
Abstract
DEAD-box proteins are found in all domains of life and participate in almost all cellular processes that involve RNA. The presence of DEAD and Helicase_C conserved domains distinguish these proteins. DEAD-box proteins exhibit RNA-dependent ATPase activity in vitro, and several also show RNA helicase activity. In this study, we analyzed the distribution and architecture of DEAD-box proteins among bacterial genomes to gain insight into the evolutionary pathways that have shaped their history. We identified 1,848 unique DEAD-box proteins from 563 bacterial genomes. Bacterial genomes can possess a single copy DEAD-box gene, or up to 12 copies of the gene, such as in Shewanella. The alignment of 1,208 sequences allowed us to perform a robust analysis of the hallmark motifs of DEAD-box proteins and determine the residues that occur at high frequency, some of which were previously overlooked. Bacterial DEAD-box proteins do not generally contain a conserved C-terminal domain, with the exception of some members that possess a DbpA RNA-binding domain (RBD). Phylogenetic analysis showed a separation of DbpA-RBD-containing and DbpA-RBD-lacking sequences and revealed a group of DEAD-box protein genes that expanded mainly in the Proteobacteria. Analysis of DEAD-box proteins from Firmicutes and γ-Proteobacteria, was used to deduce orthologous relationships of the well-studied DEAD-box proteins from Escherichia coli and Bacillus subtilis. These analyses suggest that DbpA-RBD is an ancestral domain that most likely emerged as a specialized domain of the RNA-dependent ATPases. Moreover, these data revealed numerous events of gene family expansion and reduction following speciation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
DEAD-box proteins are highly conserved in bacteria, archaea, and eukaryotes. They participate in many cellular processes involving RNA, such as transcription, translation, editing, mRNA degradation, and ribosomal assembly (Linder and Daugeron 2000; Patel and Donmez 2006). DEAD-box proteins are classified within the SF2 superfamily of helicases based on the presence of two characteristic domains, DEAD and Helicase_C. The SF2 superfamily includes 10 distinct families that can be differentiated based on sequence comparison and on the conservation of particular motifs (see Fairman-Williams et al. 2010, for a detailed analysis of the classification of SF1 and SF2 helicases). In vitro, DEAD-box proteins exhibit an RNA-dependent ATPase activity and some also exhibit ATP-dependent RNA helicase activity (Cordin et al. 2006). Other DEAD-box proteins can assist in RNA folding and displace proteins from RNA (Jankowsky and Fairman 2007). In this study, we refer to all members of this family as DEAD-box proteins, regardless of their assayed activity, since we focus on their evolutionary history and sequence homology. It is important to note that although a large number of proteins contain DEAD and Helicase_C domains, these proteins may belong to other families and not necessarily to the DEAD-box family.
All members of the DEAD-box family feature 10 characteristic conserved motifs. Crystallographic studies of specific DEAD-box proteins have revealed that these exhibit the same protein fold with two alpha–beta-RecA-like domains. For this reason, many studies refer to the DEAD and Helicase_C domains as RecA-like 1 and RecA-like 2 domains, respectively. These studies have allowed for the identification of specific residues that interact with ATP and RNA (Bleichert and Baserga 2007; Rocak et al. 2005; Tanner et al. 2003).
The C-terminal region of some eukaryotic DEAD-box proteins may contain specific domains that are required for cellular localization and for RNA or protein–protein interactions (Linder and Daugeron 2000; Tuteja and Tuteja 2004). However, in bacteria, most DEAD-box proteins lack recognizable motifs or domains at the C-terminal end. Interestingly, some bacterial DEAD-box proteins have a C-terminal domain known as DbpA RNA-binding domain (RBD) (PF03880), which has been shown to mediate in some DEAD-box proteins the specific recognition of hairpin 92 of the 23S ribosomal RNA (rRNA).
In bacteria, the best studied DEAD-box proteins belong to E. coli and B. subtilis (Table 1). E. coli possess five DEAD-box proteins that are involved in ribosome assembly, translation (DeaD/CsdA, RhlE, SrmB, and DbpA) (Charollais et al. 2003a; Karginov and Uhlenbeck 2004; Moll et al. 2002), and RNA degradation (RhlB) (Py et al. 1996). B. subtilis possess four DEAD-box proteins (DeaD/YxiN, CshA, CshB, and YfmL). Most research efforts on this species have centered on elucidating the biochemical properties and on identifying protein–protein interactions (Ando and Nakamura 2006; Hunger et al. 2006).
Homolog proteins DbpA and DeaD/YxiN from the model organisms E. coli and B. subtilis, respectively, bind hairpin 92 of 23S rRNA with high specificity (Tsu et al. 2001; Kossen et al. 2002). Moreover, the YxiN C-terminal domain was found to bind RNA with the same affinity as the full-length protein, and the catalytic domain to retain ATPase activity, which suggests that YxiN is a modular protein where the catalytic and RBDs are combined (Karginov et al. 2005). The specificity for rRNA binding and activation of ATPase activity was further demonstrated by the construction of a chimeric SrmB protein containing the DbpA-RBD from YxiN (Kossen et al. 2002). The DbpA-RBD of DeaD/YxiN has been crystallized and its structure has been solved to 1.7 Å resolution (Wang et al. 2006). The structure has an RNA recognition motif (RRM), similar to that found in many eukaryotic RBDs, although there is no similarity at the level. Interestingly, the Thermus thermophilus DEAD-box protein Hera also binds hairpin 92 from the 23S rRNA. However, the C-terminal domain (Hera-RBD) revealed the fold of an altered RRM that has limited structural homology to the RBD of the DEAD-box protein DeaD/YxiN from B. subtilis (Rudolph and Klostermeier 2009).
Since the DEAD-box family was first described by Linder et al. in 1989, efforts have focused on uncovering the biochemical properties of these proteins and understanding their role and association with particular processes in RNA metabolism. The purpose of this study is to take advantage of the large number of completely sequenced bacterial genomes to gain an understanding of the number of DEAD-box proteins present in different bacteria, their phylogenetic distribution, and the features that distinguish them in different phyla. We examined the genetic redundancy of the DEAD-box proteins and assessed the conservation of the residues that comprise the DEAD-Helicase_C domains from a large number of bacterial sequences. To explore the evolutionary implications of the maintenance and possible functional specialization of members from this protein family, we also examined the evolutionary history of DEAD-box proteins, their orthologous relationships, and lineage-specific expansions and reductions (paralogies), particularly among Firmicutes and γ-Proteobacteria, the phyla to which B. subtilis and E. coli belong, respectively.
Materials and Methods
Sequences Used for the Analysis and Hidden Markov Models Used to Detect DEAD, Helicase_C, and DbpA-RBD Domains
We analyzed 563 complete bacterial genomes (see Supplemental Table S1). The collection of DEAD-box protein sequences was obtained using a Hidden Markov Model (HMM) (Eddy 1998). The HMM model was built and calibrated from the alignment of DEAD (PF00270) and Helicase_C (PF00271) domains from 250 sequences retrieved with PSI-Blast (Altschul and Koonin 1998) against the RefSeq database, using the DeaD/CsdA (NP_417631.2) and DeaD/YxiN (NP_391790.1) protein sequences from E. coli and B. subtilis as queries against Proteobacteria and Firmicutes, respectively. The PSI-Blast was conducted for a total of three iterations. Sequence redundancy was reduced using FSA-Blast (Cameron et al. 2004). This procedure yielded a set of 1,211 DEAD-box sequences (see Supplemental Table S2) that were representative of 1,848 protein sequences (see Supplemental Table S3). The DbpA-RBD (PF03880) identification at the C-terminal region was conducted using the Pfam protocol (Finn et al. 2010) (see Supplemental Table S7). E values from 10−5 to 10−46 were obtained for the DbpA-RBD sequences that were identified. An alignment of a sample of DbpA-RBD from different taxa is shown in Supplemental Figure 5S. We examined the alignments to confirm that the C-terminal domain in these protein sequences were bona fide DbpA-RBD homologs. This was observed from the alignments, where a few stretches of amino acids are generally conserved, mainly the G residue at positions corresponding to YxiN 404, 423, 430, and 462, charged residues that accompany the first and last G residues, the relatively well-conserved aromatic residues at positions 407 and 447, and the group of conserved amino acids that is located between the amino acid stretch that constitutes the YxiN β1 and α1, and the stretch between α2 and β4, as observed by Wang et al. (2006).
Alignment and Phylogenetic Information Analysis
Sequences containing only DEAD (PF00270) and Helicase_C (PF00271) domains were first aligned with ClustalW (v. 1.83) (Thompson et al. 1994) and then manually curated. We used Gblocks to extract informative positions from the protein alignment (Talavera and Castresana 2007). A total of 323 non-ambiguous positions were used to conduct the phylogenetic analysis. A complete alignment of protein sequences without Gblocks filtering was used in the maximum likelihood analysis. A complete alignment of nucleotide and amino acid sequences without Gblocks filtering was used in the Bayesian inference.
