al. 1997; Carlsen and Croat 2013).
The chloroplast is a self-replicating organelle that plays a vital role in photosynthesis and in the synthesis of fatty acids and amino acids (Cooper 2000). In most plant lineages, the chloroplast contains its own circular double-stranded genome and has a primarily quadripartite structure in which a pair of long inverted repeat regions (IRa and IRb) separate the large single-copy (LSC) and small single-copy (SSC) regions (Palmer 1985). However, linear chloroplast genomes have also been reported (Oldenburg and Bendich 2016) in some species. Moreover, a quadripartite structure has not been observed in the chloroplast genomes of various species, such as Pinaceae (Wu et al. 2011), Cephalotaxaceae (Yi et al. 2013), and Taxodiaceae (Hirao et al. 2008). The size of the chloroplast genome of photosynthetic plants varies from 107 kb (Cathaya argyrophylla Chun & Kuang) to 218 kb (Pelargonium x hortorum L.H.Bailey) (Daniell et al. 2016). Chloroplast genomes are inherited from a single parent and show significant polymorphism (Daniell 2007; Daniell et al. 2016), which makes them well-suited for studies on phylogenetics and population genetics (Ahmed et al. 2013; Henriquez et al. 2014; Ahmed 2015).
Despite a relatively conserved structure, including gene organization, gene content, and intron content within genes (Iram et al. 2019; Mehmood et al. 2020; Shahzadi et al. 2020), chloroplast genomes have also undergone gene loss, intron loss, gene rearrangement, pseudogenization, gene duplication, and uneven expansion and contraction of IR regions. These events have led to a variable number of genes in the chloroplast genomes of angiosperms (Menezes et al. 2018; Abdullah et al. 2020b; Henriquez et al. 2020b). Moreover, the shifting of genes to single-copy regions from IR or vice versa due to IR contraction and expansion also affect the rate of DNA sequence evolution; the phenomenon is known as rate heterotachy (Lockhart et al. 2006). Previous studies of subfamilies Lemnoideae and Aroideae revealed unique and uneven contraction and expansion of IR regions, which led to a variable number of genes and gene rearrangements in the chloroplast genomes of several of their respective taxa (Wang and Messing 2011; Choi et al. 2017; Tian et al. 2018; Kim et al. 2019; Henriquez et al. 2020b). The aforementioned studies did not include species of the subfamily Pothoideae.
In this study, a comparison of the de novo assembled chloroplast genomes of A. huixtlense Matuda and P. scandens L. with chloroplast genomes of other Araceae species confirmed unique events of IR contraction and expansion in the chloroplast genome of P. scandens. The results revealed the transfer of IR genes to the LSC region at the junction of JLB (LSC/IRb) and the transfer of all SSC genes (except rps15 and ycf1) to the IR region at the junction of JSB (IRb/SSC). This transfer decreased the size of SSC region to 6779 bp and promotes heterotachy in Pothoideae by affecting the rate of evolution of these genes. To the best of our knowledge, the shortening of the SSC region to such an extent and the effects on genes evolution rate are reported here in Araceae for the first time. These results improve our understanding of the evolution of chloroplast genomes in Araceae.
Materials and Methods
DNA Extraction and Sequencing
We collected fresh healthy leaves of P. scandens and A. huixtlense from the Aroid Greenhouse at the Missouri Botanical Garden in St. Louis, Missouri. Total genomic DNA was extracted from these leaves using a Qiagen DNeasy Minikit (Qiagen, Germantown, Maryland, USA) following Henriquez et al. (2020a). Confirmation of the quality and quantity of DNA was performed using 1% gel electrophoresis and Nanodrop (ThermoScientific, Delaware, USA). Library preparation and sequencing were performed using TruSeq kits (Illumina, Inc., San Diego, California) in the Pires lab at the University of Missouri, Columbia following Henriquez et al. (2020a).
De novo Assembly and Annotation
The quality of raw reads was analyzed by FastQC (Andrews 2017) and MultiQC (Ewels et al. 2016) for comparison. After quality confirmation, the Fast-Plast v. 1.2.2 pipeline (https://github.com/mrmckain/Fast-Plast) was initially used to assemble the raw reads following similar parameters previously employed for the assembly of chloroplast genomes of subfamilies Aroideae and Monsteroideae (Henriquez et al. 2020a,b). The resulting assembly from Fast-Plast was further confirmed by de novo assembly using Velvet v.1.2.10 following Abdullah et al. (2019a, 2020b) using Kmer values of 61, 71, and 81. Validation and coverage depth analyses of de novo assembled genomes were performed by mapping short reads to their respective assembled chloroplast genomes. The assembled chloroplast genomes were annotated using GeSeq (Tillich et al. 2017) and the circular diagrams of the annotated genomes were drawn using OrganellarGenomeDRAW (OGDRAW v.13.1) (Greiner et al. 2019). The five-column tab-delimited tables were generated for de novo assembled chloroplast genomes using GB2sequin (Lehwark and Greiner 2019) and were submitted to the National Center for Biotechnology Information (NCBI) under accession number MN046891 (P. scandens) and MN996266 (A. huixtlense). The raw reads were also submitted to the sequence read archive (SRA) of NCBI under BioProject number PRJNA547619.
Characterization and Comparative Analyses of Chloroplast Genomes
Characterization of the chloroplast genomes of P. scandens and A. huixtlense and analyses of amino acid frequency and codon usage were performed in Geneious R8.1 (Kearse et al. 2012). Oligonucleotide repeats were determined using REPuter (Kurtz et al. 2001) by setting the parameter of minimum repeat size ≥ 30 and with minimum repeat similarity of 90%.
