
Abstract
Purpose
Molecular biomarkers are important for classifying intracranial gliomas, prompting research into correlating imaging with genotype (“radiogenomics”). A limitation of the existing radiogenomics literature is the paucity of studies specifically characterizing grade 2–3 gliomas into the three key molecular subtypes. Our study investigated the accuracy of multiple different conventional MRI features for genotype prediction.
Methods
Grade 2–3 gliomas diagnosed between 2007 and 2013 were identified. Two neuroradiologists independently assessed nine conventional MRI features. Features with better inter-observer agreement (κ ≥ 0.6) proceeded to consensus assessment. MRI features were correlated with genotype, classified as IDH-mutant and 1p/19q-codeleted (IDHmut/1p19qcodel), IDH-mutant and 1p/19q-intact (IDHmut/1p19qint), or IDH-wildtype (IDHwt). For IDHwt tumors, additional molecular markers of glioblastoma were noted.
Results
One hundred nineteen patients were included. T2-FLAIR mismatch (stratified as > 50%, 25–50%, or < 25%) was the most predictive feature across genotypes (p < 0.001). All 30 tumors with > 50% mismatch were IDHmut/1p19qint, and all seven with 25–50% mismatch. Well-defined margins correlated with IDHmut/1p19qint status on univariate analysis (p < 0.001), but this related to correlation with T2-FLAIR mismatch; there was no longer an association when considering only tumors with < 25% mismatch (p = 0.386). Enhancement (p = 0.001), necrosis (p = 0.002), and hemorrhage (p = 0.027) correlated with IDHwt status (especially “molecular glioblastoma”). Calcification correlated with IDHmut/1p19qcodel status (p = 0.003). A simple, step-wise algorithm incorporating these features, when present, correctly predicted genotype with a positive predictive value 91.8%.
Conclusion
T2-FLAIR mismatch strongly predicts IDHmut/1p19qint even with a lower threshold of ≥ 25% mismatch and outweighs other features. Secondary features include enhancement, necrosis and hemorrhage (predicting IDHwt, especially “molecular glioblastoma”), and calcification (predicting IDHmut/1p19qcodel).
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The addition of molecular biomarkers as a key component of glioma characterization in the 2016 update of the World Health Organization (WHO) Classification of Tumors of the Central Nervous System [1] (WHO 2016) has led to research into correlating imaging features with these molecular markers, known as radiogenomics or imaging genomics. Radiogenomics can aid clinical management in a variety of scenarios [2], and arguably, its potential value is increasing further as a greater number of molecular biomarkers enter clinical practice, based on the recommendations of cIMPACT-NOW [3] and the recent 2021 WHO classification [4] (WHO 2021). Two molecular markers have provided the basis of classification of lower grade (WHO grades 2–3) diffuse gliomas (LGG) since WHO 2016: isocitrate dehydrogenase-1 or -2 variant status (IDH; either IDH-mutant or IDH-wildtype) and 1p/19q-codeletion (combined loss of both the short arm of chromosome 1 and the long arm of chromosome 19) [1]. These two markers allow classification of LGG into one of three subtypes: IDH-mutant with 1p/19q-codeletion (IDHmut/1p19qcodel), IDH-mutant with intact 1p/19q (IDHmut/1p19qint), and IDH-wildtype (IDHwt) [1]. As such, these markers have been the focus of substantial research. A notable addition in WHO 2021 has been the addition of molecular criteria to the diagnosis of glioblastoma (“molecular GBM”), which allows a phenotypic grade 2–3 IDHwt glioma to be diagnosed as a glioblastoma if at least one of three additional molecular features is demonstrated [4]. If these molecular features are absent, the tumor is labeled IDHwt NEC (not elsewhere classified) [4]. Given the recency of this classification, however, there is a paucity of radiogenomic research examining these additional molecular markers.
Radiogenomics research on routine clinical MRI scans can be broadly split into conventional visual assessment by radiologists (conventional radiogenomics) and analysis utilizing artificial intelligence (AI) techniques (AI radiogenomics). As would be expected, there has been a recent shift towards research into AI techniques, but these have not yet reached clinical practice and there remain some limitations [5]. In contrast, predicting genotype by conventional imaging assessment can be immediately incorporated into routine clinical practice [2]. In addition, results of conventional radiogenomics research provide a basis for correlations identified by AI. For example, a study utilizing deep learning and multiparametric imaging found that the addition of CT improved the prediction obtained by using only MRI and positron emission tomography [6], attributed to the correlation between calcifications and 1p/19q-codeletion [7, 8]. Similarly, conventional radiogenomics can suggest which sequences and techniques are likely to be most helpful for incorporation into AI radiogenomics research.