Motif Consensus and HMM Logo Design
Sequence alignment of 1,208 sequences of DEAD-box proteins was used to build and calibrate the HMM model (HMMer v. 2.0) (Eddy 1998). The HMM logo was plotted with LogoMat-P (v. 0.78) (Schuster-Bockler and Bateman 2005).
Evolutionary Analysis
Phylogenetic reconstructions were done using the PHYLIP suite (version 3.67-1) (Felsenstein 2005) with the following parameters: method Neighbor-Joining (NJ), substitution model = JTT, gamma = 1, and 1,000 bootstrap replicates. We also conducted a Maximum likelihood analysis using FastTree 2 software (Price et al. 2010) to evaluate the reproducibility of the grouping of the DEAD-box sequences (see Supplemental Figure 1S). For the orthologs distribution (Fig. 4), we conducted a Maximum Likelihood analysis of 16S rRNA from selected bacteria using PhyML software (Guindon and Gascuel 2003) with the following parameters: method GTR + I + Γ and 1,000 bootstrap replicates.
To assess the evolutionary history of DEAD-box protein coding genes within γ-Proteobacteria and Firmicutes, we conducted a Bayesian inference with Mr. Bayes (version 3.1.2) (Huelsenbeck and Ronquist 2001), using 773 and 348 nucleotide and amino acid sequences from γ-Proteobacteria and Firmicutes, respectively. We used the general-time-reversible model (GTR + I + Γ) for the nucleotide substitution with unlinked parameters, nucleotide frequency, shape of the gamma distribution, and proportion of invariable sites. We evaluated two independent runs with four incrementally heated Markov chains (temp = 0.2) until 5 × 106 generations were reached with tree sampling every 200 generations. For the amino acid sequence analyses, we used the WAG and Mtmam models for Firmicutes and γ-Proteobacteria, respectively. In this case, we evaluated three independent runs with four incrementally heated Markov chains (temp = 0.2) until 5 × 106 generations were reached sampling trees every 200 generations. Plots visualized on Tracer (version 1.4.1) (Drummond and Rambaut 2007) of Ln likelihood versus generation number indicated the stabilization of likelihood values before generation 250,000. The initial 20% of trees were discarded as a burn in. Posterior probabilities and the marginal posterior estimates of the evolutionary parameters were plotted throughout the final tree. The sequence used as the outgroup for Firmicutes was from a Cyanobacteria (Nostoc sp., GI: 1108319/17232210), while for γ-Proteobacteria we used a sequence from δ-Proteobacteria (Myxococcus xanthus DK 1622, GI: 40104787/108757460). Both outgroups were selected from the grouping seen in the larger phylogenetic analysis previously performed by Neighbor joining. The final topology visualizations were edited with iTOL (http://itol.embl.de) (Letunic and Bork 2007).
Results
The Big Picture: The Bacterial DEAD-Helicase_C Tree
We studied 563 complete bacterial genomes to obtain sequences possessing all ten motifs that have been defined by Rocak and Linder (2004) and by Jankowsky and Putnam (2010) for the DEAD-box family (see Supplemental Table S1). Figure 1 shows the overall bioinformatics work flow, which included the construction of a Hidden Markov Model and multiple alignments of the retrieved sequences to only keep bona fide DEAD-box protein members. Only the DEAD and Helicase_C domains were used for the analysis, while the N- and C-terminal end sequences, which were attached to these two main domains, were excluded. Our procedure selected a set of 1,211 DEAD-box sequence relatives. The isolated DEAD-Helicase_C domain sequences were aligned and their phylogenetic distribution allowed us to observe the diversity per genome and redundancy of this protein family. As a first approximation, an amino-acid-based phylogenetic tree was obtained using Neighbor-Joining. The tree topology exhibited a separate branch for DEAD-box proteins. Upon detailed analysis of the sequences through Pfam, we found that the majority of the members of this branch (66%) possessed the DbpA-RBD (shown in yellow in Fig. 2). A second group contained sequence members who almost all lacked the DbpA-RBD (shown in blue in Fig. 2) (for details on the sequences, see Supplemental Tables S4 and S5). Each of these two groups contained members from all bacterial phyla. Additionally, a large proportion of DEAD-box protein sequences branched out into a group that was composed mainly of proteobacterial sequences. We refer to the latter group as RhlE-like, since RhlE from E. coli is the only studied member in this group (shown in green in Fig. 2). The DEAD-box protein coding genes in this group seemed to have arisen from a duplication that allowed a member lacking DbpA-RBD to expand in the Proteobacteria. A divergent branch that was basal to all others was composed of sequences from Actinobacteria, all of which lacked the DbpA-RBD (shown in brown in Fig. 2). DbpA-RBD (approximately 80 amino acids) is the only conserved domain recognized in the C-terminal end of some DEAD-box protein sequences, and although it was not included in the amino acid sequences used to construct this phylogeny, it turned out to be an outstanding feature that allowed for the distinction of members that clearly fell into separate clades.

Overview of the methodology used in this work to identify and analyze bacterial DEAD-box protein genes

Phylogenetic tree of the DEAD-Helicase_C domains in 1211 bacterial DEAD-box proteins. Sequences containing only DEAD and Helicase_C domains were first aligned and then manually curated. We used Gblocks to extract informative positions from the protein alignment and a total of 323 non-ambiguous positions were used to conduct the phylogenetic analysis by Neighbor joining (see “Materials and methods”). The grouping of classes was determined according to clades layout. Three major groups are identified and colored yellow, blue, and green. Some Actinobacteria DEAD-box protein sequences branch in a separate divergent group (brown). The presence and absence of a DbpA RNA-binding domain (RBD) is the main feature distinguishing yellow and blue DEAD-box protein sequences, respectively, while green section sequences form a separate branch that is dominated by proteobacterial members. Location of known RNA helicases from E. coli and B. subtilis are indicated in the phylogeny. Information about the genomes and the sequences IDs used to generate this phylogeny can be found in Supplemental Tables S4 and S5
The observation of the DbpA-RBD in members of all examined phyla revealed that it was present prior to speciation. Since it was suggested to have an important role in 23S rRNA binding and aid in rRNA assembly, we wondered whether it was already part of the architecture of DEAD-box proteins of ancestral origin. To answer this question, we analyzed two presumed representatives of early branching bacteria, Petrotoga mobilis SJ95 and Aquifex aeolicus VF5, both of which possess a single DEAD-box protein. Only the DEAD-box protein from Petrotoga mobilis SJ95 was found to have a DbpA-RBD. The Planctomycetes phylum has previously been suggested to be ancient (Brochier and Philippe 2002). One of its members, Rhodopirellula baltica SH 1, contains 3 DEAD-box proteins genes, one of which possesses a DbpA-RBD. This analysis already suggested that DEAD-box protein genes possessing or lacking a DbpA-RBD shared the same lineage, and the genes most likely originated from a duplication that occurred before the speciation of these bacterial families. Other strategies of phylogeny reconstruction were used, including the selection of fewer genomes to reduce the bias toward Firmicutes and Proteobacteria (data not shown). Again, DbpA-RBD-containing and DbpA-RBD-lacking sequences were observed separated from the proteobacterial-dominated RhlE-like branch, which is consistent with the hypothesis that DbpA-RBD-containing and DbpA-RBD-lacking DEAD-box proteins originated from a duplication and subsequently followed separate lineages. Moreover, a third class, which lacks the DpbA-RBD at the C-terminal, prospered mainly in the Proteobacteria. Given the large bias of available genome sequences toward Proteobacteria and Firmicutes as well as the large phylogenetic distance between them, it is difficult to conduct a fine evolutionary analysis combining these groups. To further analyze these possible evolutionary scenarios, we carried out a separate analysis for the Firmicutes and the Proteobacteria.
Evolutionary History of DEAD-Box Protein Coding Genes in Firmicutes
A Bayesian analysis at the nucleotide/codon and amino acid level was carried out to obtain phylogenetic reconstructions for the DEAD-box protein family in Firmicutes. Both analyses were capable of resolving very similar topologies (see “Materials and methods”), although the amino-acid-based phylogeny resolved the basal branches better. Figure 3 shows the phylogeny obtained at the amino acid level, and Supplemental Figure 2S shows the phylogeny at the nucleotide level. A cyanobacterial protein (Nostoc sp., GI: 17232210) was used as outgroup in this analysis. The topology of a multi-gene family, where genes appeared before the speciation of the Firmicutes, which is the case for DEAD-box proteins, has to be interpreted by clade since each of the vertically inherited genes has followed a different history. For each clade, we observed that the phylogenetic distribution of the members in all cases correlated with the genus, which suggested an absence of horizontal gene transfer among these family members. The basal proteins belonged to Acholeplasma laidlawii PG8A (GI: 162447374) and Clostridium phytofermentans ISDg (GI: 160880140), followed by two sequences of Alkaliphilus metalliredigens QYMF (GI: 150390712) and Desulfitobacterium hafniense Y51 (GI: 89896500), and one group of C. botulinum.