The chloroplast genome structure and gene content of P. scandens and A. huixtlense were compared with eight previously reported chloroplast genomes of Araceae, including Anchomanes hookeri (Kunth) Schott, Anubias heterophylla Engler, Zantedeschia aethiopica (L.) Spreng. (Henriquez et al. 2020b), Epipremnum amplissimum (Schott) Engl. (Henriquez et al. 2020a), Spathiphyllum kochii Engl. & K. Krause (Han et al. 2016), Spirodela polyrrhiza (L.) Schleid., Wolffiella lingulata Hegelm. (Wang and Messing 2011), and Symplocarpus renifolius Schott ex Tzvelev (Choi et al. 2017). The gene content and rearrangement of the genome were compared by integrated Mauve alignment (Darling et al. 2004) in Geneious R8.1 based on collinear blocks analyses. IR contraction and expansion were studied among these species using IRscope (Amiryousefi et al. 2018a).
We also analyzed synonymous (Ks) and non-synonymous (Ka) substitutions and their ratio (Ka/Ks). Symplocarpus renifolius, a species from the early diverging subfamily Orontioideae, was used as a reference and 75 protein-coding genes of P. scandens and A. huixtlense were aligned to the protein-coding genes of S. renifolius by MAFFT alignment (Katoh et al. 2005). These alignments were analyzed for the determination of Ks and Ka substitutions and Ka/Ks using DnaSP (Rozas et al. 2017) as reported previously (Abdullah et al. 2019a, 2020b; Henriquez et al. 2020a). We further elucidated selective pressure on protein-coding genes using additional codon models such as branch-site unrestricted statistical test for episodic diversification (BUSTED) (Murrell et al. 2015) and fast unconstrained Bayesian AppRoximation (FUBAR) (Murrell et al. 2013) with HyPhy (Pond et al. 2005) using the Datamonkey server (Delport et al. 2010). BUSTED was used with default parameters to investigate diversifying selection on selected genes. FUBAR was used with posterior probability of > 0.9 to identify episodic/diversifying selection on codons sites.
We also determined the extent of transition (Ts) and transversion (Tv) substitutions linked with Ks and Ka substitutions. For this purpose, we selected 11 genes from the genome of P. scandens that had various Ka/Ks values and analyzed the extent of transition and transversion types of substitutions with Ks and Ka substitutions in Geneious R8.1 (Kearse et al. 2012) following Abdullah et al. (2019a).
We analyze the effect of rate heterotachy on the evolution of protein-coding genes, using S. renifolius as a reference. We considered genes of LSC, SSC, and IR of S. renifolius and determined the rate of evolution of the respective genes in the chloroplast genomes of P. scandens and A. huixtlense. We also separately determined the rate of evolution of protein-coding genes that were transferred from IRs to LSC or from SSC to IR to elucidate the changes in evolution rate. We concatenated genes of each region and aligned using MAFFT (Katoh et al. 2005). The types of transition and transversion substitutions in P. scandens and A. huixtlense were also determined from the alignment of genes from LSC, SSC, and IR.
Phylogenetic Inference
The phylogenetic tree was inferred using 31 species of Araceae, with Acorus americanus (Acoraceae) as the outgroup (Table S1). The complete chloroplast genomes, excluding IRa, were aligned by MAFFT (Katoh et al. 2005) and the phylogeny was inferred using the IQ-tree program (Nguyen et al. 2015; Kalyaanamoorthy et al. 2017; Hoang et al. 2018) with default parameters, as reported previously (Abdullah et al. 2019a, 2020b).
Results
Assembly and Characterization of Chloroplast Genomes
The sequencing of 100 bp single-end reads generated 3.69 GB data (14.13 million reads) for A. huixtlense and 5.8 GB data (22.2 million reads) for P. scandens. Whole-genome shotgun reads contained 0.22 million reads in A. huixtlense and 0.77 million reads of chloroplast origin in P. scandens. These chloroplast reads were used for de novo assembly and provided average coverage depths of 468 × for P. scandens and 138 × for A. huixtlense.
The sizes of the complete chloroplast genomes were 163,116 bp for A. huixtlense and 164,719 bp for P. scandens. The sizes of the LSC and SSC regions showed a high level of variation between the two species due to unique IR contraction and expansion in the P. scandens chloroplast genome (Table 1). The GC content was highest in IR regions, followed by LSC and SSC regions. A high level of variation exists in the GC content of the chloroplast genome of both species.
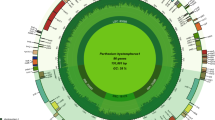
We found 113 unique functional genes in both species, including 79 protein-coding genes, 30 tRNA genes, and 4 rRNA genes (Fig. 1a, b). The infA gene was observed as a pseudogene in both species, whereas the rpl23 gene was observed as pseudogene in P. scandens due to the generation of an internal stop codon. The total number of genes varied between the two species due to IR contraction and expansion. We found 130 genes in A. huixtlense, including 37 tRNA genes, 85 protein-coding genes, and 8 rRNA genes. We also observed 17 genes that were duplicated in the IR regions in A. huixtlense, including 7 tRNA genes (2 genes also contain introns), 4 rRNA genes, and 6 protein-coding genes (3 genes also contain introns) (Fig. 1a). In P. scandens, we found 135 genes due to expansion of the IR region, including 36 tRNA genes, 90 protein-coding genes, and 8 rRNA genes (Fig. 1b). We found 22 genes that were duplicated in the IR regions in P. scandens, including 6 tRNA genes (2 genes also contain introns), 4 rRNA genes, and 12 protein-coding genes (2 genes also contain introns) (Fig. 1b).

Circular diagram of chloroplast genomes of A. huixtlense and P. scandens. LSC, SSC, and IR represent large single-copy, small single-copy, and inverted repeat regions, respectively. The genes located inside are transcribed counterclockwise, whereas the genes located outside are transcribed clockwise
Amino Acid Frequency and Codon Usage
The highest frequency observed was for leucine followed by iso-leucine, whereas the lowest frequency observed was for cysteine (Fig. 2). Relative synonymous codon usage (RSCU) analyses revealed high encoding frequency for codons containing A or T at the 3′ end and having an RSCU value of ≥ 1, whereas low encoding frequency was observed for codons having C or G at the 3′ and having RSCU < 1 (Table S2).

