
Abstract
Gene duplication and divergence are overwhelmingly considered to be the primary mechanisms by which cytochrome P450 monooxygenases (P450s) have radiated into a large and diverse gene superfamily. To address how environmental stress drives the fixation and diversification of gene duplications, we have analyzed Cyp12d1 and Cyp12d3, a pair of duplicated genes found in the sequenced Drosophila genomes of the melanogaster group. The paralog Cyp12d3, which is not found in Drosophila melanogaster, is basal to the melanogaster group, after it split from the obscura group (ca. 50 mya), and has a significant signature of positive selection in two species (D. sechellia and D. ananassae). Examination of the Cyp12d1 region in D. melanogaster wildtype and isoline populations revealed variation both in copy number and sequence, including splice-site variations, which certainly alter gene function. Further investigations of several strains have identified three cases in which differences in the Cyp12d1 gene region are associated with the differences in transcript abundance and transcriptional responses to the environmental stresses that have not been seen for other detoxificative loci. Together, these data highlight the value of using both macro- and microevolutionary approaches in studying the duplication and divergence events associated with detoxification genes and lay important groundwork for future studies in the field of evolutionary toxicogenomics, which uses the principles of phylogenetic analysis to predict possible enzymatic functions.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Genetic variation allows organisms to adapt to changes in the environment that cause stress, such as xenobiotics, temperature and prooxidants. An important source of genetic variation for adapting to environmental stresses is that of gene duplication, a process by which most new genes are gained (Ohno 1970; Zhou et al. 2008; Zhang et al. 2009). Despite the fact that the rate of gene duplication is of the same order of magnitude as or higher than the rate of nucleotide mutation, duplicate genes are more likely to be lost than preserved (Li 1999; Sebat et al. 2004; Lupski 2007; Emerson et al. 2008). Multiple models have been proposed to describe how the process of positive selection drives the emergence, retention, and diversification of duplicated genes, but the mechanisms of fixation and maintenance of gene duplications are unknown (Innan and Kondrashov 2010). To fill these voids, Innan and Kondrashov (2010) have suggested that future studies not focus exclusively on the evolutionary divergence of duplicated gene copies and instead combine comparative genomic studies with detailed copy number and expression analyses.
Gene duplication and divergence are considered to be the primary mechanisms by which the cytochrome P450 monooxygenase (P450) genes have radiated into a large and diverse superfamily (Nelson 1998, 2009). P450s perform a variety of heme-dependent oxidative reactions, often adding hydroxyl groups to lipophilic molecules and allowing them to serve as substrates for subsequent conversions and modifications. While this basic type of oxidative reaction is conserved across the many taxa containing P450 genes (bacteria, yeast, plants, invertebrates, and vertebrates), the functions performed differ from gene to gene and range from the biosynthesis of hormones, fatty acids, and pheromones to the detoxification of drugs, phytochemicals, and insecticides (Guengerich 2005; Kelly et al. 2005; Schuler et al. 2006; Li et al. 2007; Mizutani and Ohta 2010; Bak et al. 2011; Feyereisen 2011; Schuler 2011). With their multiple roles in development and growth, as well as in response to environmental assaults, their involvement in the initial stages of the detoxification process makes understanding their evolution important to future ecological and toxicological studies.
Within the large superfamily of cytochrome P450s, eukaryotic mitochondrial P450s, nuclear-encoded P450s targeted to the mitochondria, seem to have evolved from microsomal, membrane-bound P450s, despite their resemblance to bacterial soluble P450s (Feyereisen 2012). Whereas in vertebrates mitochrondrial P450s are restricted to performing essential physiological functions, insect mitochondrial P450s are involved in both ecdysteroid production and xenobiotic metabolism. Members of the CYP12 family, which include Cyp12d1 (Drosophila melanogaster), CYP12A1 (Musca domestica), and CYP12F1 (Anopheles gambiae), are primarily involved in xenobiotic metabolism.
In D. melanogaster, Cyp12d1 is differentially expressed in response to a diverse array of xenobiotics, including DDT (Festucci-Buselli et al. 2005), phenobarbital (Le Goff et al. 2006; Sun et al. 2006), atrazine (LeGoff et al. 2006), caffeine (Willoughby et al. 2006), piperonyl butoxide (Willoughby et al. 2007), hydrogen peroxide (Li et al. 2008), and pepper (Piper nigrum) extract (Jensen et al. 2006), suggesting that it has an important role in the general response to xenobiotics. Field and laboratory selections for DDT resistance in D. melanogaster have resulted in constitutively higher levels of Cyp12d1 transcript in one DDT-resistant strain (Wisconsin) and in higher inducibility in response to DDT in two DDT-resistant strains (91-R and Wisconsin) compared with a susceptible strain (Canton-S) (Pedra et al. 2004; Festucci-Buselli et al. 2005).
Although its exact substrates and inducers are still being defined, comparing the conservation of Cyp12d1 across species with the relaxed selection of Cyp12d3 can provide predictions about whether their substrates are also conserved or unique. Within the P450 superfamily, rates of gene gain and loss are not necessarily equal across all families and higher-order clans. Comparisons across ten vertebrate species have shown that P450 gene duplications or losses are more frequent for P450s with xenobiotic substrates than for P450s with endogenous substrates (Thomas 2007). Thus, efforts aimed at characterizing P450s with xenobiotic substrates important in the ecotoxicology of an organism would benefit from combining comparative genomics and evolutionary toxicogenomics approaches to discriminate between enzymes conserved across species that potentially metabolize ubiquitous xenobiotics and those conserved only in closely related species that potentially detoxify xenobiotics particular to species-specific interactions.
In addition to macroevolutionary changes of the Cyp12d1 duplication, microevolutionary analyses of the Cyp12d1 duplication in D. melanogaster can test predictions regarding transcriptional expression profiles and copy number polymorphism of duplicated genes. The Cyp12d1 duplication has been estimated to occur within ~80 % of the individuals within a wild population, but the proportion of individuals carrying the duplication was not different between unexposed field populations and survivors of DDT exposure (Schmidt et al. 2010). If the Cyp12d1 duplication does not seem to increase detoxification activity toward DDT, as posited by the positive dosage model (Kondrashov et al. 2002), then why does it seem to be so prevalent in the wild? An alternative hypothesis is that duplication of Cyp12d1 provides for the acquisition of novel transcriptional responses to environmental stresses. We provide the first evidence of the induction of a single insect P450 gene by a broad range of environmental stresses other than insecticides, including heat, CO2, starvation, and hydrogen peroxide, notably within a strain containing the Cyp12d1 duplication.
To better understand the ecological and toxicology issues associated with such P450 evolutionary events, the changes that have occurred across closely related species and in populations within a given species must be identified. In the present study, we employed comparative genomic data of twelve Drosophila genomes (Drosophila 12 Genomes Consortium et al. 2007) along with sequence and expression data of several Drosophila melanogaster strains to identify traces of positive selection and possible gene conversion with a long-term goal of addressing how environmental stress drives the fixation and diversification of the Cyp12d1 gene duplication.
Materials and Methods
Sequence Data Sets
Sequence data were downloaded from FlyBase (http://flybase.org/) by means of the following genome builds: D. ananassae (R1.3), D. erecta (R1.3), D. grimshawi (R1.3), D. mojavensis (R1.3), D. persimilis (R1.3), D. pseudoobscura (R2.6), D. sechellia (R1.3), D. simulans (R1.3), D. virilis (R1.2), D. willistoni (R1.3), and D. yakuba (R1.3). Genomic and predicted amino acid sequences of Cyp12d1 orthologs were collected from each genome by the BLASTn function and the cDNA sequence of Cyp12d1-p from D. melanogaster and the tBLASTn function to capture the outgroup sequence from M. domestica.