Bayesian inference of DEAD-box protein from Firmicutes. Complete alignment of 348 amino acid sequences from Firmicutes, which corresponds to the DEAD and Helicase_C domains without Gblocks filtering used in the Bayesian inference (see Fig. 1 and “Materials and methods”). DEAD-box proteins from B. subtilis are localized as reference sequences across the phylogram. The presence of the DbpA domain is shown with a red box on the perimeter. Notably it only occurs in a few genera, such as in Bacillus, Lysteria, and Clostridium. Proposed gene duplications are shown in the main clusters as duplication 1 and duplication 2. Interestingly, the Clostridium species appears to diverge earlier from other Firmicutes. The general clusters are shown in different colors and only values above 0.80 of posterior probability are shown. DbpA-RBD lacking branches next to DpbA-RBD containing branches are indicated
Clostridium sequences appeared to have separated earlier when compared to the other Firmicutes sequences analyzed. In this phylogeny, DbpA-RBD-containing sequences clustered into two branches. Interestingly, the DbpA-RBD domain was not represented in all genera of Firmicutes, as only the sequences of early branching genes belonging to the Clostridium, Bacillus, and Listeria genera possessed this domain. The basal sequence of those possessing a DbpA-RBD was that from Mycoplasma mobile 163K (GI: 47459115). The Clostridium genomes exhibited five clades, four of which dominated the earlier branching group. A DEAD-box protein sequence that lacked the DbpA-RBD and was basal to all sequences was present in all of the C. botulinum genomes. Although all the Clostridia had orthologs for the B. subtilis DeaD/YxiN protein, they also possessed an additional divergent DbpA-RBD-containing protein. Most Clostridium genomes also had CshA ortholog proteins; however, they all lacked orthologs for both the YfmL and CshB genes (see Fig. 4).

Relationship between phylogeny and the presence of putative ortholog sets of DEAD box proteins. The phylogenetic tree was based on the maximum likelihood method with the 16S rRNA gene (sequences from the RDP database) using the PhyML model (GTR + I) with 1,000 bootstrap replicates. The rows represent reference DEAD-box protein genes from E. coli (for γ-Proteobacteria) and from B. subtilis (for Firmicutes). The columns represent protein conservation using symbols to denote the presence of a putative ortholog. Red symbols are used for DEAD-box proteins containing the DbpA RNA-binding domain (RBD), while blue symbols are used for DEAD-box proteins lacking the DbpA domain. The conservation data were obtained from the phylogenetic reconstruction shown in Figs. 3 and 5, which is only based on the DEAD-Helicase_C N-terminal domain. D1 and D2 refer to the duplications 1 and 2 observed for the DEAD-box proteins from the Firmicutes. For each putative ortholog set, the length size difference is generally less than 20%, with a few exceptions (see Supplemental Tables S10 and S11 for the identification number of each protein as well as a comparison of the length for each of the sequences shown in this figure)
Several genera from Firmicutes, such as Staphylococcus, Lactococcus, Streptococcus, and Lactobacillus, only possessed DEAD-box proteins that lacked the DbpA-RBD. Within B. subtilis, we found that the earliest branching DEAD-box protein corresponded to the DeaD/YxiN protein, followed by the CshA, YfmL, and CshB. The YfmL and CshB proteins were each located in one of two large branches that we termed duplication 1 and duplication 2. These branches seemed to have resulted from an early duplication event, since this duplication was observed in almost all the members of the clade, including the Listeria, Streptococcus, and Lactobacillus genera. In addition, some genera lacked members from duplication 1, such as the possible orthologs of YfmL from B. subtilis. This was also true for Lactococcus and Staphylococcus, which only contained DEAD-box proteins that branched from duplication 2. All Streptococcus genomes examined (8 species, 28 genomes) had members within duplication 2 (possible orthologs of CshB from B. subtilis), while some genomes, including Streptococcus pyogenes, S. agalactiae NEM316, and S. agalactiae A909 had no members from duplication 1. Other Streptococcus genomes, including those from Streptococcus agalactiae (2603 V/R), also had a member within this duplicated branch. Although the Lactobacillus genus had more members within duplication 2, it still possessed members from duplication 1, which suggested that the presence of members from the two duplications may have occurred before speciation. In addition, these observations suggested that members from duplication 1 were later lost and that only members from duplication 2 (GI: 116514512, 104774414, and 2518569 from L. delbrueckii subsp. bulgaricus ATCC BAA 365, and ATCC 11842 from L. johnsonii NCC 533, respectively) remained.
Of the 22 Bacillus genomes analyzed (including the close relatives Oceanobacillus, Geobacillus, and Lysinbacillus), only the species that were phylogenetically closest to B. subtilis (B. pumilus and B. licheniformis) shared orthologs of the four DEAD-box proteins. In addition, some Bacillus species lacked one or two of the reference B. subtilis genes, with YfmL being the least conserved. For instance, the B. amyloliquefaciens FZB42 genome lacked both CshA and CshB protein coding genes. In contrast, the B. coahuilensis and O. iheyensis HTE831 genomes did not possess any DEAD-Box protein with a DbpA-RBD. Since there is no common element of DEAD-box protein coding genes shared by all Bacillus, we suggest that the different DEAD-box proteins are adaptable proteins that can participate in the same or similar pathways. Figure 4 summarizes the occurrence of possible orthologs for the B. subtilis DEAD-box proteins in other genera based on a Bayesian reconstruction and displays the losses and expansions of particular genes from different genera. In B. subtilis, only one DEAD-box protein, DeaD/YxiN, contains a DbpA-RBD. DeaD/YxiN appears in a different branch from CshA that lacks this C-terminal domain. Since CshA orthologs already appeared in several genera basal to the Bacillus, it is possible that CshA arose from an ancestral duplication of a DbpA-RBD-containing member and lost the DbpA-RBD. Alternatively, DeaD/YxiN and CshA may have been the result of ancestral duplications followed by the acquisition of new functions. In fact, the DbpA-RBD may have already evolved within DEAD-box proteins. B. subtilis has another pair of DEAD-box proteins, CshB and YfmL, which appear in sister branches and seem to have originated from ancestral duplication (see Figs. 3, 4).
Evolutionary History of DEAD-Box Proteins in γ-Proteobacteria
The DEAD-box protein family in the γ-Proteobacteria was also analyzed at the nucleotide/codon and amino acid level using a Bayesian analysis to obtain phylogenetic reconstructions (see “Materials and methods”). The tree reconstruction based on amino acids resulted in the same clades and general topology as that obtained with nucleotides. Figure 5 shows the phylogeny obtained at the amino acid level and Supplemental Figure 3S shows the nucleotide-based phylogeny. A DEAD-box protein sequence from the β-Proteobacteria Myxococcus xanthus DK 1622 (GI: 108757460) was used as an outgroup. One feature that was immediately observed in this tree was the separation of a branch of DEAD-box proteins containing the C-terminal DbpA-RBD (DbpA-RBD-containing branch) and another branch that lacked this domain (DbpA-RBD-lacking branch). Interestingly, each of these branches was further divided into two branches. The DbpA-RBD was present in most of the genera analyzed, with the exception of Acinetobacter, Psycrobacter, Coxiella, and some genes from obligate symbionts, including Wolbachia. From the topology of the tree, we deduced that the occurrence of certain DEAD-box protein coding genes in γ-Proteobacteria was a result of a recent expansion. Based on their location within the tree topology, many of these expansions appear to have arisen from independent events. This could be better explained if the γ-Proteobacteria originated from an ancestor that possessed two main classes of DEAD-box proteins, with one class containing the DbpA-RBD and one class lacking the domain. The tree topology shows sequences grouped according to their genus affiliation, at each of the main branches, which suggests that these genes were present in the common ancestor and that their diversification occurred after speciation events. Moreover, each of the clades closely follows a γ-Proteobacteria species phylogeny.

Bayesian inference of γ-Proteobacteria DEAD-box proteins. Complete alignment of 773 amino acid sequences from γ-Proteobacteria, which correspond to the DEAD and Helicase_C domains without Gblocks filtering used in the Bayesian inference (see Fig. 1 and “Materials and methods”). DEAD-box proteins from E. coli are localized as reference sequences across the phylogram. Note the defined group containing a DbpA-RBD that is shown with a red box around the perimeter. Noticeable gene expansion occurred in the Shewanella and Vibrio genera. From the topology, we inferred that the γ-Proteobacteria ancestor possessed both DbpA-RBD-containing and DbpA-RBD-lacking DEAD-box proteins. Genera are depicted with different colors and only posterior probability with values of 0.60 and above are shown. Three DbpA-RBD-lacking branches and two DpbA-RBD-containing branches are indicated
A striking expansion of this gene family occurred in the Vibrio and Shewanella genera. Based on this observation, we conducted a further analysis of the Shewanella DEAD-box protein coding genes. A synteny analysis was done to relate the evolutionary history of DEAD-box protein coding genes to their location in the genomes (data not shown). Surprisingly, within the large number of Shewanella sequences analyzed, only a pair of genes in 4 of the 17 analyzed species seemed to have resulted from a recent duplication event.