Amino acid frequency in A. huixtlense and P. scandens. The x-axis shows amino acids, whereas the Y-axis shows percentage of amino acid frequency
Repeats Analyses
REPuter detected four types of oligonucleotide repeats in the chloroplast genomes of A. huixtlense and P. scandens. The number of repeats and types varied in both species to a high degree. We observed 37 repeats in A. huixtlense and 85 repeats in P. scandens. We observed 9 forward, 12 palindromic, 6 complementary, and 10 reverse repeats in A. huixtlense. In P. scandens we observed 21 forward, 33 palindromic, 8 complementary, and 23 reverse repeats (Fig. 3a). Most of the repeats were found in LSC regions instead of SSC and IR regions (Fig. 3b). Most of the repeats ranged in size from 40 to 44 bp in A. huixtlense. In P. scandens, most of the repeats varied in size from 35 to 39 bp (Fig. 3c). Details are provided in Table S3.

Analyses of repeats in A. huixtlense and P. scandens. a Types of repeats. b Distribution of repeats in the chloroplast genomes. c The size of repeats in the genome. F Forward, C complementary, R reverse, P palindromic, LSC large single copy, SSC small single copy, IR inverted repeat, LSC/SSC and LSC/IR represent shared repeats between regions
Evolutionary Rate of Protein-Coding Genes
The evolution rate of protein-coding genes revealed strong purifying selection on these genes and that none of the genes are under positive selection pressure. Except for a few genes that showed neutral selection, all other genes showed purifying selection (Table S4) (average Ks = 0.16, Ka = 0.026, and Ka/Ks = 0.18). As expected, the highest purifying selection pressure was observed for genes that are involved in photosynthesis.
The results of codon models were similar to the Ka/Ks method, and none of the genes was found to be under positive selection in the subfamily Pothoideae using BUSTED. The mixture model implemented in BUSTED needs a relatively high fraction of sites (5–10%) to be under positive selection for accurate detection. At a next step, we implemented FUBAR to detect rare sites that might be under positive selection. These tests showed a few codons under positive selection in genes rpl22 (2 codons), ycf1 (8 codons), and ycf2 (5 codons) (Table S5).
In the protein-coding genes of P. scandens, we found 4061 substitutions in comparison with Symplocarpus renifolius reference genome. Of these, 2814 contained transition (Ts) substitutions and 1247 contained transversion (Tv) substitutions; the Ts/Tv ratio was 2.26. In A. huixtlense, we recorded 3960 substitutions, of which 2690 were Ts and 1270 were Tv; the Ts/Tv ratio was 2.12 (Table 2). Examination of 11 protein-coding genes revealed a Ts/Tv of 2.79 for synonymous substitutions and a Ts/Tv of 1.43 for non-synonymous substitutions. Hence, a higher number of Tv leads to non-synonymous substitutions as compared to Ts and vice versa.
Gene Arrangement and Inverted Repeats Contraction and Expansion
The genomes of Pothoideae show unique gene and structural rearrangements. The P. scandens chloroplast genome showed unique IR contraction and expansion, which led to a variable number of genes and also a change in gene arrangement. At the JLB junction (LSC/IRb), the contraction of IR resulted in expansion of the LSC region, whereas at the JSB (IRb/SSC) junction, the expansion of IR resulted in contraction of the SSC region. Hence, genes including rpl2, rpl23, trnM, ycf2, trnL, ndhB that are usually duplicated in the IRs became single copy after their transfer to LSC. In contrast, genes such as ndhH, ndhA, ndhI, ndhG, ndhE, psaC, ndhD, ccsA, trnL, rpl32, and ndhF that usually exist as single copy in SSC were duplicated after their transfer to IR regions (Fig. 1b). The arrangement of genes in LSC in both A. huixtlense and P. scandens did not change due to contraction of IR regions and gene arrangement was found to be similar to other species (Spathiphyllum kochii, E. amplissimum, S. renifolius, and A. heterophylla), as shown in Colinear block of Mauve alignment (Fig. 4). However, the genes of the SSC region showed variation in gene arrangement (Fig. 4). In the genome of A. huixtlense, the SSC was inverted when compared to other species of Aroideae. However, this could not be considered an important evolutionary event as chloroplast genomes exist in two equimolar states and can be differentiated by orientation of the SSC region (Walker et al. 2015).

Analyses of gene arrangements among the species of Araceae based on Mauve alignment. White boxes indicate protein-coding genes, red indicate rRNA, black indicate tRNA, green indicate intron-containing tRNA, and the line between two white boxes indicates intron-containing protein-coding genes
The contraction and expansion of IR regions at the junctions JLB (LSC/IRb), JSB (IRb/SSC), JSA (SSC/IRa), and JLA (IRa/LSC) were analyzed among the species of Araceae. We observed five types of variation in the junctions (Fig. 5). Type A included P. scandens, type B included A. huixtlense, E. amplissimum, S. kochii, S. renifolius, and A. heterophylla, type C included Wolffiella lingulata and Spirodela polyrhiza, type D included Z. aethiopica, and Type E included A. hookeri. These results show that the P. scandens chloroplast genome displays a novel type of IR contraction and expansion.