A recent systematic review found that the T2-FLAIR mismatch sign was the most distinctive conventional radiogenomic feature of LGG [5]. High specificity, approaching 100%, has been demonstrated in several cohorts, with good inter-rater agreement [7, 9,10,11,12]. An acknowledged limitation of the T2-FLAIR mismatch sign, however, is its limited sensitivity, being less than 50% in a recent pooled meta-analysis [12]. A variety of other conventional MRI features have been investigated, including tumor location, tumor margin appearances, internal signal characteristics, enhancement, cortical involvement, and magnetic susceptibility (including calcifications and hemorrhage), with varying results [5]. In addition, some features have been investigated in the context of glioblastoma, in particular assessing the morphology of noncontrast-enhancing tumor as a predictor of IDH status [7, 13], but these have not yet been investigated in the context of LGG to our knowledge.
The above systematic review also identified several limitations of the existing literature [5]. For example, only a minority of studies have reported inter-observer comparisons, and most studies investigated either IDH or 1p/19q status, or both independently, with classification into the three molecular subtypes according to the 2016 WHO criteria being less common [5]. A further limitation of the existing literature is that it is unclear how best to incorporate and weight multiple imaging features into a conventional radiogenomic prediction that can be applied in everyday clinical practice. Algorithmic approaches are less common in the literature and have generally not allowed for a prediction of all three molecular subtypes [7, 9, 14, 15].
The purpose of this study was to investigate the accuracy of multiple different conventional MRI features for the prediction of the three molecular subtypes of LGG, as well as distinguishing between molecular GBM and IDHwt NEC, with the goal of developing a user-friendly and robust algorithm for the prediction of LGG molecular subtype based on conventional MRI features.
Methods
Patient identification
Institutional Human Research Ethics Committee approval was obtained (HREC number QA2017093). Patients with a new diagnosis of a grade 2–3 intracranial glioma between September 2007 and December 2013 (to allow for long term survival data) were identified through the Central Nervous System Tumor Database at our hospital and previous research at our institution [7, 16]. All were adult patients.
Imaging assessment
Only patients with at least T2WI, FLAIR, and pre- and post-contrast T1WI were included. MRIs had been performed on a variety of scanners, including at external institutions. Relevant CT images were also reviewed to assess for calcification, preferentially pre-operative, or otherwise post-operative where relevant (and accounting for interval resection). Susceptibility-sensitive sequences were also reviewed, including phase images where available.
Imaging features
The following imaging features were assessed for all patients, based on prior published definitions where possible:
-
Tumor location, recorded as either frontal (all/predominantly frontal, but not insular/temporal), temporal (all/predominantly temporal, but not insular/frontal); fronto-temporal (+ / − insular) or none of the above (no/minimal frontal and/or temporal lobe involvement)
-
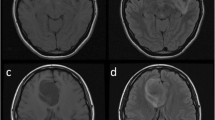
T2-FLAIR mismatch [11], stratified as either > 50% of the tumor volume (distinguished from edema [13]), 25–50% or < 25% (Fig. 1)
-
Well-defined tumor margins, defined as > 75% of the tumor circumference considered geographically marginated [17]
-
Homogeneous (on both T1WI and T2WI)
-
Magnetic susceptibility, recorded as either none, consistent with calcification, convincingly representing hemorrhage or indeterminate between calcification and hemorrhage
-
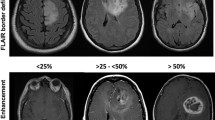
Enhancement, stratified as either none, wispy (subtle and ill-defined, without discrete nodularity) and distinct (Fig. 2)
-
Presence of central necrosis (requiring a central non-enhancing area surrounded by a complete enhancing ring)
-
The presence of eccentric FLAIR hyperintensity consistent with infiltrative noncontrast-enhancing tumor (nCET) [13]
-
The presence of a mass-like morphology of nCET [7]
-
Predominant cortical involvement of nCET

Axial T2WI (top row) and FLAIR (bottom row) images of two patients with IDHmut/1p19qint LGG. The first patient (a, b) has a tumor centered on the right lentiform nucleus, demonstrating 25–50% T2-FLAIR mismatch. The arrows highlight areas with high T2 signal and substantially lower signal on FLAIR. The second patient (c, d) has a left frontal lobe tumor with > 50% T2-FLAIR mismatch and well-defined tumor margins

Axial pre-contrast T1WI (top row) and post-contrast T1WI (bottom row) demonstrating different patterns of enhancement. The first patient (a, b) has an IDHmut/1p19qint LGG demonstrating wispy enhancement, shown by the arrows. The second patient (c & d) has a right parietal lobe tumor with nodular enhancement, evidence of necrosis (arrowhead), and ill-defined tumor margins
Assessment was performed independently by two neuroradiologists from different institutions with subspecialty expertise in neuro-oncology (having 8 and 13 years of experience), blinded to molecular status. After this read, inter-rater agreement was calculated for all features and directed the features to be assessed through consensus review, based on inter-observer agreement yielding κ ≥ 0.6. Features with lower inter-rater agreement (κ < 0.6) were not assessed further.