It is interesting to note that basal to the DEAD-box proteins that clustered in the DbpA-RBD-containing branch (Fig. 5), a sequence was found that belonged to Francisella philomiragia subsp. philomiragia ATCC 25017 (GI: 167627672), which lacked the DbpA-RBD. There are a few genes within the DbpA-RBD-containing group that lacked this domain (GI: 77166069, 120555341, and 33519584 from Nitrosococcus oceani ATCC 19707, Marinobacter aquaeolei VT8, and Candidatus Blochmannia floridanus, respectively) and we propose that this domain was lost in these members.
The DEAD-box proteins RhlB, SrmB, and RhlE from E. coli str. K12 substr. DH10B were found in the DbpA-RBD-lacking branches 1, 2, and 3, respectively. The E. coli proteins DbpA and DeaD/CsdA, which contained the DbpA-RBD, were apparently the result of an ancient duplication, since these appeared in separate branches and were conserved in all γ-Proteobacteria. The exception to this was the genus Salmonella, which lacked the E. coli DeaD/CsdA ortholog, and Coxiella, which lacked the E. coli DbpA ortholog. Figure 4 summarizes the occurrence of possible orthologs for the E. coli DEAD-box protein coding genes based on the Bayesian reconstruction and displays the losses and expansions of particular genes from different genera. DEAD-box protein SrmB seemed to lack an ortholog in Xanthomonas, which might have been compensated by the presence of a possible paralog of RhlE. The RhlE and RhlB were found to be present in all the γ-Proteobacteria analyzed, with the exception of species within the Buchnera and Haemophilus genera. As noted above, the RhlE protein from E. coli defined a particular class of DEAD-box proteins that was exclusive to the monophyletic group present in the Proteobacteria, and which had expanded in Shewanella, Vibrio, and Photobacterium. Similar to the Firmicutes sequences, none of the five DEAD-box protein ortholog families of γ-Proteobacteria was absolutely conserved, and the presence of each DEAD-box protein coding gene varied among the different genera. These findings suggested that DEAD-box proteins are flexible and can adapt to participate in the necessary cellular functions.
We also examined in detail the E. coli DEAD-box proteins in the phylogenetic reconstructions. DbpA and DeaD/CsdA are the DbpA-RBD-containing DEAD-box proteins and appear in sister branches, suggesting that they originated from an ancestral duplication of a DbpA-RBD-containing member (see Figs. 4, 5). The gene pairs coding for two more DEAD-box proteins, SrmB and RhlB, seem to also be the result of a duplication of a gene that already encoded a DbpA-RBD-lacking DEAD-box protein (see Figs. 4, 5). Lastly, the E. coli gene encoding RhlE is part of the branch that seems to have expanded in the Proteobacteria. If the Proteobacteria arose later than Firmicutes, as some studies have suggested (Wu et al. 2009), then a duplication of a DEAD-box protein coding gene most likely occurred in the early lineages to give rise to the RhlE coding gene, since most Proteobacteria have corresponding orthologs (see Fig. 4).
Architecture, Abundance, and Phylogenetic Distribution of Bacterial DEAD-Box Protein Genes
Among the genomes analyzed, those from Firmicutes and γ-Proteobacteria had the largest representation, with 133 and 162 genomes, respectively. Table 2 shows a selected sample of genomes from different phyla and includes an abbreviated description of the number of DEAD-box proteins and their classification based on protein architecture. Upon broad analysis, DbpA-RBD-containing and DbpA-RBD-lacking members appear in every phylum, with the exception of Acidobacteria, which only appeared in the Proteobacteria-derived group defined by the RhlE-like members. In contrast, the RhlE-like group contained mostly DEAD-box proteins from genomes belonging to the Proteobacteria, with exceptions coming from Planctomycetes, Chlorobi, Bacteroidetes, Acidobacteria, and Verrucomicrobia. In addition, this class also contained some DEAD-box proteins from Cyanobacteria as well as two sequences from Firmicutes that belonged to Clostridium botulinum B str. Eklund 17B (GI: 187934290) and C. phytofermentans ISDg (GI: 160880140). These other bacteria, which branched before the Proteobacteria, may have been the source of this divergent lineage. Notably, phyla that were taxonomically distant from the Proteobacteria generally lacked DEAD-box proteins from this group (Deinococcus, Thermotogae, Fusobacteria, Firmicutes, and Chloroflexi). However, we noted that some members of the Actinobacteria had genes that belonged solely to the divergent group.
Variation in the Number of DEAD-Box Proteins Genes Per Genome
The abundance of this protein family and the distribution of their members led us to analyze the number of DEAD-box proteins per genome and assess their taxonomic affiliation (see Table 2 for data on a selected number of genomes and Supplemental Table S1 for the complete set of data). We found a few examples of recently sequenced genomes from obligate endo-mutualists that lack DEAD-box proteins. Examples of these are Candidatus Sulcia muelleri GWSS (NC_01018), Candidatus Sulcia muelleri SMDSEM (NC_013123), and Carsonella ruddii PV (NC_008512). In contrast, other endo-mutualists, such as Buchnera aphidicola and Wigglesworthia glossinidia, contained DEAD-box protein genes. Interestingly, we only found one plasmid-encoded DEAD-box protein, which was present in Sinorhizobium medicae WSM 419 (NC_009620, YP_001313217). The genomes with the largest number of DEAD-box proteins (10–12) were all within the γ-Proteobacteria phylum and belonged to the Shewanella, Vibrio, and Photobacterium genera. In contrast, the genomes with the smallest number of DEAD-box protein genes were distributed among different phyla. A single DEAD-box protein gene was found in each of 116 genomes, and interestingly, approximately 33% of these contained the DbpA-RBD (see Supplemental Table S6). Therefore, the genomes that carry a single DEAD-box protein lacking DbpA-RBD may bind the 23S rRNA through a different mechanism. In some bacterial genomes that contained several DEAD-box proteins, such as Streptomyces coelicolor A3(2), Sulfurimonas denitrificans DSM 1251, and Sulfurovum sp. NBC37-1, none of the five different DEAD-box proteins had a DbpA-RBD. Interestingly, all five S. coelicolor A3(2) DEAD-box protein sequences clustered in the same group (depicted in brown in Fig. 2), while those from Sulfurimonas and Sulfurovum appeared in different branches.
We evaluated a possible correlation between the number of DEAD-box protein genes and the number of coding genes or GC content (see Supplemental Figure 4S) and found no significant correlation between genomes with more DEAD-box proteins and those with the maximum number of coding genes (e.g., Sorangium cellulosum “So ce 56”). Similarly, the GC content did not correlate with the number of DEAD-box proteins, since the genomes with the largest number of DEAD-box genes had a GC contents ranging from 38 to 45%. However, we observed great variability in the content of DEAD-box proteins (1–9) in genomes that had a GC content in the range of 50 to 74%.
We also explored a possible correlation between the gene copy number of rRNA genes and the number of DEAD-box proteins. A correlation was observed (Supplemental Figure 2S) with genomes harboring eight to twelve DEAD-box proteins, which showed the largest copy numbers of 23S rRNA genes. The maximum number of rRNA operon copies is 15 for Photobacterium profundum SS9 (Alm et al. 2006), which contains 10 genes coding for DEAD-box proteins. This correlation makes sense given the involvement of some DEAD-box proteins in the assembly of rRNA.
Variability of the C-Terminal Region of Bacterial DEAD-Box Proteins
The architecture of DEAD-box proteins is distinguished by an N-terminal region of approximately 350 amino acids in length that comprises the DEAD-Helicase_C domains. We found that in most bacterial DEAD-box proteins, this region is preceded by approximately 50 residues (mean length) (Fig. 6). The C-terminal is the most variable region. A large number of bacterial DEAD-box proteins possess approximately 30 amino acids at the ends in addition to the DEAD-Helicase_C domains, while a few possess upwards of 766 amino acids, mainly as C-terminal extensions. The smallest sequence included in this analysis was that from Onion yellows phytoplasma OY-M, which had 357 amino acids (NP_950705.1) and the largest sequence corresponded to Nocardia farcinica IFM-1152, which had 1148 amino acids (YP_120788).