Comparative analysis of junction sites in Araceae chloroplast genomes. Abbreviations denote junction site of the plastid genome JLA (IRa/LSC), JLB (IRb/LSC), JSA (SSC/IRa) and JSB (IRb/SSC). Genes are represented by colored boxes while arrows are showing the coordinate positions of each gene near the junctions. Genes displayed on the top appear on the negative strand, while genes present bellow and found on the positive strand of the genome
Effects of Rate Heterotachy
Contraction and expansion in IRs affected the rate of evolution in protein-coding genes. The genes that were transferred from the SSC region to IR regions showed a decrease in the rate of evolution, whereas genes that were transferred from IR regions to the LSC region showed an increase in the rate of evolution. In P. scandens, we found 2454 (5.67%) substitutions in the genes located in LSC, 269 substitutions (2.64%) in the genes present in IRs, and 1338 (9.27%) substitutions in the genes found in SSC. In A. huixtlense, we found 2428 (5.62%) substitutions in genes of LSC, 205 (2.0%) substitutions in genes of IRs, and 1327 (9.16%) in genes of SSC. We found a higher rate of evolution in P. scandens genes than in A. huixtlense and observed a difference of 0.043% in genes of LSC, 0.64% in genes of IRs, and 0.11% in genes of SSC. We observed the highest difference in evolution rate between P. scandens and A. huixtlense in IRs. This might be due to transfer of most of the IR genes of P. scandens to LSC region. To further verify the effect of rate heterotachy, we separately compared the rate of evolution of those genes that transferred from SSC to IRs in P. scandens. Genes of P. scandens that were transferred from SSC to IRs showed 0.43% less evolution than genes of A. huixtlense, whereas average rate of evolution of the genes of all regions were found higher in P. scandens than in A. huixtlense. This confirmed that the transfer of the genes from single-copy regions to IRs is responsible for decreased evolution rates.
Phylogenetic Inference of the Family Araceae
The phylogenetic tree was reconstructed with the best fitting Model GTR + F + I + G4. The nucleotide alignment contained a total of 94,654 sites in which 65,927 were invariable, 14,393 were parsimony informative, and 9262 sites showed a distinct pattern. The phylogenetic tree supported the monophyly of the five subfamilies that were included in the study with 100% bootstrap support (Fig. 6). However, at subfamily level, the low bootstrap support was also observed for some nodes, specifically among the species of subfamily Aroideae (Fig. 6). The subfamily Pothoideae showed sister relationship with the subfamily Monsteroideae. The subfamily Orontioideae was the basal group, whereas Aroideae was the crown group.

Maximum likelihood phylogenetic tree of the Araceae family reconstructed from plastid genome data. a Phylogenetic tree and b cladogram
Discussion
In the current study, we assembled the chloroplast genomes of two species from subfamily Pothoideae of Araceae. The chloroplast genomes of both P. scandens and A. huixtlense were found to be unique among Araceae species and showed a unique type of IR contraction and expansion that affected the rate of evolution in P. scandens.
In the current study, the chloroplast genome of P. scandens showed uneven IR contraction and expansion, which led to a variable number of genes. IR contraction and expansion is very common in chloroplast genomes and leads to variation in the number of genes in various plant lineages, including Araceae (Ahmed et al. 2012; Menezes et al. 2018; Cho et al. 2018; Lee et al. 2018; Abdullah et al. 2020b; Henriquez et al. 2020b). IR contraction and expansion also results in new combinations of genes in the IR regions, which in turn leads to rearrangement of genes in the SSC region, as previously reported in Araceae (Wang and Messing 2011; Ahmed et al. 2012; Henriquez et al. 2020b). However, in P. scandens, we observed the formation of a new combination of genes in IRs but not an accompanying rearrangement of the genes. A similar effect of IR contraction and expansion was also reported in other plant lineages without any effect on the arrangement of genes (Wang et al. 2016; Cho et al. 2018; Lee et al. 2018). In P. scandens, the SSC region showed significant shortening and contained only two genes. Similar shortening of the SSC region was also reported in other angiosperms and even smaller SSC regions have been reported (Cho et al. 2018; Lee et al. 2018). Previously, four types of gene arrangements were observed in Araceae. Two types of gene arrangements were observed at IR junctions in one comparison of Araceae species (Choi et al. 2017) and two other types of gene arrangements at the junctions were reported in the chloroplast genomes of two species of subfamily Aroideae, including A. hookeri and Z. aethiopica (Henriquez et al. 2020b). In the current study, we reported a fifth type of gene arrangement at the junctions in the chloroplast genome of P. scandens. Further genomic resources from the genus Pothos and subfamily Pothoideae might be helpful to gain insight into chloroplast genome structure and to discern whether this uneven IR contraction and expansion occurs only in P. scandens or in the genus Pothos as a whole. The reason of IR contraction and expansion might be due to double strand break model and illegitimate recombination, as previously suggested in Mimosoideae (Wang et al. 2017).
The expansion of IR regions in the P. scandens genome reported here decreased the evolutionary rate of protein-coding genes that shifted from SSC to IR, whereas an increase in the evolutionary rate was observed in the genes that transferred from IR to LSC. Similar results were reported in the chloroplast genomes of other species and a higher rate was observed in the regions that exist in the single-copy region instead of IR region (Zhu et al. 2016). In contrast, the effect on evolutionary rate in Pelargonium was not observed due to IR contraction and expansion (Weng et al. 2017). This phenomenon might have been due to incipient transfer of genes in Pelargonium. The low rate of evolution in genes that exist in IR regions might be due to a repairing mechanism (Zhu et al. 2016).
The variations in chloroplast genomes are important for the studies of population genetics and phylogenetics and provide insight into evolutionary relationship of various taxa (Ahmed 2014; Henriquez et al. 2014). The polymorphisms of chloroplast genomes are also helpful to gain insight into the origin, geographical distribution, domestication and adaptation of plants to various climatic conditions (Daniell et al. 2016). Moreover, the polymorphisms of chloroplast genomes are useful in the identification of commercial cultivars (Suzuki et al. 2017), and identification of closely related and genetically compatible species for breeding (Daniell et al. 2016). Previously, ycf1 region was used for phylogenetic inference in the subfamily Monsteroideae (Zuluaga et al. 2019). Our study demonstrates that the rate of evolution for the genes at the junctions of single-copy and IR regions are affected by rate heterotachy. The utilization of such regions is amenable to misleading results in drawing phylogenetic inferences (Lockhart et al. 2006; Zhong et al. 2011). Hence, such genes should be avoided while studying phylogenetic relationships and determining times of divergence when using few loci. Genes which are important in domestication and adaption remain functional during stress conditions, despite occurrence of long inversion and high level of IR contraction and expansion (Daniell et al. 2016). In our study, no gene loss was evident despite significant contraction of SSC and expansion of IRs in P. scandens which provide insight into the important role of these genes in adaptation.