Fly Lines
The 53 wild-type strains of D. melanogaster used in this study were obtained from the Bloomington Drosophila Stock Center (http://flystocks.bio.indiana.edu/). Strains included: Amherst 3, BER 2, Berlin K, BOG 2, BOG 3, Canton-S, Canton-S-iso2B, CO 4, CO 7, Crimea, EV, Florida-9, Harwich, Hikone-A-S, Hikone-A-W, Hikone-R, KSA 3, KSA 4, Lausanne-S, MO 1, MWA 1, NO 1, Oregon-R, Oregon-R-modENCODE, Oregon-R-P2, Oregon-R-S, Oregon-R-SNPiso2, pi2<P>, PYR 3, RC 1, Reids 1, Reids 2, Reids 3, RVC 2, RVC 4, Samarkand, Swedish-C, TW 1, TW 2, TW 3, Urbana-S, VAG 2, VAG 3, Wild 10E, Wild 11C, Wild 11D, Wild 1A, Wild 1B, Wild 2A, Wild 3B, Wild 5A, Wild 5B, and Wild 5C. The laboratory strains w 1118 and y; cn bw sp were also obtained from the Bloomington Drosophila Stock Center. The Wisconsin strain was collected in Door County, Wisconsin (Brandt et al. 2002). The 91-R and 91-C strains, derived from a common population founded from several hundred individuals collected in St. Paul, Minnesota in 1952 (Dapkus and Merrell 1977), were selected for DDT resistance (91-R) or were never exposed to DDT (91-C) and were provided to us in 2000 by Dr. Ranjan Ganguly (University of Tennessee). From 2000 to 2003, 91-R was periodically selected for DDT resistance by collecting survivors exposed to 4,000 μg of DDT (Festucci-Buselli et al. 2005).
Fly Rearing
91-C and Oregon-R Drosophila stocks were reared on both Formula 4-24® Instant Drosophila Medium (Carolina Biological Supply Co., Burlington, NC) and Jazz-Mix® Drosophila Food (Cat. No. AS153; Fisher Scientific, Hanover Park, IL) at 25 °C and 40 % relative humidity on a 12:12 h photoperiod. Phenobarbital-treated flies and their respective controls were reared and treated on Formula 4-24® as 4-24 is prepared without having to heat the media. All other flies were reared similarly on Jazz-Mix® from bottle populations, to virgin isolation vials, to treatment scintillation vials. Adults were transferred to new bottles every 2 weeks.
DNA Extraction and Polymerase Chain Reaction Analysis
Genomic DNA was extracted from 10 to 15 adult flies using the DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA). DNA was quantified by spectrophotometry using Nanodrop 1000 (Thermo Scientific, Wilmington, DE). The Cyp12d1 region, based on the reference genome, was amplified from 50 ng of genomic DNA using 2-μL of primers (10 pmol/μL), 5 U of TaKaRa Long Amplification Taq polymerase (Otsu, Shiga, Japan) with a final concentration of 1× LA PCRTM Mg2+-free buffer II, 2.5 mM MgCl2, and 2.5 mM of each dNTP in a 50-μL volume, according to manufacturer’s instructions. Thermal cycling started at 94 °C for 1 min; followed by 30 cycles of 98 °C for 10 s and a combined annealing and extension step of 68 °C for 12 min; and finished with 72 °C for 15 min.
Based on the D. melanogaster reference genome (y; cn bw sp), primers were designed to amplify the regions spanning Cyp12d1-p and Cyp12d1-d, which included duplicated noncoding regions as well as coding regions. Additional primers were designed to amplify sequences ~1.2 kb upstream and ~0.58 and 5.8 kb downstream of the duplicated region in the reference genome (Supplemental Table 1).
Sequencing and Alignment of Cyp12d1 Genomic Region from 91-C and 91-R
All amplification products were purified using the QIAquick PCR purification kit (Qiagen, Valencia, CA). The amplification products of the Cyp12d1 genomic region for the 91-C and 91-R strains were sequenced with 2× coverage by primer walking at the Core DNA Sequencing Facility of the W. M. Keck Center for Comparative and Functional Genomics at the University of Illinois Urbana-Champaign. Nucleotide alignments were performed with Clustal W (3.2) using Biology Workbench (http://workbench.sdsc.edu/).
Phylogenetic Analysis
The phylogeny of Cyp12d1/Cyp12d3 was inferred by Maximum Likelihood (ML) with the Jones et al. (1992) (JTT) amino acid substitution model (MEGA5; Tamura et al. 2011) using the best-hit sequence from M. domestica as an outgroup sequence. This method also allowed us to test for possible departures of constancy in rates of evolution across the phylogeny (i.e., molecular clock). We also investigated rate constancy between paralogs after duplication by means of the branch-model implemented in the program codeml of PAML v4.4 (Yang 1997; Nielsen and Yang 1998; Yang et al. 2000), where a model allowing different ratios of nonsynonymous to synonymous substitution rates (ω = dN/dS) for Cyp12d1 and Cyp12d3 is compared to a model with constant w by a likelihood-ratio tests (d.f. = 1).
To investigate the possible action of positive selection at the level of amino acid sequence change, we applied the site and branch-site models implemented in codeml: site models allow for testing the presence of positive selection on a subset of sites (codons) taking into account the whole phylogeny, while branch-site models allow for a direct test of positive selection (on a subset of codons) on the lineages of interest. In particular, we applied a likelihood-ratio test comparing the models M1a (nearly neutral) and M2a (positive selection) in both site and branch-site models with d.f. = 2. Model M1a assumes two classes of sites, conserved sites with ω estimated from the data (ω < 1) and neutral sites (ω = 1), while M2a considers an extra third class of sites under positive selection (ω > 1) (Wong et al. 2004). In all cases, we used equilibrium codon frequencies calculated from the average nucleotide frequencies at third codon positions (CodonFreq = 2). We also identified positively selected sites at which nonsynonymous substitutions occur at a higher rate than synonymous ones by the Bayes Empirical Bayes (BEB) calculation of posterior probabilities (Yang et al. 2005) as implemented in codeml (PAML) for model M2a.
Analysis of DGRP Strains
We generated genomic sequences for 34 highly inbred lines from the Drosophila Genetic Reference Panel, DGRP (http://www.hgsc.bcm.tmc.edu/project-species-i-Drosophila_genRefPanel.hgsc) using publicly available 454-Roche and Illumina next-generation sequencing reads. Our analysis was limited to the Cyp12d1-p/Cyp12d1-d genomic region in accordance with the Fort Lauderdale agreement on Community Resource Projects and the resulting NHGRI policy statement (see also DGRP Data Release policy; http://www.hgsc.bcm.tmc.edu/project-species-i-Drosophila_genRefPanel.hgsc). Sequencing reads (raw data) were obtained from NCBI short read archive (SRA) study SRP000694. The availability of long reads is particularly important when analyzing duplicated regions, and therefore we restricted the analysis to the 34 DGRP strains with 454-Roche as well as Illumina sequencing reads which provide us high mapping quality and deep coverage. Filtering of reads, mapping, and generation of consensus sequences were carried out using the FASTX-toolkit (http://hannonlab.cshl.edu/fastx_toolkit/), BWA (Li and Durbin 2010), SAMtools v1.4 (Li et al. 2009), and custom scripts. SNPs were called when the phred-scaled likelihood that the genotype is identical to the reference (‘SNP quality’) was Q40 or greater (a probability of being equal to the reference of 0.0001).