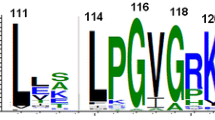
Conservation of motifs across DEAD and Helicase_C domains of 1208 bacterial DEAD-box proteins. Revised motifs obtained from the alignment of 1,208 sequences in comparison with the consensus motifs from DEAD-box proteins originally proposed by Rocak and Linder (2004) and Jankowsky and Putnam (2010). A traditional consensus residue format and a HMM-logo description are shown. The N- and C-terminal amino acid length distribution is also shown. Included for comparison are the following selected eukaryotic DEAD-box protein sequences: Dm: Drosophila melanogaster (NP_723899.1); Mm: eIF4AII Mus musculus (NP_001116510.1); Sc: Ded1 Saccharomyces cerevisiae (NP_014847.1); and Mj: Methanocaldococcus jannaschii DSM 2661(NP_247653.1)
However, we found that most DEAD-box protein sequences with short C-terminal sequences contained K- and R-rich segments within these regions. Accordingly, the isoelectric point of the C-terminal end is generally basic (see Supplemental Tables S8 and S9), which is a feature shared among proteins that interact with nucleic acids (Adhikari et al. 2010). In general, the DbpA-RBD-containing group contained members with the longest C-termini. To determine whether the presence of particular domains/motifs were the source of variability in protein size, we looked for possible domains at the C-termini using Pfam (see Supplemental Table S7). The only domain that appeared with a significant score (E value of 10−5 to 10−46) was DbpA-RBD. A few sequences were found to have a duplicated DbpA-RBD and belonged to β-Proteobacteria (Janthinobacterium sp. Marseille: YP_001355159.1; Herminiimonas arsenicoxydans; YP_001101466.1) and δ-Proteobacteria (Myxococcus xanthus DK 1622: YP_634008.1). This observation suggested that these domain duplications occurred independently in these genomes.
Revised Motifs Within the DeaD and Helicase_C Domains
Given the large number of compared DEAD-box protein sequence we revisited the motifs that had been previously described for the DEAD and Helicase_C domains (Jankowsky and Putnam 2010; Rocak and Linder 2004). We aligned 1,208 sequences and obtained HMM models to determine the amino acid frequency at each position within these domains. This approach allowed us to evaluate the conservation profile of the residues in the 10 known motifs (Rocak and Linder 2004) (Fig. 6). Based on these findings, we propose a revised consensus motif that incorporates extensions to the already defined motifs. The advantage of the HMM-logo description of conserved residues is that it allows for the identification of particular amino acid residues that may be less conserved for some sequences, and yet important in others.
The F residue of the motif F (Fig. 6) exhibited high conservation, although it is absent in some DEAD-box sequences. The significance of this apparent absence is difficult to interpret, since in these cases, variation exists in the location of the protein start site and therefore it is difficult to assess whether another F residue in the vicinity is substituted in place of it. In other DEAD-box sequences, a stretch of amino acids at the N-terminus end were absent. However, in some cases this could be an annotation problem. In a few cases, the F was substituted by a different residue, such as in Colwellia psychrerythraea 34H (YP_268332) and Psychromonas ingrahamii 37 (YP_943183), where there was a V substitution. As expected, the Q residue was the most conserved within motif Q, followed by the I residue that preceded it. At the beginning of motif Q, we observed a GY pair that had not been previously described in DEAD-box protein sequences. For motif I, we found that the consensus sequence AxTGSGKTxxFxxP was invariable in the examined DEAD-box proteins. However, the first T residue was often substituted for an S in eukaryotic and archaeal DEAD-box proteins (Fig. 6). For motif Ia, the PTRELA stretch of amino acid residues is highly conserved in the bacterial DEAD-box proteins; however, we also included a remarkably conserved Q that was found two residues downstream of this sequence at a very high frequency. In fact, there were no exceptions to this observation, and only a single exception for the presence of the P within the examined DEAD-box proteins. Motif Ib was the next most highly conserved motif in bacterial DEAD-box proteins. The motif was identified by a GG doublet, and although it had been previously described (Linder et al. 1989), it was ignored since it was not conserved in all DEAD-box proteins. However, the importance of this residue has been demonstrated in the eukaryotic DEAD-box protein eIF4AII, where mutations in these residues abolish the function of the protein (Zakowicz et al. 2005). Within this motif, we included a Q residue that occurred downstream with high frequency among bacterial DEAD-box proteins. This residue had not been previously described, and was conserved in sequences from archaea and eukaryotes (Fig. 6). In motif Ic, the reported sequence remained the same as that previously described. Motif II included the DEAD sequence, which is the hallmark of this family, and it is known that the A residue can vary. Interestingly, a number of different amino acids were able to replace it, including V, M, S, G, T, F, and C. We extended this motif by six residues to include R/E, M, L, D, M, G, and F. The M residue located at the middle of the proposed motif was extremely well conserved (92% of 1,208 sequences examined), followed by the GF pair (95.7%). Motif III was defined by the sequence FSAT. In our alignment, we found that the F that preceded the motif was as well conserved as the SAT, which is not the case in previously described motifs. Motif IV was the least conserved and contained the sequence xIIFxx, where F was the most conserved residue and each residue varied. The next motif was named IVa. It has previously been identified by some authors as QxxR (Banroques et al. 2008) and we have extended this motif to include residues HG, which are as conserved as the Q residue. Motif V has T and D residues that are highly conserved at the end of the motif (Jankowsky and Putnam 2010). However, we also identified two A residues, one positioned before the T and the other positioned after the D residue. Motif Va (ARGID) had the same amino acid frequency and has already been reported. In the motif Vb (VxxVINxx) in bacterial DEAD-box proteins, we observed a slight modification, where the first V residue seemed to be absent, while a P residue at the end of the motif had a higher frequency than VIN residues, which had not been previously described. Finally, residues HRxGRTGR in the motif VI were highly conserved in the bacterial DEAD-box proteins. In fact, the first residue in this motif has been shown to be an identifier of DEAD-box proteins, since it is replaced with Q in DEAH proteins (Rocak et al. 2005; Yao et al. 1997). We have included other residues as part of this motif that occurred at high frequency: a Y and G at the beginning and end of the motif, respectively. A few bacterial species, such as Mycoplasma and Ureaplasma, possess DEAD-box protein sequences that deviated from several of these motifs. This observation suggests that rapid divergence occurred.
To assess if the newly described and edited motifs existed beyond bacteria, we looked for these new amino acid signatures in some well-studied eukaryotic DEAD-box proteins, such as Vasa (NP_723899; Drosophila melanogaster), eIF4AII (NP_001116510; Mus musculus), and Ded1 (NP_014847; Saccharomyces cerevisiae). We found that most of the new signatures proposed in this study are not limited to bacterial DEAD-box proteins. The specific role of the uncovered residues from these new signatures remains to be tested. They may contribute to the binding of RNA or may help establish interactions between these proteins and their substrates.
Discussion
In this study, we have described the diversity of bacterial DEAD-box proteins from 563 genomes to gain insight into the evolutionary history of this protein family in the Firmicutes and γ-Proteobacteria phyla, which include the best studied bacterial models, E. coli and B. subtilis. Gene duplication is the primary source of new genes. Duplicate genes that are stably preserved in genomes usually have divergent functions. The general rules governing the functional divergence, however, are controversial and not well understood. The neofunctionalization hypothesis asserts that after duplication, one daughter gene retains the ancestral function, while the other acquires new functions. The DEAD-box protein gene family is probably the result of more than one duplication and neofunctionalization event. For instance, among the five encoded DEAD-box proteins that were identified in E. coli, four most likely play a role in ribosome assembly (DeaD/CsdA, DbpA, SrmB, and RhlE) (Charollais et al. 2003b; Iost and Dreyfus 2006; Jain 2008; Moll et al. 2002), while one (RhlB) has been shown to be involved in RNA degradation (Py et al. 1996). DeaD/CsdA and DbpA, which both contain a DbpA-RBD, originated from what seemed to be duplicated branches. SrmB and RhlB seemed to have a common origin, but most likely diverged before the speciation of γ-Proteobacteria. Since these two proteins have different functions, it is likely that these two DEAD-box protein genes are an example of neofunctionalization after duplication. The phylogenetic relationship between these two DEAD-box protein genes had been previously suggested by Iost and Dreyfus on the basis of Blast scores (Iost and Dreyfus 2006). One or both genes that code for SrmB and RhlB are absent in some genera from the γ-Proteobacteria, and only the gene coding for SrmB has been observed as a duplication. However, RhlE seems to have originated in a Proteobacterial ancestor, since it has evolved and expanded in sequences that form a Proteobacterial-specific branch and is, therefore, almost exclusively present in this phylum. The observed expansions in Shewanella, Photobacterium, Vibrio, and Cowellia emerge specifically from the RhlE protein branch. Silander and Ackermann (2009) analyzed the level of gene conservation for different orthologous genes. In their study, the E. coli DEAD-box proteins showed high levels of ortholog loss (ROL). They also noticed that many of the genes that exhibit a loss may be recent innovations and have a distribution that is restricted to the Proteobacteria. This is exactly the case for the rhlE gene. The ROL is also thought to be inversely correlated with gene essentiality, which is in agreement with the fact that none of the E. coli DEAD-box proteins are essential in E. coli.