Chloroplast genomes are mostly conserved in terms of gene content and organization, and GC content of LSC, SSC and IR regions (Wang and Messing 2011; Ahmed et al. 2012; Iram et al. 2019; Abdullah et al. 2020b; Henriquez et al. 2020a,b; Shahzadi et al. 2020). We observed high GC content in the IR regions when compared with the LSC and SSC regions, consistent with previous reports. However, the IR regions of the P. scandens genome showed a decrease in GC content up to 5% when compared with A. huixtlense genome. This was due to expansion of the IR regions and subsequent inclusion of most of the genes of SSC region (which has low GC content). Average GC content of the regions of SSC that were transferred to IRs was 29.3%, whereas the average GC content of IRs genes was as high as 43%.
In our study, leucine and iso-leucine remained the most frequent, while cysteine remained the least frequent amino acid. Higher RSCU values (≥ 1) were found for codons with A or T at the 3′ position and showed high encoding efficacy. Similar results for amino acid frequency and codon usage have also been reported in the chloroplast genomes of other angiosperms, which might be due to the high overall AT content in the chloroplast genome (Amiryousefi et al. 2018b; Menezes et al. 2018; Abdullah et al. 2019b; Mehmood et al. 2020). The analyses of oligonucleotide repeats showed the existence of four types of repeats, but the repeats varied in size and types between the two species. The variation in the types and size of repeats were also previously reported in the chloroplast genomes of angiosperms and in other species of Araceae (Abdullah et al. 2020b; Henriquez et al. 2020b; Mehmood et al. 2020). These repeats might be useful as a proxy to identify mutational hotspots (Ahmed et al. 2012; Abdullah et al. 2020c) and design molecular markers for phylogenetic and population genetics studies (Ahmed et al. 2013).
Our finding of increased transition substitutions compared to transversion substitutions is consistent with other findings (Wakeley 1996; Cao et al. 2018). Contradictory findings of more transversion than transition findings have also been documented previously (Cai et al. 2015; Abdullah et al. 2019a; Shahzadi et al. 2020). This bias of higher transversions might be due to the composition of genomes and the genetic characteristics of codons (Morton et al. 1997). We observed higher transition substitutions linked to synonymous substitutions and vice versa, as reported in the chloroplast genomes of Firmiana, a genus of family Malvaceae (Abdullah et al. 2019a).
The higher rate of synonymous substitutions than non-synonymous substitutions is indicative of strong purifying selection pressure acting on these genes during the course of evolution (Matsuoka et al. 2002). The observation of purifying selection in our study are consistent with previous studies on angiosperm chloroplast genomes, including other aroid (Menezes et al. 2018; Abdullah et al. 2019a, 2020a, b; Henriquez et al. 2020a; Shahzadi et al. 2020). In current study, some codons sites in three protein-coding genes (rpl22, ycf1, and ycf2) were found under positive selection pressure in the subfamily Pothoideae based on the results of FUBAR. The rpl22 is encoding the large subunit of ribosomes (Mache 1990) while ycf1 and ycf2 are the largest protein-coding genes in the chloroplast genome that are part of the chloroplast inner envelope membrane protein translocon (Kikuchi et al. 2013). These genes were also found under positive selection in species of Araceae and other angiosperms (Fan et al. 2018; Liu et al. 2018; Zhong et al. 2019; Henriquez et al. 2020a; Abdullah et al. 2020a). The presence of positively selected codons revealed the role of these genes in adaptation of the species in their ecological niches. Previously, only seven genes in subfamily Monsteroideae were found under positive selection (Henriquez et al. 2020a), whereas in comparison of chloroplast genomes of four species from the three subfamilies of Araceae three genes were reported under positive selection (Abdullah et al. 2020a). In another study of Araceae, most genes (62/71) were reported under positive selection (Kim et al. 2019) based on the evaluation of Ka/Ks values in DnaSP. We also followed the Ka/Ks methodological approach of Kim et al. (2019). However, Kim et al. (2019) included 13 other species from four subfamilies of Araceae (Orontioideae, Lemnoideae, Monsteroideae, and Aroideae) and a misidentified taxon, Alocasia macrorrhizos (L.) G.Don (KR296655) in their comparison. The inclusion of these additional taxa might be the reason for the observation of unusually high number of genes undergoing positive selection, reported by Kim et al. (2019); as positive selection was also reported on 1/3 protein-coding genes of grasses (Piot et al. 2018).
The phylogenetic inference among the species of five subfamilies (Orontioideae, Lemnoideae, Pothoideae, Monsteroideae, and Aroideae) of Araceae are in agreement with previous findings (Cabrera et al. 2008; Cusimano et al. 2011; Nauheimer et al. 2012; Henriquez et al. 2014). Kim et al. (2019) showed an unusual sister relationship between Epipremnum (Monsteroideae) and Dieffenbachia (Aroideae). In our study, Epipremnum was confirmed to share a sister relationship with the genus Monstera in subfamily Monsteroideae instead of Aroideae. Hence, phylogenetic inference based on complete chloroplast genome confirmed the placement of genus Epipremnum in Monsteroideae as reported previously (Cusimano et al. 2011; Henriquez et al. 2014; Chartier et al. 2014; Zuluaga et al. 2019). Kim et al. (2019) included Epipremnum aureum (MK286107) in their comparison, hence, this shows that E. aureum is either mislabeled or is a case of chloroplast genome capture from another species as hybridization and polyploidy is common in Araceae (Ahmed 2014).
In conclusion, our study provides insight into the evolution of chloroplast genomes of Pothoideae (Araceae). Our study shows unique IR contraction and expansion affecting the number of genes and rate of evolution in P. scandens. We observed a two-fold higher transition substitution rate than transversion substitutions and found higher transversion substitutions linked with non-synonymous substitutions when compared with transition substitutions.