Induction of Cyp12d1 Expression by Environmental Stresses
All bioassays were performed using male and female virgin flies, 3 days post-eclosion at the beginning of experiments. 91-C and Oregon-R fly lines were tested in parallel for each biological replicate with three biological replicates performed for each experiment in this study. All bioassays were performed by placing five insects of each sex (for a total of 10 insects) in glass scintillation vials.
Phenobarbital
Bioassays for phenobarbital (PB) were performed according to Sun et al. (2006). Flies are raised on F-24 diets from larvae to adult without PB. The control and treated flies were then transferred to PB-containing F-24 diet for 24 h. At the end of the 24 h period, flies were flash-frozen in liquid nitrogen.
Cold, Heat, and CO2 Exposure
Three separate experiments were performed to assess stress reactions to cold, heat, and CO2 exposure. Treated and control flies had access to 5 % sucrose solution on a cotton plug throughout the experiment. The cold-treated flies were held at 4 °C, the heat-treated flies were held at 37 °C, and the CO2-treated flies were exposed to 10 L/min of CO2 as administered using the Flystuff Flow Buddy Flow Regulator (Genesee Scientific, San Diego, CA) on a 0.3175-cm porous polyethylene Flystuff Flypad (Genesee Scientific). Treated flies were subjected to 30 min of cold, heat, and CO2 exposure. All exposure times were followed by two recovery times to insure only live flies were recovered for RNA extraction (i.e., insects that survived the exposure) as well as to observe Cyp12d1 expression at 1 h and 2 h timeframes. Flies were deemed “recovered” upon exhibiting the ability to walk on the wall of the treatment vial. Flies treated at 1 h and 2 h timeframes had respective recovery times of 30 and 90 min after the initial 30 min of exposure. Flies were flash-frozen in liquid nitrogen at the end of the recovery period and stored at −80 °C. Control flies were maintained in vials for the same time as that of the respective treatments and were flash-frozen at the same time as that of the treatments.
Starvation, Supplemental Sucrose, and Hydrogen Peroxide
Flies were exposed to starvation, supplemental sucrose, and hydrogen peroxide for 24 h. For all three experiments, control flies were held with access to a 5 % sucrose solution on a cotton plug. Control and treated flies were held in respective vials for 24 h before being flash-frozen in liquid nitrogen as described. Starvation treatment flies were given access to an equal volume of water with no added sucrose. Supplemental sucrose treatment flies were exposed to a 10 % sucrose solution instead of the 5 % control amount. Finally, H2O2 treatment flies were exposed to a 13.4 % H2O2 solution, including a 5 % sucrose solution. Based on dose–response curve data for 91-C (Sun 2009), we used a H2O2 dosage that caused approximately 10 % mortality in the fly population at 24 h of exposure for both the 91-C and Oregon-R strains.
RNA Extraction and cDNA Synthesis
RNA was extracted from 10 to 15 adult flies using the RNeasy Mini Kit (Qiagen, Valencia, CA). Genomic DNA was cleaved and removed from samples using RNase-Free DNase Set (Qiagen). RNA was quantified by spectrophotometry using Nanodrop 1000 (Thermo Scientific, Wilmington, DE). First strand cDNA was synthesized using the iScript cDNA Synthesis Kit (BIO-RAD, Hercules, CA) on an MJ Research Peltier Thermal Cycler-200 (GMI, Ramsey, MN) by initially incubating 1 μg of RNA in 5X cDNA synthesis kit buffer, iScript enzyme mixture, and nuclease-free water for 5 min at 25 °C, 30 min at 42 °C, and 5 min at 85 °C, according to manufacturer’s instructions. Diluted cDNA samples (1:25) were held at 4 °C before quantitative real-time polymerase chain reaction (qRT-PCR) experiments.
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
qRT-PCR reactions were performed using Fast SYBR Green Master Mix (Applied Biosystems, Foster City, CA) and GoTaq qPCR Master Mix (Promega, Madison, WI) on a Step One Real-Time PCR System. Each 20 μL amplification reaction was technically replicated for a minimum of three times. The individual threshold cycle (CT) was calculated by means of a Step One Software (2.0). Step One data outputs were evaluated by comparative amplification curves as well as melting curves to assess the performance of primer and master mix reactions. If melting curves provided multiple peaks, annealing temperature titrations were performed using both Fast SYBR Green and GoTaq qPCR master mixes at 0.5 °C increments to discern the optimal reaction formulae. All cDNA samples, control and treated, for a given experiment were run on the same plate and subjected to the same reagents and annealing temperatures (Supplemental Table 1). Relative expression levels were calculated as 2^(CT TARGET GENE—CT rp49) in which the CT value for the rp49 reference gene is subtracted from the TARGET GENE average CT value. The relative gene expression levels of Cyp12d1 in the 91-C samples were compared with those of Oregon-R.
Quantification of Transcript Abundance by Absolute qRT-PCR
In order to evaluate the expression differences between the Cyp12d1-p and Cyp12d1-d transcript, absolute quantitative real-time PCR was performed. Gene-specific primers were designed for the 3′UTR of each gene (Supplemental Table 1). First strand complementary DNA was synthesized using GoScript reverse transcription system by Promega. Real time PCR was performed at an annealing temperature of 62 °C, which had been optimized by gradient RT-PCR. For each gene, a single amplification product was confirmed by a single peak melting curve and a single band on an agarose gel. The amplified PCR product for each gene was purified using a Qiagen PCR clean-up kit and sequenced to insure correct amplification. Amplicon sizes were 245 nt and 254 nt for Cyp12d1-d and Cyp12d1-p, respectively. The purified amplicons were quantified using a Nano drop 1000 spectrophotometer and tenfold dilution series (from 1 nM to 1 fM) were prepared for each as standards for RT-PCR analysis. Six biological replicates of adult flies (1-3-days old) of mixed sexes for strain y; cn bw sp were tested.
Results
Identification of Cyp12d1 and Cyp12d3 Genes Across Twelve Drosophila Genomes
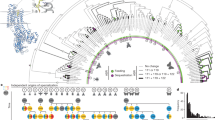
Comparisons among the twelve available Drosophila genomes demonstrate that Cyp12d1 duplicated into two genes before the divergence of D. melanogaster from other members of the melanogaster subgroup. In the six sequenced species of the melanogaster group, Cyp12d1 was tandemly duplicated in the genome (Fig. 1), and in all of these species, except D. melanogaster, amino acid identity was below the 97 % limit needed to apply individual names to P450 genes (Nelson et al. 1996). Therefore, we propose designating the second gene in most of these strains as Cyp12d3, as the gene name Cyp12d2 has been given to a P450 gene in Lucilia cuprina, and designating the highly conserved genes in the D. melanogaster reference strain as Cyp12d1-p and Cyp12d1-d (99.4 % identical). In D. simulans, D. erecta, D. ananassae, and D. sechellia, Cyp12d1 and Cyp12d3 display between 49 and 66 % amino acid identity (Table 1). In D. yakuba, Cyp12d1 and Cyp12d3 display only 12 % amino acid identity with this large degree of divergence likely due to a premature stop codon that truncates the Cyp12d3 sequence after 169 residues in middle of the second exon creating a pseudogene.