Regarding B. subtilis, the DeaD/YxiN protein bears a DbpA-RBD at the C-terminal end and has been suggested to be a member of a subfamily that is involved in ribosome assembly. Indeed, it has been shown that the 23S rRNA stimulates its ATPase and unwinding activities (Kossen and Uhlenbeck 1999). CshA, which lacks DbpA-RBD, has also been shown to exhibit ATPase activity and associate with the ribosomal fraction (Ando and Nakamura 2006). Our evolutionary study suggests that both DeaD/YxiN and CshA genes could have originated from DbpA-RBD-containing helicases, and CshA could have lost this domain. YfmL and CshB appear to have arisen from another duplication. In addition, YfmL from Bacillus seems to have orthologs in other Firmicutes genera, such as Staphylococcus, Lactobacillus, and Streptococcus, where there is at least one species that has lost this particular DEAD-box ortholog.
While some sequenced genomes from obligate endo-symbionts lack DEAD-box protein, all the analyzed genomes of free-living bacteria assessed in this study possessed at least one DEAD-box coding gene, which is what would be expected for a protein that is involved in multiple aspects of the metabolism of RNA. Some genomes can have up to 12 copies of the genes. The expansion of DEAD-box proteins in some genera may be similar to what has been observed for histidine kinase genes that have been found in the genomes that have the highest enrichment of signaling proteins, such as early branching Proteobacteria, suggesting that divergence in domain structure and changes in expression patterns are hallmarks of recent expansions (Alm et al. 2006). Interestingly, the large number of DEAD-box proteins in the Shewanella genus extends to other protein families, such as diguanylate cyclases (51 proteins) and phosphodiesterases (27 proteins). For this reason, it has been suggested that these particular families may be participating in the post-transcriptional control of different cellular processes (Fredrickson et al. 2008) where DEAD-box proteins may also be involved.
It is not clear why some DEAD-box proteins in E. coli and B. subtilis are dispensable for growth under laboratory conditions, whereas in yeast, most DEAD-box proteins are essential (Bernstein et al. 2006; Granneman et al. 2006). For many genes, duplication is accompanied by changes in regulation. It is possible that bacterial DEAD-box proteins are only necessary under certain growth conditions but also that there is some redundancy. In support of this hypothesis, it has been shown that low temperature induces the expression of DEAD-box protein genes in several bacteria and archaea (Chamot and Owttrim 2000; Lim et al. 2000; Söderberg and Cianciotto 2010). A recent study showed that DEAD-box protein CrhR regulated the expression of groEL1 and groEL2 during acclimatization to low temperature in Synechocystis sp. PCC6803 (Prakash et al. 2010).
DEAD-box proteins may have RBDs that provide high specificity to dedicated helicase functions (Kossen et al. 2002; Linden et al. 2008), or low specificity to general RNA chaperone functions (Mohr et al. 2008; Russell 2008; Del Campo et al. 2009). The function of RBDs is to position neighboring RNA regions for unwinding by the helicase core (Diges and Uhlenbeck 2005; Tijerina et al. 2006). In bacterial DEAD-box proteins, the differences in length of the amino acid sequences found between the different classes are explained predominantly by differences in their C-terminal region. The absence of conserved motifs at the C-terminal region and the prevalence of high isoelectric points and low hydrophobicity suggest that this region may be involved in RNA binding and complex formation in bacterial DEAD-box proteins, which has been reported for some eukaryotic DEAD-box proteins (Mss116p, Cyt-19, and eIF4A) (Mohr et al. 2008; Schutz et al. 2008). The only recognizable, conserved domain to date in the C-terminal region of some members is DbpA-RBD, which is the best example of target specificity for DEAD-box proteins. The presence of this domain is thought to limit these proteins to a particular function, which is most likely that of ribosomal RNA assembly. The crystallographic structure of the DeaD/YxiN RBD showed that, despite the lack of apparent sequence similarity, it has the same tertiary fold as the RNA recognition motifs (RRM) that are prevalent in eukaryotes. However, RNA binding assays of mutant DeaD/YxiN RBD suggest that the mode in which this domain binds RNA differs substantially from that of the eukaryotic RRMs (Wang et al. 2006). It is possible that the DbpA-RBD has had an evolutionary constrain to maintain its role in rRNA assembly, since the RRM in eukaryotic proteins is one of the most abundant motifs and exhibits great variation and flexibility in function (Maris et al. 2005).
Most bacterial genomes have at least one gene coding for a DbpA-RBD-containing DEAD-box protein. However, there are numerous bacterial genomes that completely lack the DbpA-RBD, and therefore the mechanism by which 23S rRNA assembly takes place remains elusive. Hairpin 92 in the 23S rRNA is highly conserved (Pei et al. 2009), and therefore a change in the DbpA-RBD cannot be explained as an adjustment to accommodate variant 23S structures. However, the presence of a DbpA does not dictate that a protein will be targeted to the hairpin 92 of the 23S rRNA. For instance, the E. coli DeaD/CsdA protein has a DbpA-RBD, but it does not exhibit binding specificity for this hairpin (Jones et al. 1996). It is possible that its location within a larger C-terminal region, flanked by an R-rich region upstream and five conserved R-, E-, G-rich nonamers downstream (GGERRGGGR; data not shown) influence its RNA binding features. Interestingly, the Thermus thermophilus DEAD-box protein Hera has a C-terminal RBD that also binds hairpin 92 in the 23S rRNA, although it does not have significant sequence homology to the DbpA-RBD. In contrast to B. subtilis DeaD/YxiN RBD, which displays a high specificity for hairpin 92, Hera is promiscuous and also binds to RNase P RNA (Linden et al. 2008). The Hera-RBD and DeaD/YxiN RBD have no significant sequence homology, but it has been suggested that a functional homology in 23S rRNA binding is likely to be significant (Rudolph and Klostermeier 2009).
Another DEAD-box protein involved in ribosome assembly is the E. coli SrmB (Nishi et al. 1988). A recent report from Trubetskoy et al. (2009) showed that SrmB was able to interact with L24 and L4 and suggested that these proteins serve as a bridge for the interaction with its target RNA. However, the C-terminal end did not mediate this interaction and was found to be dispensable, suggesting that it provides stability to the complex. Interestingly, the genome of Synechocystis sp. PCC6803 has a single gene coding for a DEAD-box protein and it lacks a DbpA-RBD. We believe that these data provide an example of a bacterium with a single DEAD-box protein that lacks a DbpA-RBD, but which can cover multiple functions.
The origin of the DbpA-RBD is intriguing. It does not occur as a separate module in other proteins, as it is only found associated to DEAD-Helicase_C domains, and the only exceptions occur in three Treponema species and one Gemmatimonas aurantiaca species (data not shown) in small genes that predominantly consist of this domain. Therefore, we suggest that in mesophilic bacteria the DbpA-RBD originated as part of the DEAD-Helicase_C as a specialization. This specialization occurred after a duplication of these proteins (neofunctionalization), which would explain the occurrence of both DbpA-RBD-containing and DbpA-RBD-lacking proteins. However, we cannot exclude the possibility that this domain was gained (i.e., by fusion) from another protein that does not exist in current genomes.
Finally, alignment of 1,208 non-redundant DEAD-box proteins allowed us to revise the consensus sequence of the motifs that define this protein family in bacteria. We delineated a revised consensus for the bacterial DEAD-box protein family using the largest number of members ever analyzed. We obtained an HMM-logo that allowed us to determine the conservation frequency of different residues and establish particular trademarks in bacteria for this protein family. The HMM-logo includes previously unrecognized amino acids that were within or extended the known motifs. As expected for a highly conserved gene family, the bacterial motifs have several similarities with a eukaryotic and archaeal model DEAD-box protein (Fig. 6). Numerous biochemical studies have been carried out to elucidate the functions of these conserved motifs (Caruthers and McKay 2002; Cordin et al. 2004; Rogers et al. 2002). For this reason, residues that occur at a high frequency in or proximal to bacterial DEAD-box protein sequences at these motifs seem particularly interesting for future studies, since they may influence the biochemical functions of the protein.
Conclusions
In this study, we observed numerous examples of the gain and loss of DEAD-box protein genes following speciation in bacteria. Few taxonomic incongruencies were detected, suggesting that these genes are not frequently transferred. Moreover, several gene members had good phylogeny information content, since they were congruent with species phylogenies. In addition, a few examples of the loss of the DbpA-RBD were also observed. We propose that DEAD-box proteins in extant bacteria evolved from an ancient progenitor that already possessed a duplicated gene, of which one copy possessed a DbpA-RBD. The flexible nature of the DEAD-Helicase_C domains facilitated its expansion and specialization by multiple evolutionary events that included the gain and loss of genes. Finally, given that a single DEAD-box protein seems to be sufficient for multiple tasks in some bacteria, further research should focus on analyzing the contribution of redundancy to the fitness of bacteria that possess multiple DEAD-box proteins.