References
Abdullah, Shahzadi I, Mehmood F et al (2019a) Comparative analyses of chloroplast genomes among three Firmiana species: identification of mutational hotspots and phylogenetic relationship with other species of Malvaceae. Plant Gene 19:100199. https://doi.org/10.1016/J.PLGENE.2019.100199
Abdullah, Waseem S, Mirza B et al (2019b) Comparative analyses of chloroplast genome in Theobroma cacao and Theobroma grandiflorum. Biologia (Bratisl). https://doi.org/10.2478/s11756-019-00388-8
Abdullah, Henriquez CL, Mehmood F et al (2020a) Comparison of chloroplast genomes among species of unisexual and bisexual clades of the monocot family Araceae. Plants 9:773. https://doi.org/10.3390/plants9060737
Abdullah, Mehmood F, Shahzadi I et al (2020b) Chloroplast genome of Hibiscus rosa-sinensis (Malvaceae): comparative analyses and identification of mutational hotspots. Genomics 112:581–591. https://doi.org/10.1016/j.ygeno.2019.04.010
Abdullah, Mehmood F, Shahzadi I et al (2020c) Correlations among oligonucleotide repeats, nucleotide substitutions and insertion-deletion mutations in chloroplast genomes of plant family Malvaceae. J Syst Evol. https://doi.org/10.1111/jse.12585
Ahmed I (2014) Evolutionary dynamics in taro. PhD dissertation, Massey University, Palmerston North, New Zealand
Ahmed I (2015) Chloroplast genome sequencing: some reflections. J Next Gener Seq Appl 02:119. https://doi.org/10.4172/2469-9853.1000119
Ahmed I, Biggs PJ, Matthews PJ et al (2012) Mutational dynamics of aroid chloroplast genomes. Genome Biol Evol 4:1316–1323. https://doi.org/10.1093/gbe/evs110
Ahmed I, Matthews PJ, Biggs PJ et al (2013) Identification of chloroplast genome loci suitable for high-resolution phylogeographic studies of Colocasia esculenta (L.) Schott (Araceae) and closely related taxa. Mol Ecol Resour 13:929–937. https://doi.org/10.1111/1755-0998.12128
Amiryousefi A, Hyvönen J, Poczai P (2018a) IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34:3030–3031. https://doi.org/10.1093/bioinformatics/bty220
Amiryousefi A, Hyvönen J, Poczai P (2018b) The chloroplast genome sequence of bittersweet (Solanum dulcamara): plastid genome structure evolution in Solanaceae. PLoS ONE 13:1–23. https://doi.org/10.1371/journal.pone.0196069
Andrews S (2017) FastQC: a quality control tool for high throughput sequence data. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 15 Sep 2019
Boyce PC, Croat TB (2018) The Überlist of Araceae, totals for published and estimated number of species in aroid genera
Cabrera LI, Salazar GA, Chase MW et al (2008) Phylogenetic relationships of aroids and duckweeds (Araceae) inferred from coding and noncoding plastid DNA. Am J Bot 95:1153–1165. https://doi.org/10.3732/ajb.0800073
Cai J, Ma PF, Li HT, Li DZ (2015) Complete plastid genome sequencing of four Tilia species (Malvaceae): a comparative analysis and phylogenetic implications. PLoS ONE 10:1–13. https://doi.org/10.1371/journal.pone.0142705
Cao J, Jiang D, Zhao Z et al (2018) Development of chloroplast genomic resources in chinese Yam (Dioscorea polystachya). Biomed Res Int. https://doi.org/10.1155/2018/6293847
Carlsen MM, Croat TB (2013) A molecular phylogeny of the species-rich neotropical genus Anthurium (Araceae) based on combined chloroplast and nuclear DNA. Syst Bot 38:576–588. https://doi.org/10.1600/036364413x670287
Chartier M, Gibernau M, Renner SS (2014) the evolution of pollinator-plant interaction types in the Araceae. Evolution 68:1533–1543. https://doi.org/10.1111/evo.12318
Cho WB, Han EK, Choi G, Lee JH (2018) The complete chloroplast genome of Daphne kiusiana, an evergreen broad-leaved shrub on Jeju Island. Conserv Genet Resour 10:103–106. https://doi.org/10.1007/s12686-017-0774-5
Choi KS, Park KT, Park S (2017) The chloroplast genome of Symplocarpus renifolius: a comparison of chloroplast genome structure in Araceae. Genes (Basel) 8:324. https://doi.org/10.3390/genes8110324
Cooper G (2000) Chloroplasts and other plastids in the cell: a molecular approach, 2nd edn. Sinauer Associates, Sunderland
Cusimano N, Bogner J, Mayo SJ et al (2011) Relationships within the Araceae: comparison of morphological patterns with molecular phylogenies. Am J Bot 98:654–668. https://doi.org/10.3732/ajb.1000158
Daniell H (2007) Transgene containment by maternal inheritance: effective or elusive? Proc Natl Acad Sci USA 104:6879–6880. https://doi.org/10.1073/pnas.0702219104
Daniell H, Lin C-S, Yu M, Chang W-J (2016) Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol 17:134. https://doi.org/10.1186/s13059-016-1004-2
Darling ACE, Mau B, Blattner FR, Perna NT (2004) Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res 14:1394–1403. https://doi.org/10.1101/gr.2289704
Delport W, Poon AFY, Frost SDW, Pond SL (2010) Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 26:2455–2457. https://doi.org/10.1093/bioinformatics/btq429
Ewels P, Magnusson M, Lundin S, Käller M (2016) MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32:3047–3048. https://doi.org/10.1093/bioinformatics/btw354
Fan WB, Wu Y, Yang J et al (2018) Comparative chloroplast genomics of Dipsacales species: insights into sequence variation, adaptive evolution, and phylogenetic relationships. Front Plant Sci 9:689. https://doi.org/10.3389/fpls.2018.00689