Histological assessment
Histological diagnoses were obtained from the database and confirmed with the formal pathology report (in particular, confirming the diagnosis of a grade 2–3 diffuse glioma). Where available, IDH1-R132H immunohistochemistry and 1p/19q-codeletion results (the latter typically performed by fluorescence in situ hybridization) were obtained from hospital pathology results (performed as part of routine patient care) and previous research performed at our institution [7, 16]. IDH pyrosequencing results were also available for a small number of patients through prior research [18]. Complete molecular characterization required IDH1 codon 132 and IDH2 codon 172 sequencing if IDH1-R132H immunohistochemistry was negative, and determination of 1p/19q-codeletion status for patients with IDH mutation detected by any method. For patients without complete molecular characterization based on existing data, next-generation sequencing (NGS) was performed to determine IDH and 1p/19q-codeletion status, utilizing previously reported methodology [19]. For patients undergoing NGS, the output also included TERT promoter variant status, + 7/ − 10 and EGFR copy number status according to the updated criteria for glioblastoma in WHO 2021 [4].
Based on the above results, all tumors were classified as either IDHmut/1p19qcodel, IDHmut/1p19qint, or IDHwt. Patients in whom definitive molecular characterization as one of these three genotypes was not possible—most commonly due to insufficient or inadequate material being available for NGS—were excluded. In addition, IDHwt tumors were further divided into molecular GBM and IDHwt NEC [4].
Statistical analysis
All statistical analyses were performed in R (R version 4.0.3 (2020–10-10)) using standard and validated statistical procedures. Descriptive statistics of baseline characteristics of all included patients were reported. Continuous variables were described as median, minimum, and maximum, and categorical variables were described as counts and percentages. Cohen’s κ-coefficient (κ) was used to measure the degree of agreement between a pair of variables. Logistic regression analysis was used to assess for associations between MRI features and molecular subtype. Odds ratios (OR) with 95% confidence intervals (CI) and p-values were calculated. Multivariable logistic regression analysis included all important risk factors with p < 0.1. A p value less than 0.05 was defined as statistically significant.
Results
Patient results
Twenty-three patients were excluded due to incomplete molecular characterization. A further three patients were excluded due to the identification of H3 histone mutations (two K27M, one G34R). Thus, the study population of 119 patients included 58 (48.7%) IDHmut/1p19qint tumors, 32 (26.9%) IDHwt and 29 (24.4%) IDHmut/1p19qcodel. The patients had a median age of 38 years (range 17–83 years), and 62% were male. NGS was performed on the majority of patients (108 of 119), with only 11 tumors already adequately characterized molecularly based on existing data. Only one patient had had IDHwt status confirmed through IDH pyrosequencing; the remaining 31 IDHwt tumors underwent NGS. Twenty-four (77.4%) of these met the criteria for molecular GBM [4].
After single-reader assessment, the following features had at least moderate inter-observer agreement (κ ≥ 0.6) and proceeded to the consensus read: location (κ = 0.939), necrosis (κ = 0.766), well-defined tumor margins (κ = 0.723), enhancement (κ = 0.706), magnetic susceptibility (κ = 0.622), and T2-FLAIR mismatch (κ = 0.616). The following features had a lower inter-observer agreement and were excluded from the subsequent assessment: eccentric nCET (κ = 0.51), homogeneous signal (κ = 0.481), cortical involvement (κ = 0.272), and mass-like nCET (κ = 0.231).