References
Adhikari S, Manthena PV, Sajwan K, Kota KK, Roy R (2010) A unified method for purification of basic proteins. Anal Biochem 400:203–206
Alm E, Huang K, Arkin A (2006) The evolution of two-component systems in bacteria reveals different strategies for niche adaptation. PLoS Comput Biol 2:1329–1342
Altschul SF, Koonin EV (1998) Iterated profile searches with PSI-BLAST—a tool for discovery in protein databases. Trends Biochem Sci 23:444–447
Ando Y, Nakamura K (2006) Bacillus subtilis DEAD protein YdbR possesses ATPase, RNA binding, and RNA unwinding activities. Biosci Biotechnol Biochem 70:1606–1615
Banroques J, Cordin O, Doere M, Linder P, Tanner NK (2008) A conserved phenylalanine of motif IV in superfamily 2 helicases is required for cooperative, ATP-dependent binding of RNA substrates in DEAD-box proteins. Mol Cell Biol 28:3359–3371
Bernstein KA, Granneman S, Lee AV, Manickam S, Baserga SJ (2006) Comprehensive mutational analysis of yeast DEXD/H box RNA helicases involved in large ribosomal subunit biogenesis. Mol Cell Biol 26:1195–1208
Bleichert F, Baserga SJ (2007) The long unwinding road of RNA helicases. Mol Cell 27:339–352
Brochier C, Philippe H (2002) Phylogeny: a non-hyperthermophilic ancestor for bacteria. Nature 417:244
Cameron M, Williams HE, Cannane A (2004) Improved gapped alignment in BLAST. IEEE/ACM Trans Comput Biol Bioinform 1:116–129
Caruthers JM, McKay DB (2002) Helicase structure and mechanism. Curr Opin Struct Biol 12:123–133
Chamot D, Owttrim GW (2000) Regulation of cold shock-induced RNA helicase gene expression in the Cyanobacterium anabaena sp. strain PCC 7120. J Bacteriol 182:1251–1256
Charollais J, Pflieger D, Vinh J, Dreyfus M, Iost I (2003a) The DEAD-box RNA helicase SrmB is involved in the assembly of 50S ribosomal subunits in Escherichia coli. Mol Microbiol 48:1253–1265
Charollais J, Pflieger D, Vinh J, Dreyfus M, Iost I (2003b) The DEAD-box RNA helicase SrmB is involved in the assembly of 50S ribosomal subunits in Escherichia coli. Mol Microbiol 48:1253–1265
Cordin O, Tanner NK, Doere M, Linder P, Banroques J (2004) The newly discovered Q motif of DEAD-box RNA helicases regulates RNA-binding and helicase activity. EMBO J 23:2478–2487
Cordin O, Banroques J, Tanner NK, Linder P (2006) The DEAD-box protein family of RNA helicases. Gene 367:17–37
Del Campo M, Mohr S, Jiang Y, Jia H, Jankowsky E, Lambowitz AM (2009) Unwinding by local strand separation is critical for the function of DEAD-box proteins as RNA chaperones. J Mol Biol 389:674–693
Diges CM, Uhlenbeck OC (2005) Escherichia coli DbpA is a 3′ → 5′ RNA helicase. Biochemistry 44:7903–7911
Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214
Eddy SR (1998) Profile hidden Markov models. Bioinformatics 14:755–763
Fairman-Williams ME, Guenther UP, Jankowsky E (2010) SF1 and SF2 helicases: family matters. Curr Opin Struct Biol 20:313–324
Felsenstein J (2005) PHYLIP (Phylogeny Inference Package) version 3.67-1. Department of Genome Sciences. University of Washington, Seattle. Distributed by the author
Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, Holm L, Sonnhammer EL, Eddy SR, Bateman A (2010) The Pfam protein families database. Nucleic Acids Res 38:D211–D222
Fredrickson JK, Romine MF, Beliaev AS, Auchtung JM, Driscoll ME, Gardner TS, Nealson KH, Osterman AL, Pinchuk G, Reed JL, Rodionov DA, Rodrigues JL, Saffarini DA, Serres MH, Spormann AM, Zhulin IB, Tiedje JM (2008) Towards environmental systems biology of Shewanella. Nat Rev Microbiol 6:592–603
Fuller-Pace FV, Nicol SM, Reid AD, Lane DP (1993) DbpA: a DEAD box protein specifically activated by 23 s rRNA. EMBO J 12:3619–3626
Gibson TJ, Thompson JD (1994) Detection of dsRNA-binding domains in RNA helicase A and Drosophila maleless: implications for monomeric RNA helicases. Nucleic Acids Res 22:2552–2556
Granneman S, Bernstein KA, Bleichert F, Baserga SJ (2006) Comprehensive mutational analysis of yeast DEXD/H box RNA helicases required for small ribosomal subunit synthesis. Mol Cell Biol 26:1183–1194
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Huelsenbeck JP, Ronquist F (2001) MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755
Hunger K, Beckering CL, Wiegeshoff F, Graumann PL, Marahiel MA (2006) Cold-induced putative DEAD box RNA helicases CshA and CshB are essential for cold adaptation and interact with cold shock protein B in Bacillus subtilis. J Bacteriol 188:240–248
Iost I, Dreyfus M (2006) DEAD-box RNA helicases in Escherichia coli. Nucleic Acids Res 34:4189–4197
Jain C (2008) The E. coli RhlE RNA helicase regulates the function of related RNA helicases during ribosome assembly. RNA 14:381–389
Jankowsky E, Fairman ME (2007) RNA helicases—one fold for many functions. Curr Opin Struct Biol 17:316–324
Jankowsky E, Putnam A (2010) Duplex unwinding with DEAD-box proteins. Methods Mol Biol 587:245–264
Jones PG, Mitta M, Kim Y, Jiang W, Inouye M (1996) Cold shock induces a major ribosomal-associated protein that unwinds double-stranded RNA in Escherichia coli. Proc Natl Acad Sci USA 93:76–80
Karginov FV, Uhlenbeck OC (2004) Interaction of Escherichia coli DbpA with 23S rRNA in different functional states of the enzyme. Nucleic Acids Res 32:3028–3032
Karginov FV, Caruthers JM, Hu Y, McKay DB, Uhlenbeck OC (2005) YxiN is a modular protein combining a DEx(D/H) core and a specific RNA-binding domain. J Biol Chem 280:35499–35505
Kossen K, Uhlenbeck OC (1999) Cloning and biochemical characterization of Bacillus subtilis YxiN, a DEAD protein specifically activated by 23S rRNA: delineation of a novel sub-family of bacterial DEAD proteins. Nucleic Acids Res 27:3811–3820
Kossen K, Karginov FV, Uhlenbeck OC (2002) The carboxy-terminal domain of the DExDH protein YxiN is sufficient to confer specificity for 23S rRNA. J Mol Biol 324:625–636
Letunic I, Bork P (2007) Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23:127–128
Lim J, Thomas T, Cavicchioli R (2000) Low temperature regulated DEAD-box RNA helicase from the Antarctic archaeon, Methanococcoides burtonii. J Mol Biol 297:553–567
Linden MH, Hartmann RK, Klostermeier D (2008) The putative RNase P motif in the DEAD box helicase Hera is dispensable for efficient interaction with RNA and helicase activity. Nucleic Acids Res 36:5800–5811
Linder P, Daugeron MC (2000) Are DEAD-box proteins becoming respectable helicases? Nat Struct Biol 7:97–99
Linder P, Lasko PF, Ashburner M, Leroy P, Nielsen PJ, Nishi K, Schnier J, Slonimski PP (1989) Birth of the D-E-A-D box. Nature 337:121–122
Maris C, Dominguez C, Allain FH (2005) The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. Febs J 272:2118–2131
Mohr G, Del Campo M, Mohr S, Yang Q, Jia H, Jankowsky E, Lambowitz AM (2008) Function of the C-terminal domain of the DEAD-box protein Mss116p analyzed in vivo and in vitro. J Mol Biol 375:1344–1364
Moll I, Grill S, Grundling A, Blasi U (2002) Effects of ribosomal proteins S1, S2 and the DeaD/CsdA DEAD-box helicase on translation of leaderless and canonical mRNAs in Escherichia coli. Mol Microbiol 44:1387–1396
Nishi K, Morel-Deville F, Hershey JW, Leighton T, Schnier J (1988) An eIF-4A-like protein is a suppressor of an Escherichia coli mutant defective in 50S ribosomal subunit assembly. Nature 336:496–498
Patel SS, Donmez I (2006) Mechanisms of helicases. J Biol Chem 281:18265–18268
Pei A, Nossa CW, Chokshi P, Blaser MJ, Yang L, Rosmarin DM, Pei Z (2009) Diversity of 23S rRNA genes within individual prokaryotic genomes. PLoS One 4:e5437
Prakash JS, Krishna PS, Sirisha K, Kanesaki Y, Suzuki I, Shivaji S, Murata N (2010) An RNA helicase, CrhR, regulates the low-temperature-inducible expression of heat-shock genes groES, groEL1 and groEL2 in Synechocystis sp. PCC 6803. Microbiology 156:442–451
Price MN, Dehal PS, Arkin AP (2010) FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490
Py B, Higgins CF, Krisch HM, Carpousis AJ (1996) A DEAD-box RNA helicase in the Escherichia coli RNA degradosome. Nature 381:169–172
Rocak S, Linder P (2004) DEAD-box proteins: the driving forces behind RNA metabolism. Nat Rev Mol Cell Biol 5:232–241
Rocak S, Emery B, Tanner NK, Linder P (2005) Characterization of the ATPase and unwinding activities of the yeast DEAD-box protein Has1p and the analysis of the roles of the conserved motifs. Nucleic Acids Res 33:999–1009