Greiner S, Lehwark P, Bock R (2019) OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res 47:W59–W64. https://doi.org/10.1093/nar/gkz238
Gunawardena AHLAN, Dengler NG (2006) Alternative modes of leaf dissection in monocotyledons. Bot J Linn Soc 150:25–44. https://doi.org/10.1111/j.1095-8339.2006.00487.x
Han L, Wang B, Wang ZZ (2016) The complete chloroplast genome sequence of Spathiphyllum kochii. Mitochondrial DNA 27:2973–2974. https://doi.org/10.3109/19401736.2015.1060466
Henriquez CL, Abdullah, Ahmed I et al (2020a) Molecular evolution of chloroplast genomes in Monsteroideae (Araceae). Planta 251:72. https://doi.org/10.1007/s00425-020-03365-7
Henriquez CL, Abdullah, Ahmed I et al (2020b) Evolutionary dynamics of chloroplast genomes in subfamily Aroideae (Araceae). Genomics 112:2349–2360. https://doi.org/10.1016/j.ygeno.2020.01.006
Henriquez CL, Arias T, Pires JC et al (2014) Phylogenomics of the plant family Araceae. Mol Phylogenet Evol 75:91–102. https://doi.org/10.1016/j.ympev.2014.02.017
Hirao T, Watanabe A, Kurita M et al (2008) Complete nucleotide sequence of the Cryptomeria japonica D. Don. chloroplast genome and comparative chloroplast genomics: diversified genomic structure of coniferous species. BMC Plant Biol. https://doi.org/10.1186/1471-2229-8-70
Hoang DT, Chernomor O, von Haeseler A et al (2018) UFBoot2: Improving the ultrafast bootstrap approximation. Mol Biol Evol 35:518–522. https://doi.org/10.1093/molbev/msx281
Iram S, Hayat MQ, Tahir M et al (2019) Chloroplast genome sequence of Artemisia scoparia: comparative analyses and screening of mutational hotspots. Plants 8:476. https://doi.org/10.3390/plants8110476
Kalyaanamoorthy S, Minh BQ, Wong TKF et al (2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14:587–589. https://doi.org/10.1038/nmeth.4285
Katoh K, Kuma KI, Toh H, Miyata T (2005) MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res 33:511–518. https://doi.org/10.1093/nar/gki198
Kearse M, Moir R, Wilson A et al (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. https://doi.org/10.1093/bioinformatics/bts199
Kim S-H, Yang J, Park J et al (2019) Comparison of whole plastome sequences between thermogenic skunk cabbage Symplocarpus renifolius and nonthermogenic S. nipponicus (Orontioideae; Araceae) in East Asia. Int J Mol Sci 20:4678. https://doi.org/10.3390/ijms20194678
Kikuchi S, Bédard J, Hirano M et al (2013) Uncovering the protein translocon at the chloroplast inner envelope membrane. Science 339:571–574. https://doi.org/10.1126/science.1229262
Kurtz S, Choudhuri JV, Ohlebusch E et al (2001) REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res 29:4633–4642
Lee SY, Ng WL, Mohamed R, Terhem R (2018) The complete chloroplast genome of Aquilaria malaccensis Lam. (Thymelaeaceae), an important and threatened agarwood-producing tree species. Mitochondrial DNA Part B 3:1120–1121. https://doi.org/10.1080/23802359.2018.1519382
Lehwark P, Greiner S (2019) GB2sequin—a file converter preparing custom GenBank files for database submission. Genomics 111:759–761. https://doi.org/10.1016/J.YGENO.2018.05.003
Liu ML, Fan WB, Wang N et al (2018) Evolutionary analysis of plastid genomes of seven lonicera L. species: implications for sequence divergence and phylogenetic relationships. Int J Mol Sci 19:4039. https://doi.org/10.3390/ijms19124039
Lockhart P, Novis P, Milligan BG et al (2006) Heterotachy and tree building: a case study with plastids and eubacteria. Mol Biol Evol 23:40–45. https://doi.org/10.1093/molbev/msj005
Mache R (1990) Chloroplast ribosomal proteins and their genes. Plant Sci 72:1–12
Matsuoka Y, Yamazaki Y, Ogihara Y, Tsunewaki K (2002) Whole chloroplast genome comparison of rice, maize, and wheat: implications for chloroplast gene diversification and phylogeny of cereals. Mol Biol Evol 19:2084–2091
Mayo SJ, Bogner J, Catherine E, Boyce PJ (1997) The genera of araceae. Royal Botanic Gardens, Kew, London
Mehmood F, Abdullah, Shahzadi I et al (2020) Characterization of Withania somnifera chloroplast genome and its comparison with other selected species of Solanaceae. Genomics 112:1522–1530. https://doi.org/10.1016/j.ygeno.2019.08.024
Menezes APA, Resende-Moreira LC, Buzatti RSO et al (2018) Chloroplast genomes of Byrsonima species (Malpighiaceae): comparative analysis and screening of high divergence sequences. Sci Rep 8:1–12. https://doi.org/10.1038/s41598-018-20189-4
Morton BR, Oberholzer VM, Clegg MT (1997) The influence of specific neighboring bases on substitution bias in noncoding regions of the plant chloroplast genome. J Mol Evol 45:227–231
Murrell B, Moola S, Mabona A et al (2013) FUBAR: a fast, unconstrained bayesian AppRoximation for inferring selection. Mol Biol Evol. https://doi.org/10.1093/molbev/mst030
Murrell B, Weaver S, Smith MD et al (2015) Gene-wide identification of episodic selection. Mol Biol Evol 32:1365–1371. https://doi.org/10.1093/molbev/msv035
Nauheimer L, Metzler D, Renner SS (2012) Global history of the ancient monocot family Araceae inferred with models accounting for past continental positions and previous ranges based on fossils. New Phytol 195:938–950. https://doi.org/10.1111/j.1469-8137.2012.04220.x
Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating Maximum-likelihood phylogenies. Mol Biol Evol 32:268–274. https://doi.org/10.1093/molbev/msu300