Cyp12d1 and Cyp12d3 evolution across 12 Drosophila species. Phylogeny of the 12 Drosophila species with sequenced genomes shows a single duplication event of Cyp12d1 within Drosophila evolution, its chromosomal location (2R = the second right; 3 = the third; nd = not determined, nd), and the next closest gene on the chromosome. The 5′ and 3′ ends of the genes indicated by the heads and tails of the arrows show that despite their conservation in chromosome position, large-scale chromosomal inversions have occurred in some species. Based on amino acid sequence identity between all of the species, Cyp12d1 (in blue) is the ancestral gene, while Cyp12d3 (in orange) is most likely the result of a duplication and divergence event. The shading of each gene in blue or orange represents the level of sequence identity between each gene and its ortholog in D. simulans; genes with a similar shade showed the greatest sequence similarity. In D. yakuba, Cyp12d3 (marked by a red X) has become a pseudogene encoding a truncated protein, and in the other species Cyp12d3 encodes a full-length protein
In D. pseudoobscura, D. persimilis, and D. willistoni, the most closely related basal species within the Sophophora, a single copy of Cyp12d1 has been retained at the same location on the chromosome approximately 10 kb from the BBS4 gene. In the more distantly related D. virilis, D. mojavensis, and D. grimshawi (within the Drosophila subgenus), a single copy of Cyp12d1 is located in a different, non-homologous chromosome (Fig. 1), suggesting that a relocation event occurred after divergence of the Sophophora and Drosophila subgenera but basal, before radiation, in either Sophophora or Drosophila.
Phylogenetic Analysis of Cyp12d1 Orthologs and Cyp12d3 Paralogs
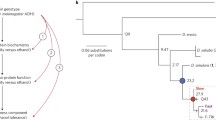
The phylogeny of Cyp12d1 (Fig. 2) recapitulates the proposed Drosophila phylogeny (Drosophila 12 Genomes Consortium 2007; Singh et al. 2009). We also observed that the duplication event that gave rise to its paralog Cyp12d3 was basal to the melanogaster group, after it split from the obscura group (ca. 50 mya). After duplication, Cyp12d3 was recently lost independently in the D. melanogaster lineage (after the split from the D. simulans lineage) and became a pseudogene in the D. yakuba lineage (after the split from D. erecta lineage).

Phylogenetic analysis of Cyp12d1/Cyp12d3 in Drosophila. Phylogeny inferred by Maximum Likelihood (ML) by means of the Jones et al. (1992) (JTT) amino acid substitution model. The tree with the highest log likelihood (−5381.2767) is shown. Bootstrap values, as percentage of trees in which the associated taxa clustered together, are shown next to the branches. The tree is drawn to scale with branch lengths measured in the number of substitutions per site. All positions containing gaps and missing data were eliminated. There were a total of 369 positions in the final dataset. Evolutionary analyses were conducted in MEGA5 (Tamura et al. 2011)
Our tests of the molecular clock for Cyp12d1/Cyp12d3 revealed a significant acceleration in the rate of amino acid substitution for Cyp12d3 based on the two different maximum likelihood methods. First, we applied a Jones et al. (1992) model as implemented in MEGA5 (Tamura et al. 2011), rejecting rate constancy, by either the complete tree (with Musca domestica as outgroup) or only the species of the Sophophora group with D. pseudoobsura/D. persimilis as outgroup species, ancestral to the duplication event (P < 1 × 10−8 in both cases; Table 2). Rate constancy between the paralogs after duplication can also be rejected when using the branch-model implemented in PAML (codeml), where a model allowing different dN/dS (w) ratios for Cyp12d1 and Cyp12d3 provides a better fit to the data than one with constant w (2Δ ln L = 45.75; P = 0.00018; d.f. = 1).
This latter analysis also shows that w for Cyp12d3 (0.2738) is almost four times higher than that for Cyp12d1 (0.0697), suggesting either relaxed constraints or the action of positive selection on amino acid evolution for Cyp12d3. To distinguish between these two possibilities, we applied a likelihood-ratio test comparing models with and without the possibility of positive selection (M1a against M2a implemented in PAML; see “Materials and Methods” for details). When all Cyp12d3 lineages are analyzed together (site model), there is no evidence for positive selection (P > 0.5), suggesting a general trend of relaxed selection on amino acid changes after duplication. A branch-site model, however, reveals the significant signature of positive selection acting in the lineages leading to D. ananassae and the sister species D. simulans/D. sechellia (P < 0.001 in both cases). The latter signature of selection can be attributed to the D. sechellia lineage after it split from D. simulans (2Δ ln L = 101.02; d.f. = 2, P < 0.0001) with 21 amino acid sites under positive selection having posterior probabilities greater than 0.95 based on Bayes Empirical Bayes (BEB) analysis (Yang et al. 2005). The position of these 21 amino acid sites is 104, 105, 106, 107, 109, 111, 113, 114, 119, 120, 123, 125, 142, 144, 146, 148, 149, 151, 152, 155, and 193 according to the Cyp12d1 D. melanogaster protein.
Copy Number Polymorphism in the Cyp12d1 Region Across D. melanogaster Strains
According to the published reference genome, D. melanogaster Cyp12d1-p and Cyp12d1-d are 99.4 % identical in their coding regions with differences occurring in only three amino acid positions. The flanking regions of the coding sequence, extending from 1,867 nucleotides upstream of the transcription start site to 136 nucleotides downstream of the stop codon, are 100 % identical between the two copies (http://flybase.org/, D. melanogaster R5.23). To amplify the entire Cyp12d1 region for comparisons among 58 strains, Cyp12d1 region primers were designed to flank the entire duplicated region as shown in Fig. 3. For the reference strain (y; cn bw sp) and nine other strains, amplification with the Cyp12d1 region-spanning primers (Cyp12d1 region for and Cyp12d1 region rev) produced the expected 7.7-kb band (Fig. 4); this set included three of the five strains of Oregon-R (Oregon-R, Oregon-R-S, and Oregon-R-SNPiso2). For most strains (47 of 58), however, amplification with the Cyp12d1 region-spanning primers produced a smaller 3.5-kb band, suggesting that most of these tested strains contain only a single Cyp12d1 gene (Fig. 4). One strain, PYR3 from the Pyrenees, produced a single band of ~10 kb.