Rogers GW Jr, Komar AA, Merrick WC (2002) eIF4A: the godfather of the DEAD box helicases. Prog Nucleic Acid Res Mol Biol 72:307–331
Rudolph MG, Klostermeier D (2009) The Thermus thermophilus DEAD box helicase Hera contains a modified RNA recognition motif domain loosely connected to the helicase core. RNA 15:1993–2001
Russell R (2008) RNA misfolding and the action of chaperones. Front Biosci 13:1–20
Schuster-Bockler B, Bateman A (2005) Visualizing profile-profile alignment: pairwise HMM logos. Bioinformatics 21:2912–2913
Schutz P, Bumann M, Oberholzer AE, Bieniossek C, Trachsel H, Altmann M, Baumann U (2008) Crystal structure of the yeast eIF4A-eIF4G complex: an RNA-helicase controlled by protein-protein interactions. Proc Natl Acad Sci USA 105:9564–9569
Silander OK, Ackermann M (2009) The constancy of gene conservation across divergent bacterial orders. BMC Res Notes 2:2
Söderberg MA, Cianciotto NP (2010) Mediators of lipid a modification, RNA degradation, and central intermediary metabolism facilitate the growth of Legionella pneumophila at low temperatures. Curr Microbiol 60:59–65
Talavera G, Castresana J (2007) Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol 56:564–577
Tanner NK, Cordin O, Banroques J, Doere M, Linder P (2003) The Q motif: a newly identified motif in DEAD box helicases may regulate ATP binding and hydrolysis. Mol Cell 11:127–138
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Tijerina P, Bhaskaran H, Russell R (2006) Nonspecific binding to structured RNA and preferential unwinding of an exposed helix by the CYT-19 protein, a DEAD-box RNA chaperone. Proc Natl Acad Sci USA 103:16698–16703
Trubetskoy D, Proux F, Allemand F, Dreyfus M, Iost I (2009) SrmB, a DEAD-box helicase involved in Escherichia coli ribosome assembly, is specifically targeted to 23S rRNA in vivo. Nucleic Acids Res 37:6540–6549
Tsu CA, Kossen K, Uhlenbeck OC (2001) The Escherichia coli DEAD protein DbpA recognizes a small RNA hairpin in 23S rRNA. Rna 7:702–709
Tuteja N, Tuteja R (2004) Unraveling DNA helicases. Motif, structure, mechanism and function. Eur J Biochem 271:1849–1863
Wang S, Hu Y, Overgaard MT, Karginov FV, Uhlenbeck OC, McKay DB (2006) The domain of the Bacillus subtilis DEAD-box helicase YxiN that is responsible for specific binding of 23S rRNA has an RNA recognition motif fold. RNA 12:959–967
Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, Kunin V, Goodwin L, Wu M, Tindall BJ, Hooper SD, Pati A, Lykidis A, Spring S, Anderson IJ, D’Haeseleer P, Zemla A, Singer M, Lapidus A, Nolan M, Copeland A, Han C, Chen F, Cheng JF, Lucas S, Kerfeld C, Lang E, Gronow S, Chain P, Bruce D, Rubin EM, Kyrpides NC, Klenk HP, Eisen JA (2009) A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature 462:1056–1060
Yao N, Hesson T, Cable M, Hong Z, Kwong AD, Le HV, Weber PC (1997) Structure of the hepatitis C virus RNA helicase domain. Nat Struct Biol 4:463–467
Zakowicz H, Yang HS, Stark C, Wlodawer A, Laronde-Leblanc N, Colburn NH (2005) Mutational analysis of the DEAD-box RNA helicase eIF4AII characterizes its interaction with transformation suppressor Pdcd4 and eIF4GI. RNA 11:261–274
Acknowledgments
We acknowledge Amanda Castillo for her suggestions on the Bayesian phylogenies, Luis Delaye for the discussion and comments made to our manuscript, Ismael L. Hernández-González for the analysis of the Shewanella DEAD-box protein genes, and to Mauricio Carrillo Tripp for his help in running some analysis in the Cinvestav-Langebio cluster. Varinia López-Ramírez and Luis David Alcaraz were supported by a CONACYT fellowship. Gabriel Moreno received support from CONACYT for his academic visit to Cinvestav-Irapuato (MOD-ORD-3-09-PCI-009-03-09). This work was supported by CONACYT Grants 79927 (2007-2008) and 102712 (2008–2011) and a Multidisciplinary grant from Cinvestav to GO. We also acknowledge the anonymous reviewers for their contributions to the improvement of this work.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Figure 1S
Phylogenetic tree of the DEAD-Helicase_C domain in 1211 bacterial DEAD-box proteins. Sequences containing only DEAD and Helicase_C domains were first aligned and then manually curated. We used Gblocks to extract informative positions from the protein alignment and a total of 323 non-ambiguous positions were used to conduct the phylogenetic analysis by Maximum likelihood (see Materials and Methods). The grouping of classes was determined according to clades layout. The group of sequences containing DbpA-RBD was identified and colored green. Location of known RNA helicases from E. coli and B. subtilis are indicated in the phylogeny. Information about the genomes and the sequences IDs used to generate this phylogeny can be found in Supplemental Tables S4 and S5 (Talavera G. and Castresana J. 2007. Syst Biol 56:564-77). (PDF 2326 kb)
Figure 2S
Bayesian inference of DEAD-box protein from Firmicutes. Complete alignment of 345 nucleotide sequences from Firmicutes, which corresponds to the DEAD and Helicase_C domains (without Gblocks filtering) was used to obtain a Bayesian inference (see Figure 1 and Materials and Methods). DEAD-box proteins from B. subtilis are indicated as reference sequences across the phylogram. The presence of the DbpA-RBD is shown with a red box on the perimeter. Notably it only occurs in a few genera, such as in Bacillus, Lysteria, and Clostridium. Proposed gene duplications are shown in the main clusters as Duplication 1 and Duplication 2. Interestingly, the Clostridium species appear to have diverged earlier from other Firmicutes. The general clusters are shown in different colors and only values above 0.80 of posterior probability are shown. DbpA-RBD lacking branches next to DpbA-RBD containing branches are indicated (PDF 1236 kb)
Figure 3S
Bayesian inference of γ-Proteobacteria DEAD-box proteins. Complete alignment of 765 nucleotide sequences from γ-Proteobacteria, which correspond to the DEAD and Helicase_C domains (without Gblocks filtering) was used in producing the Bayesian inference (see Figure 1 and Materials and Methods). DEAD-box proteins from E. coli are indicated as reference sequences across the phylogram. Note the defined group containing a DbpA-RBD that is shown with a red box around the perimeter. Noticeable gene expansion occurred in the Shewanella and Vibrio genera. From the topology, we inferred that the γ- Proteobacteria ancestor possessed both DbpA-RBD containing and DbpA-RBD lacking DEAD-box proteins. Genera are depicted with different colors and only posterior probability with values of 0.60 and above are shown. Three DbpA-RBD lacking branches and two DpbA-RBD containing branches are indicated (PDF 2457 kb)
Figure 4S
Correlation analysis of the number of DEAD-box protein genes and selected genomic features. Correlation between DEAD-box protein number and (A) Total number of coding genes, including those genes present in plasmids; (B) 23S gene copy number; and (C) GC content for a particular bacterial genome. All statistical analyses were conducted using R (2.6.2) (Team RDC. 2009 R Foundation for Statistical Computing) (PDF 608 kb)
Figure 5S
Comparative analysis of DbpA-RNA binding domain sequences from diverse bacterial taxa. The C-terminal region of many bacterial DEAD-box proteins share a conserved domain, known as DbpA-RNA binding domain (RBD) identified as the Pfam (PF03880). A complete alignment can be obtained from the Pfam site. The Bacillus subtilis DeaD/YxiN possesses the conserved DbpA-RBD whose tertiary structure has been solved and it is an α-β sandwich with four antiparallel β-strands on one face and two helices on the other. The order of secondary structure elements in the polypeptide sequence is β-α-β-β-α-β (Wang S et al., 2006. RNA. 12:959-67). The YxiN sequence shown as reference starts at residue 404 (PDF 1427 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
López-Ramírez, V., Alcaraz, L.D., Moreno-Hagelsieb, G. et al. Phylogenetic Distribution and Evolutionary History of Bacterial DEAD-Box Proteins. J Mol Evol 72, 413–431 (2011). https://doi.org/10.1007/s00239-011-9441-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-011-9441-8