Oldenburg DJ, Bendich AJ (2016) The linear plastid chromosomes of maize: terminal sequences, structures, and implications for DNA replication. Curr Genet 62:431–442. https://doi.org/10.1007/s00294-015-0548-0
Palmer JD (1985) Comparative organization of chloroplast genomes. Annu Rev Genet 19:325–354. https://doi.org/10.1146/annurev.ge.19.120185.001545
Piot A, Hackel J, Christin PA, Besnard G (2018) One-third of the plastid genes evolved under positive selection in PACMAD grasses. Planta 247:255–266. https://doi.org/10.1007/s00425-017-2781-x
Pond SLK, Frost SDW, Muse SV (2005) HyPhy: hypothesis testing using phylogenies. Bioinformatics 21:676–679. https://doi.org/10.1093/bioinformatics/bti079
Rozas J, Ferrer-Mata A, Sanchez-DelBarrio JC et al (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 34:3299–3302. https://doi.org/10.1093/molbev/msx248
Shahzadi I, Abdullah, Mehmood F et al (2020) Chloroplast genome sequences of Artemisia maritima and Artemisia absinthium: comparative analyses, mutational hotspots in genus Artemisia and phylogeny in family Asteraceae. Genomics 112:1454–1463. https://doi.org/10.1016/J.YGENO.2019.08.016
Tian N, Han L, Chen C, Wang Z (2018) The complete chloroplast genome sequence of Epipremnum aureum and its comparative analysis among eight Araceae species. PLoS ONE 13:e0192956. https://doi.org/10.1371/journal.pone.0192956
Tillich M, Lehwark P, Pellizzer T et al (2017) GeSeq—versatile and accurate annotation of organelle genomes. Nucleic Acids Res 45:W6–W11. https://doi.org/10.1093/nar/gkx391
Wakeley J (1996) The excess of transitions among nucleotide substitutions: new methods of estimating transition bias underscore its significance. Trends Ecol Evol 11:158–162
Walker JF, Jansen RK, Zanis MJ, Emery NC (2015) Sources of inversion variation in the small single copy (SSC) region of chloroplast genomes. Am J Bot 102:1751–1752
Wang W, Messing J (2011) High-throughput sequencing of three Lemnoideae (duckweeds) chloroplast genomes from total DNA. PLoS ONE. https://doi.org/10.1371/journal.pone.0024670
Wang Y, Zhan D-F, Jia X et al (2016) Complete chloroplast genome sequence of Aquilaria sinensis (Lour.) Gilg and evolution analysis within the Malvales order. Front Plant Sci 7:1–13. https://doi.org/10.3389/fpls.2016.00280
Wang YH, Qu XJ, Chen SY et al (2017) Plastomes of Mimosoideae: structural and size variation, sequence divergence, and phylogenetic implication. Tree Genet Genomes 13:1–18. https://doi.org/10.1007/s11295-017-1124-1
Weng M-L, Ruhlman TA, Jansen RK (2017) Expansion of inverted repeat does not decrease substitution rates in Pelargonium plastid genomes. New Phytol 214:842–851. https://doi.org/10.1111/nph.14375
Wu CS, Lin CP, Hsu CY et al (2011) Comparative chloroplast genomes of Pinaceae: insights into the mechanism of diversified genomic organizations. Genome Biol Evol 3:309–319. https://doi.org/10.1093/gbe/evr026
Yi X, Gao L, Wang B et al (2013) The complete chloroplast genome sequence of Cephalotaxus oliveri (Cephalotaxaceae): evolutionary comparison of Cephalotaxus chloroplast dnas and insights into the loss of inverted repeat copies in gymnosperms. Genome Biol Evol 5:688–698. https://doi.org/10.1093/gbe/evt042
Zhong B, Deusch O, Goremykin VV et al (2011) Systematic error in seed plant phylogenomics. Genome Biol Evol 3:1340–1348. https://doi.org/10.1093/gbe/evr105
Zhong Q, Yang S, Sun X et al (2019) The complete chloroplast genome of the Jerusalem artichoke (Helianthus tuberosus L.) and an adaptive evolutionary analysis of the ycf2 gene. PeerJ. https://doi.org/10.7717/peerj.7596
Zhu A, Guo W, Gupta S et al (2016) Evolutionary dynamics of the plastid inverted repeat: the effects of expansion, contraction, and loss on substitution rates. New Phytol 209:1747–1756. https://doi.org/10.1111/nph.13743
Zuluaga A, Llano M, Cameron K (2019) Systematics, biogeography, and morphological character evolution of the hemiepiphytic subfamily Monsteroideae (Araceae). Ann Missouri Bot Gard 104:33–48. https://doi.org/10.3417/2018269
Acknowledgements
Open access funding provided by University of Helsinki including Helsinki University Central Hospital. Authors thank Barbara Schaal at Washington University in St. Louis and J. Chris Pires at the University of Columbia, Missouri for laboratory support. Authors thank Tatiana Arias for help in the laboratory and data processing. Authors are also thankful to Emily Colletti in the aroid greenhouse at the Missouri Botanical Garden for provision of critical assistance with living material.
Funding
The funding for this study was provided by GAANN fellowship, the J. Chris Pires Lab, and the Rettner B. Morris Scholarship, Washington University in St. Louis, (NSF DEB 1146603).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
No conflict of interest exists.
Additional information
Handling editor: William Murphy.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abdullah, Henriquez, C.L., Mehmood, F. et al. Complete Chloroplast Genomes of Anthurium huixtlense and Pothos scandens (Pothoideae, Araceae): Unique Inverted Repeat Expansion and Contraction Affect Rate of Evolution. J Mol Evol 88, 562–574 (2020). https://doi.org/10.1007/s00239-020-09958-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-020-09958-w