Sequence polymorphism in Cyp12d1 gene locus in D. melanogaster with the Cyp12d1 region primers. PCR amplification using the Cyp12d1 region primer set and genomic DNA from the 91-C and 91-R strains revealed that the region was only half the length of the Cyp12d1 gene region in the reference genome (y; cn bw sp). Pictured are the PCR amplification products for strains 91-C, 91-R, and y; cn bw sp using the Cyp12d1 region primer set with a 1-kb ladder for comparison (New England Biolabs, Cat # N3232) (a). Based on this evidence, we predicted that 91-C and 91-R contain a single copy (b) of Cyp12d1 instead of the tandem duplication observed in the reference genome (c). To test this prediction, we used the tandem duplication primer set, which only produces an amplification product when the duplication is present, as noted by circled primer names for the reference strain. Sequencing through the Cyp12d1 gene in 91-C and 91-R revealed that in these strains Cyp12d1 lacks the conserved polyadenylation signal sequence of Cyp12d1-d (indicated by white box) and contains only the suboptimal polyadenylation signal sequence of Cyp12d1-p (indicated by black box)

Copy number and sequence polymorphisms in Cyp12d1 region of D. melanogaster (a) (Top) Proportion of strains tested that demonstrate a 3.5-, 7.7-, or 10.0-kilobase (kb) PCR amplification product using the Cyp12d1 region primers that flank the duplicated sequences. For the reference strain (y; cn bw sp) and nine other strains, amplification with the first set of primers produced the expected band size of 7.7-kb. However, the majority of strains, 47 of 58, produced a smaller band corresponding to a size of 3.5 kb with the Cyp12d1 region-spanning primer set, suggesting that most of the strains tested contained only one copy of the Cyp12d1. A single strain, PYR3 from the Pyrenees, produced a single band of ~10 kb. PCR amplification with the tandem duplication primers showed a single copy of Cyp12d1 for each strain exhibiting a 3.5-kb band and a tandem duplication for those strains exhibiting 7.7- and 10.0-kb bands. (Bottom) Within 34 sequenced genomes from the Drosophila Population Genome Project, all strains exhibited the Cyp12d1 duplications, but two of the strains were missing intron 3 and 300 nucleotides of exon 4 from both copies of Cyp12d1. (b) Based on sequence analyses, we have identified three polymorphisms in the Cyp12d1 duplication. First, the Cyp12d1 gene has been found singly, as in the 91-C and 91-R strains, and in tandem duplication, as in the reference strain. Second, there is variation in the presence of a conserved or weak polyadenylation signal within the 3′ UTR of the gene. This variation suggests that gene conversion has occurred in the DPGP isoline strains with a potential selective sweep of the strong polyadenylation signal
To determine whether the tandem duplication is restricted to those strains that produced a band of 7 kb or larger, we used a method described by Emerson et al. (2008) in which a set of primers was designed to face inversely within the Cyp12d1 gene (Fig. 3). In the presence of tandem duplications, the forward and reverse tandem duplication primers produced a 3.5-kb band; if only single copy genes is present, the primers fail to produce amplified products. Analysis of all 58 strains with this primer sets showed that all strains producing 7.7-kb bands with the Cyp12d1 region-spanning primers also produced amplified products with the tandem duplication primers, supporting the hypothesis that they contain duplications of Cyp12d1. None of the 47 strains producing a 3.5-kb band with the Cyp12d1 region-spanning primers generated an amplified product with the tandem duplication primers, demonstrating that they do not contain tandem duplications of this genomic region.
To detect additional DNA corresponding to a second Cyp12d1 gene in these strains, genomic regions upstream and downstream of the original 7.7-kb region in three strains (91-C, 91-R and reference strain) were amplified with more distal primer sets (571-nt downstream, 5,264-nt downstream, and 1,207-nt upstream primers) (Supplemental Table 1). Amplification of the reference strain with the upstream primer (1,207-nt upstream) and either the 571-nt downstream primer or the 5,264-nt downstream primer produced the expected bands of 8 kb and 12 kb. Amplification of the 91-C and 91-R strains with these same primer sets produced bands of 5.5 kb and 10 kb, respectively, which were the expected lengths for a single copy Cyp12d1 gene and its flanking regions (Table 3).
Sequence Differences in Cyp12d1-p and Cyp12d1-d in Sequenced D. melanogaster Genomes
Detailed analysis of the Cyp12d1-d and Cyp12d1-p gene region of the reference genome indicated that the Cyp12d1-d gene contains a conserved polyadenylation signal (AATAAA) 187-nt downstream of its stop codon and that this signal does not exist in the Cyp12d1-p gene. Consistent with this observation, the 3′ noncoding regions of the three full-length polyadenylated mRNA transcripts in the Genbank database (BT001433.1, AY061415.1, and BT031280.1) correspond to the 3′ noncoding region of Cyp12d1-d, but not to that of Cyp12d1-p. In addition, Cyp12d1-p and Cyp12d1-d both contain a suboptimal polyadenylation signal (AGTAAA) that is predicted to produce a transcript ~124 nt shorter than those found in Genbank (Fig. 5).

Single Cyp12d1 gene in 91-C and 91-R resembles Cyp12d1-p of reference strain. Alignment of 3′ UTR of the sequenced Cyp12d1 gene of 91-C and 91-R with the reference sequences of Cyp12d1-p and Cyp12d1-d shows their sequence similarity with Cyp12d1-p. Underlined sequences are the Cyp12d1-p 3′-UTR and Cyp12d1-d 3′-UTR reverse primers and the Cyp12d1 3′-UTR forward primer that were used to distinguish which transcripts are expressed in the reference strain. Two putative polyadenylation signal sequences are indicated in bold and underlined. The conserved polydenylation site, found only in Cyp12d1-d, occurs 188-nt downstream of the stop codon (bolded), while a suboptimal polyadenylation site, shared by Cyp12d1-d and -p, occurs 57-nt downstream of the stop codon. Based on the Cyp12d1 cDNA transcripts found in Genbank (BT001433.1, AY061415.1, BT031280.1), polyadenylation occurs 17- to 20-nt downstream of the conserved polyadenylation signal sequence
To expand our analysis of natural variation within D. melanogaster, we generated complete genome assemblies for 34 highly inbred lines from a well-studied North Carolina population included in the Drosophila Genetic Reference Panel (DGRP). Our analysis was restricted to DGRP strains where both short (Illumina) and long (454) reads were available and to the Cyp12d1-p/Cyp12d1-d genomic region (see “Materials and Methods” for details). The analysis of the Cyp12d1-p/Cyp12d1-d genomic region among DGRP strains revealed a low level of polymorphism within the exons of Cyp12d1 relative to introns, suggesting current (or recent) functionality. All the strains contain the tandem duplication of Cyp12d1, and both copies of the gene exhibit the strong polyadenylation signal of Cyp12d1-d in the reference strain. In two strains, intron 3 is missing from both the proximal and distal copies.
Sequence Changes in the Single Copy Cyp12d1 in the 91-C and 91-R Strains
The 91-C and 91-R strains, founded by a few hundred individuals from a common population in St. Paul, Minnesota in 1952, were selected for DDT resistance (91-R) or were never exposed to DDT (91-C). Individuals from the 91-R strain were then re-selected to be highly resistant to DDT (Festucci-Buselli et al. 2005). Sequencing of the genomic region of Cyp12d1 of 91-C and 91-R revealed a single allele of the gene in each strain that contains the 5′ upstream region of Cyp12d1-d in the reference genome and the three coding region SNPs and 3′ untranslated region (UTR) of Cyp12d1-p in the reference genome. As a consequence, this Cyp12d1 gene contains the suboptimal polyadenylation signal found in Cyp12d1-p and not the optimal polyadenylation signal found in Cyp12d1-d (Fig. 5).
Within the Cyp12d1 coding regions, introns 1 and 2 from the 91-C and 91-R strains were identical to the reference genome, but intron 3 differed in the 91-R strain in having a GT-to-AT change in its 5′ splice site that would prevent its recognition and cleavage. Based on the aberrant splice-site junction found in 91-R, the Cyp12d1 transcript from this strain would be predicted to retain its third intron (Fig. 6). Amplification and sequencing of the full-length coding sequence of Cyp12d1 from 91-R confirmed the retention of intron 3 in the transcript. Inclusion of intron 3 creates a premature stop codon that would eliminate 339 amino acids from this protein (Fig. 6). In addition to this change, the 5′ flanking sequences of Cyp12d1 do not greatly differ between the 91-C and 91-R strains, but do differ substantially from the reference genome in having two insertions (17 nt at 518 upstream of ATG; 40 nt at 366 upstream of ATG), one deletion (18 nt at 981 upstream of ATG) and a transversion of the TA-rich region immediately upstream of the 17-nt insertion to a GC-rich region (Supplemental Fig. 1).

Exon-intron structure of Cyp12d1 from 91-C and 91-R strains. Alignment of the sequenced Cyp12d1 genomic region from 91-C and 91-R strains with +593/+893 regions of Cyp12d1-d and Cyp12d1-p of the reference genome shows a GT-to-AT mutation (circled) of the third intron (delineated) splice-site junction in 91-R. This mutation was predicted to cause the retention of intron 3 in the Cyp12d1 transcript of the 91-R strain, which was observed when the transcript was sequenced. Retention of intron 3 introduces a premature stop codon (boxed) that, if functional, would decrease the protein length by 339 amino acids
Relative Expression of Cyp12d1- p and Cyp12d1- d Genes
To determine whether transcripts expressed in the y; cn bw sp reference strain are Cyp12d1-d or Cyp12d1-p, we designed gene-specific Cyp12d1-p 3′ UTR and Cyp12d1-d 3′ UTR primer sets in which the reverse directional primers for qRT-PCR were unique to the 3′ UTR region of either gene. In the reference strain, the Cyp12d1-d-specific primer produced, on average, 140-fold (11709.6 fM) higher expression than the Cyp12d1-p primer set (83.1 fM) (t test, p = 0.008). Thus, even with 99.4 % identity in their coding sequences, the Cyp12d1-p and Cyp12d1-p demonstrate differential patterns of constitutive expression.
Induction of Cyp12d1 Expression by Environmental Stresses
We tested a variety of stress treatments, including cold (4 °C), heat (37 °C), carbon dioxide, starvation, sucrose, hydrogen peroxide, and phenobarbital, to characterize the range of environmental stressors to which Cyp12d1 responds in single and duplicated copies.
Phenobarbital treatment of single copy strain 91-C and double copy strain Oregon-R flies showed that Cyp12d1 was highly induced at similar levels (18.1- and 14.8-fold, Fig. 7a) by this potent xenobiotic inducer in both strains. In contrast, for non-xenobiotic stresses, the single copy strain 91-C showed no or low levels of induction of Cyp12d1 and even low levels of repression (Fig. 7b), whereas, in the double copy strain Oregon-R, Cyp12d1 responds to a wider range of environmental stresses including heat both at 1 h (3.4-fold) and 2 h (fourfold) after recovery, starvation (3.4-fold), and hydrogen peroxide (3.4-fold). Sucrose and CO2 treatment caused twofold decrease of Cyp12d1 in Oregon-R, and CO2 treatment caused a slight but statistically significant 1.2-fold increase of Cyp12d1 in 91-C (Fig. 7b).

Comparison of Cyp12d1 inducible expression in single and double copy strains. Fold induction of the Cyp12d1 gene in Oregon-R (double copy) and 91-C (single copy) flies treated with a phenobarbital at 0.4 % and b various environmental stresses as compared with untreated adults using quantitative real time PCR (qRT-PCR). Significance levels are indicated by two asterisks (P < 0.0001), one asterisk (P < 0.001) or pound sign (P = 0.0016)
Discussion
Using the twelve sequenced Drosophila genomes (Drosophila 12 Genomes Consortium et al. 2007), a large pool of D. melanogaster strains of diverse origins and a smaller pool of D. melanogaster strains with recent microevolutionary changes, we have focused on the Cyp12d1 gene as an initial step toward investigating how environmental selection pressures act on the fixation and diversification of duplicated genes.
On the macroevolutionary scale, we found that the Cyp12d1 duplication in D. melanogaster is shared across the melanogaster subgroup (D. simulans, sechellia, yakuba, ananassae, melanogaster, and erecta), but the two genes have remained almost identical only in D. melanogaster. Phylogenetic analysis of maximum likelihood of the Cyp12d1/Cyp12d3 paralogs across Drosophila species showed increased rates of amino acid changes in Cyp12d3 with a highly significant signature of positive selection in the species D. sechellia, a species that has specialized to detoxify phytochemicals from its only host plant, Morinda citrifolia (R’Kha et al. 1991). Thus, in D. sechellia, positive selection of Cyp12d3 may have occurred concurrently with its ecological specialization on its toxic host plant. Based on its signature of positive selection, we predict that Cyp12d3 in D. sechellia has evolved a function unique to this species, perhaps in the metabolism of toxic compounds in its host plant, such as octanoic or hexanoic acid (Amlou et al. 1998; Legal et al. 1994; Farine et al. 1996). To test this hypothesis, the Cyp12d3 protein would need to be isolated using a heterologous expression system and the metabolism of the host plant compounds by the protein measured.
To address mechanisms of the evolution of gene duplication within a species, we identified copy number variation for Cyp12d1 across a diverse set of D. melanogaster populations. Although in the reference genome of D. melanogaster, the Cyp12d1 (Cyp12d1-p) and Cyp12d3 (Cyp12d1-d) paralogs are nearly identical and resemble Cyp12d1 genes in other Drosophila species, our extensive analyses indicate that these are not newly duplicated orthologs as previous assumed. The existence of Cyp12d3 in the melanogaster subgroup indicates that the duplication event predates the emergence of the current version of D. melanogaster. The almost complete absence of divergence between Cyp12d1-p and Cyp12d1-d in many current D. melanogaster strains suggests that these genes have undergone gene conversion, while the Cyp12d1/Cyp12d3 gene pairs in the other Drosophila species have diverged.
The apparent high rate of duplication and loss observed in D. melanogaster has a potential to provide the genetic variability needed for the evolution of novel regulatory or structural mechanisms to deal with xenobiotic or other environmental challenges. In the reference strain, Cyp12d1 is tandemly duplicated to produce two nearly identical genes that differ in their 3′ UTR with different polyadenylation signals that may affect mRNA stability. In contrast, the strong polyadenylation signal is found in both copies of all the 34 strains analyzed from the Drosophila Genetic Reference Panel; within two of these lines, a deletion of intron 3 is found in both the proximal and distal copies, which suggests the possibility that gene conversion rapidly homogenized these mutations in both copies.
To address whether the observed sequence differences in the 3′ UTR of Cyp12d1-d and Cyp12d1-p translate into expression differences, we measured the absolute levels of Cyp12d1-d and Cyp12d1-d transcript’s abundance in the reference strain y; cn bw sp. We found significant differences in basal expression levels, where Cyp12d1-d, which contains a strong polyadenylation signal, is expressed 140-fold higher than Cyp12d1-p. This suggests that duplication of Cyp12d1 led to the inclusion of a strong polyadenylation signal in the 3′ UTR of Cyp12d1-d by duplication of a sequence already found in the 5′ untranslated region (Fig. 3). Thus, even with 99.4 % nucleotide similarity, duplication of Cyp12d1 does not necessarily produce a double dose of gene expression, as even seemingly slight changes in the 3′ UTR affect transcript abundance.
Based on our comparisons between strains with genomes that contain single (91-C) and duplicated copies (Oregon-R) of Cyp12d1, duplication may instead broaden the range of environmental stresses to which Cyp12d1 responds. In Oregon-R, Cyp12d1 demonstrates higher induction levels and responses to a wide range of environmental stresses (including heat, starvation, and H2O2) than in 91-C. The observation that phenobarbital treatment induced Cyp12d1 expression in 91-C to a greater level than in Oregon-R does not support the positive dosage model of gene duplication, which further strengthens our evidence based on transcript abundance differences between Cyp12d1-p and Cyp12d1-d presented above. However, it remains to be determined if these results are unique to the Oregon-R strain or are also reflective of other strains with the duplication. Further studies need to be performed to determine if, across multiple strains, the acquisition of these responses to novel environmental stressors supports a model in which gain-of-function mutations occurs (Innan and Kondrashov 2010). Regardless, this study represents the first report showing that a broad range of non-xenobiotic environmental stresses can induce Cyp12d1.
Copy number variation for Cyp12d1 across a diversity of D. melanogaster populations showed no association with selection pressures for insecticide resistance. While the link between gene amplification and insecticide resistance is commonly encountered for esterases and glutathione S-transferases, gene duplication is often less associated with insecticide resistance and cytochrome P450 monooxygenases, though some cases have been documented (Bass and Field 2011). In D. melanogaster, Cyp6g1 has been found as a duplication in wild populations throughout Europe, Asia, and the United States, but increased transcription of Cyp6g1 cannot be dissociated from the presence of a transposable element within the 5′ promoter region, a common mechanism of overexpression of P450 genes (Schmidt et al. 2010). Overexpression of Cyp12d1 in a transgenic fly system resulted in flies with tolerance to DDT, ostensibly due to the increased metabolism of the compound, but other P450 genes associated with DDT resistance (Cyp6a2, Cyp6a8) do not seem to increase DDT metabolism (Daborn et al. 2001). Insecticide (DDT) resistance is most likely multifactorial and epigenetic interactions must be considered when evaluating its evolution within a given strain.
This complicated scenario of DDT resistance is illustrated by our finding that within the highly DDT-resistant strain 91-R there is a single copy of Cyp12d1 with a mutation in a splice-site junction that would render the protein nonfunctional, a mutation not found in the 91-C strain. The strains 91-C and 91-R were split from a single population of several hundred females 59 years ago, and the 91-R strain has undergone at least two periods of intense selection pressure for DDT resistance, once in the original population and then again in the current laboratory. To the authors’ knowledge, these lines have never been outbred, and sequencing of the PCR products of the Cyp12d1 locus resulted in a single sequencing read without significant noise. 91-R carries a mutation in the 5′ splice site of intron 3, which produces a premature stop codon in the transcript. Owing to the strain’s high metabolic activity, adaptive selection may have favored the repression of Cyp12d1 to limit the production of reactive oxidative species, a byproduct of the P450 oxidative reactions, notably in the mitochondria where Cyp12d1 is found.
By means of evolutionary approaches that encompass millions of year to decades, to study the Cyp12d1 region, we have encountered considerable variation, marked by gene duplication, loss and mutations, suggesting that this region could be a genomic hotspot for adaptive response to xenobiotics within the Drosophila species we investigated. The twelve Drosophila genomes offer an unprecedented view of the divergence of paralogous and orthologous genes. Limiting observations of the Cyp12d1 gene to one strain of one species results in a myopic view of the evolution of that gene. The comparison of xenobiotic-responsive genes across species and strains within a species, which we have named “evolutionary toxicogenomics,” has a potential for clarifying the ways in which a gene has responded to the selection in its environment and the extent to which it has been constrained by its genomic history. Indeed, the evolution of the Adh/Adhr duplication in Drosophila species has already demonstrated the power of cross-species comparisons in constructing phylogenetic relationships and connecting gene evolution with environmental selection pressure, yet the function of the Adhr is still unknown, despite Adh being one of the best studied Drosophila genes (Betran and Ashburner 2000; Oppentacht et al. 2002; Matzkin and Eanes 2003; Tamura et al. 2004). Evolutionary toxicogenomics also offers a systematic strategy for researchers to identify which enzymes, be they conserved or unique, are appropriate for further characterization. As the cost of sequencing genomes decreases, the availability of these genomic sequences should dramatically increase our ability to understand the evolution of detoxification systems by helping to identify those genes and gene families that are shaped by broad evolutionary patterns and those that are shaped by local or specific evolutionary challenges, whether abiotic or biotic.
References
Amlou M, Moreteau B, David JR (1998) Larval tolerance in the Drosophila melanogaster species complex toward the two toxic acids of the D. sechellia host plant. Hereditas 129:7–14
Bak S, Beisson F, Bishop G, Hamberger B, Hofer R, Paquette S, Werck-Reichhart D (2011) Cytochrome P450s. In: Somerville CR, Meyerowitz EM (eds) The arabidopsis book. American Society of Plant Biologists, Rockville. URL:http://www.aspb.org/publications/arabidopsis
Bass C, Field LM (2011) Gene amplification and insecticide resistance. Pest Manag Sci 67:886–890
Betran E, Ashburner M (2000) Duplication, dicistronic transcription, and subsequent evolution of the Alcohol dehydrogenase and Alcohol dehydrogenase-related genes in Drosophila. Mol Biol Evol 17:1344–1352
Brandt A, Scharf M, Pedra JHF, Holmes G, Dean A, Kreitman M, Pittendrigh BR (2002) Differential expression and induction of two Drosophila cytochrome P450 genes near the Rst(2)DDT locus. Insect Mol Biol 11:337–341
Drosophila 12 Genomes Consortium, Clark AG, Eisen MB et al (2007) Evolution of genes and genomes on the Drosophila phylogeny. Nature 450:203–218
Daborn P, Boundy S, Yen J, Pittendrigh B, ffrench-Constant R (2001) DDT resistance in Drosophila correlates with Cyp6g1 over-expression and confers cross-resistance to the neonicotinoid imidacloprid. Mol Genet Genomics 266:556–563
Dapkus D, Merrell DJ (1977) Chromosomal analysis of DDT-resistance in long-term selected population of Drosophila melanogaster. Genetics 87:685–697
Emerson JJ, Cardoso-Moreira M, Borevitz JO, Long M (2008) Natural selection shapes genome-wide patterns of copy-number polymorphism in Drosophila melanogaster. Science 320:1629–1631
Farine J, Legal L, Moreteau B, Le Quere JL (1996) Volatile components of ripe fruits of Morinda citrifolia and their effects on Drosophila. Phytochemistry 41:433–438
Festucci-Buselli RA, Carvalho-Dias AS, de Oliveira-Andrade M, Caixeta-Nunes C, Li HM, Stuart JJ, Muir W, Scharf ME, Pittendrigh BR (2005) Expression of Cyp6g1 and Cyp12d1 in DDT resistant and susceptible strains of Drosophila melanogaster. Insect Mol Biol 14:69–77
Feyereisen R (2011) Arthropod CYPomes illustrate the tempo and mode in P450 evolution. Biochim Biophys Acta 1814:19–28
Feyereisen R (2012) Insect CYP genes and P450 enzymes. In: Gilbert LI (ed) Insect molecular biology and biochemistry. Elsevier Press, Oxford, pp 236–316
Guengerich FP (2005) Human cytochrome P450 enzymes. In: Ortiz de Montellano PR (ed) Cytochrome P450: structure, mechanism, and biochemistry, 3rd edn. Kluwer Academic, New York, pp 377–530
Innan H, Kondrashov F (2010) The evolution of gene duplications: classifying and distinguishing between models. Nature Rev Genet 11:97–108
Jensen HR, Scott IM, Sims S, Trudeau VL, Arnason JT (2006) Gene expression profiles of Drosophila melanogaster exposed to an insecticidal extract of Piper nigrum. J Agric Food Chem 54:1289–1295
Jones DT, Taylor WR, Thornton JM (1992) The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci 8:275–282
Kelly SL, Kelly DE, Jackson CJ, Warrilow AGS, Lamb DC (2005) The diversity and importance of microbial cytochromes P450. In: Ortiz de Montellano PR (ed) Cytochrome P450: structure, mechanism, and biochemistry, 3rd edn. Kluwer Academic, New York, pp 585–618
Kondrashov FA, Rogozin IB, Wolf YI, Koonin EV (2002) Selection in the evolution of gene duplications. Genome Biol 3:research0008–research0008.9
Le Goff G, Hilliou F, Siegfried BD, Boundy S, Wajnberg E, Sofer L, Audant P, ffrench-Constant RH, Feyereisen R (2006) Xenobiotic response in Drosophila melanogaster: sex dependence of P450 and GST gene induction. Insect Biochem Mol Biol 36:674–682
Legal L, Chappe B, Jallon J (1994) Molecular basis of Morinda citrifolia (L.): toxicity on Drosophila. J Chem Ecol 20:1931–1943
Li WH (1999) Molecular evolution. Sinauer Associates, Sunderland
Li H, Durbin R (2010) Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26:589–595
Li X, Schuler MA, Berenbaum MR (2007) Molecular mechanisms of metabolic resistance to synthetic and natural xenobiotics. Annu Rev Entomol 52:231–253
Li HM, Buczkowski G, Mittapalli O, Xie J, Wu J, Westerman R, Schemerhorn BJ, Murdock LL, Pittendrigh BR (2008) Transcriptomic profiles of Drosophila melanogaster third instar larval midgut and responses to oxidative stress. Insect Mol Biol 17:325–339
Li H, Handsaker B, Wysoker A, Fennell T, Ruan R et al (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079
Lupski JR (2007) Genomic rearrangements and sporadic disease. Nat Genet 39:S43–S47
Matzkin LM, Eanes WF (2003) Sequence variation of alcohol dehydrogenase (Adh) paralogs in cactophilic Drosophila. Genetics 163:181–194
Mizutani M, Ohta D (2010) Diversification of P450 genes during land plant evolution. Annu Rev Plant Biol 61:291–315
Nelson DR (1998) Metazoan cytochrome P450 evolution. Comp Biochem Phyiol C Pharmacol Toxicol Endocrinol 121:15–22
Nelson DR (2009) The cytochrome P450 homepage. Hum Genomics 4:59–65
Nelson DR, Koymans L, Kamataki T, Stegeman JJ, Feyereisen R, Waxman DJ, Waterman MR, Gotoh O, Coon MJ, Estabrook RW, Gunsalus IC, Nebert DW (1996) P450 superfamily: update on new sequences, gene mapping, accession numbers and nomenclature. Pharmacogenetics 6:1–42
Nielsen R, Yang Z (1998) Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148:929–936
Ohno S (1970) Evolution by gene duplication. Springer, New York
Oppentocht JE, van Delden W, van de Zande L (2002) Isolation and characterization of the genomic region from Drosophila kuntzei containing the Adh and Adhr genes. Mol Biol Evol 19:1026–1040
Pearson WR (1990) Rapid and sensitive sequence comparison with FASTP and FASTA. Method Enzymol 183:63–98
Pedra JH, McIntyre LM, Scharf ME, Pittendrigh BR (2004) Genome-wide transcription profile of field- and laboratory-selected dichlorodiphenyltrichloroethane (DDT)-resistant Drosophila. Proc Natl Acad Sci USA 101:7034–7039
R’Kha S, Capy P, David JR (1991) Host-plant specialization in the Drosophila melanogaster species complex: a physiological, behavioral, and genetical analysis. Proc Natl Acad Sci USA 88:1835–1839
Schmidt JM, Good RT, Appleton B, Sherrard J, Raymant GC, Bogwitz MR, Martin J, Daborn PJ, Goddard ME, Batterham P, Robin C (2010) Copy number variation and transposable elements feature in recent, ongoing adaptation at the Cyp6g1 locus. PLoS Genet 6:e1000998
Schuler MA (2011) P450s in plant-insect interactions. Biochim Biophys Acta 1814:36–45
Schuler MA, Duan H, Bilgin M, Ali S (2006) Arabidopsis P450s through the looking glass: a window on plant biochemistry. Phytochem Reviews 5:205–237
Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Maner S, Massa H, Walker M, Chi M et al (2004) Large-scale copy number polymorphism in the human genome. Science 305:525–528
Singh ND, Larracuente AM, Sackton TB, Clark AG (2009) Comparative genomics on the Drosophila phylogenetic tree. Annu Rev Ecol Evol Syst 40:459–480
Sun L (2009) The response of Drosophila melanogaster to methamphetamine and oxidative stress. Ph.D. dissertation, Purdue University
Sun W, Margam VM, Sun L, Buczkowski G, Bennett GW, Schemerhorn B, Muir WM, Pittendrigh BR (2006) Genome-wide analysis of phenobarbital-inducible genes in Drosophila melanogaster. Insect Mol Biol 15:455–464
Swofford DL (2002) PAUP*. Phylogenetic analysis using parsimony (*and other methods). Version 4. Sinauer Associates, Sunderland, MA
Tamura K, Subramanian S, Kumar S (2004) Temporal patterns of fruit fly (Drosophila) evolution revealed by mutation clocks. Mol Biol Evol 21:36–44
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Thomas JH (2007) Rapid birth-death evolution specific to xenobiotic cytochrome P450 genes in vertebrates. PLoS Genet 3:e67
Willoughby L, Chung H, Lumb C, Robin C, Batterham P, Daborn PJ (2006) A comparison of Drosophila melanogaster detoxification gene induction responses for six insecticides, caffeine and phenobarbital. Insect Biochem Mol Biol 36:934–942
Willoughby L, Batterham P, Daborn PJ (2007) Piperonyl butoxide induces the expression of cytochrome P450 and glutathione S-transferase genes in Drosophila melanogaster. Pest Manag Sci 63:803–808
Wong WS, Yang Z, Goldman N, Nielsen R (2004) Accuracy and power of statistical methods for detecting adaptive evolution in protein coding sequences and for identifying positively selected sites. Genetics 168:1041–1051
Yang Z (1997) PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 13:555–556
Yang Z, Nielsen R, Goldman N, Pedersen AM (2000) Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155:431–449
Yang Z, Wong WS, Nielsen R (2005) Bayes empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol 22:1107–1118
Zhang F, Gu W, Hurles ME, Lupski JR (2009) Copy number variation in human health, disease, and evolution. Ann Rev Genom Hum G. 10:451–481
Zhou Q, Zhang G, Zhang Y, Xu S, Zhao R, Zhan Z, Li X, Ding Y, Yang S, Wang W (2008) On the origin of new genes in Drosophila. Genome Res 18:1446–1455
Acknowledgments
We thank Susan Balfe for her guidance in the laboratory. Research was funded by C.W. Kearns, C.L. Metcalf, and W.P. Flint Endowment Funds to BRP. CMM was funded by a University of Illinois Dissertation Completion Fellowship.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
McDonnell, C.M., King, D., Comeron, J.M. et al. Evolutionary Toxicogenomics: Diversification of the Cyp12d1 and Cyp12d3 Genes in Drosophila Species. J Mol Evol 74, 281–296 (2012). https://doi.org/10.1007/s00239-012-9506-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-012-9506-3