The frequency of each MRI feature based on tumor subtype is presented in Table 1. Concordant with the aforementioned literature [12], the T2-FLAIR mismatch sign was highly predictive of an IDHmut/1p19qint tumor (p < 0.001), including with a lower threshold of T2-FLAIR mismatch, and this was the single most predictive feature across molecular subtypes. All 30 tumors with > 50% mismatch were IDHmut/1p19qint, as well as all seven with 25–50% mismatch (p < 0.001). Combining both groups, all 37 tumors with ≥ 25% mismatch were IDHmut/1p19qint, producing a specificity of 100% and sensitivity of 63%. Eight (21.6%) of the 37 tumors with ≥ 25% mismatch demonstrated solid enhancement (all in the > 50% group), with a further eight demonstrating wispy enhancement. The presence of ≥ 25% mismatch predicted IDHmut/1p19qint status in both enhancing and non-enhancing tumors.
On univariate analysis, well-defined tumor margins correlated with IDHmut/1p19qint status (OR 7.7, 95% CI 3.0–19.8, p < 0.001), with 29 (50.0%) of 58 IDHmut/1p19qint tumors having well-defined margins. Most IDHwt tumors (30 of 32, 93.8%, p = 0.003) had ill-defined margins, as well as 24 (82.8%) of 29 IDHmut/1p19qcodel tumors, limiting the distinction between these two subtypes. Importantly, well-defined tumor margins strongly correlated with T2-FLAIR mismatch (p < 0.001; contingency coefficient, C = 0.72), especially with > 50% mismatch (23 of 30 were well-defined, 76.7%). There was less correlation with tumors exhibiting 25–50% mismatch (two of seven with well-defined margins, 28.6%). Given the above association, well-defined margins no longer correlated significantly with IDHmut/1p19qint status when considering only tumors with < 25% mismatch, with only four (19.0%) of the 21 IDHmut/1p19qint tumors with < 25% mismatch exhibiting well-defined margins (p = 0.386). IDHmut/1p19qint tumors were most commonly frontal (46.6%), IDHmut/1p19qcodel tumors were usually frontal (55.2%) and uncommonly temporal (6.9%), and IDHwt tumors were most commonly neither frontal nor temporal (46.9%); however, there was no statistically significant correlation between tumor location and genotype.
The predictive value of magnetic susceptibility was limited, as the majority of tumors (91 of 119, 76.5%) demonstrated no susceptibility, and for a further 13 (10.9%), the distinction between calcification and hemorrhage was unclear. Ten tumors exhibited convincing calcification, and seven of these (70%) were IDHmut/1p19qcodel (OR 8.3, 95% CI 2.0–35.0, p = 0.004). However, most (22 of 29) IDHmut/1p19qcodel tumors did not have convincing calcification. There were no other particular features of the calcified, non-codeleted tumors that would have confidently suggested an alternate subtype: no tumors demonstrated calcification and ≥ 25% mismatch, and the one tumor with both calcification and necrosis was IDHmut/1p19qcodel. Five tumors showed evidence of hemorrhage, of which four (80%) were IDHwt. Notably, an IDHmut/1p19qint tumor with hemorrhage had ≥ 25% mismatch, so all four tumors with evidence of hemorrhage and < 25% mismatch were IDHwt. As for calcification, however, most (28 of 32) IDHwt tumors did not demonstrate hemorrhage.
Enhancement and necrosis both correlated strongly with IDHwt status (OR 5.1, 95% CI 2.0–12.8, p = 0.001 for enhancement; OR 5.2, 95% 1.8–14.5, p = 0.002 for necrosis), with 19 (52.8%) of 36 enhancing tumors and 11 (57.9%) of 19 necrotic tumors being IDHwt. Of the 32 IDHwt tumors, 59.4% demonstrated enhancement and 34.4% had necrosis. Of note, necrosis was visualized in five tumors demonstrating ≥ 25% mismatch (all having > 50% mismatch), and all were IDHmut/1p19qint. Once tumors with ≥ 25% mismatch were excluded from the subgroup analysis, 78.6% of tumors demonstrating necrosis were IDHwt (of which three also demonstrated evidence of hemorrhage). Enhancement and necrosis were least common in IDHmut/1p19qcodel tumors, with only three (10.3%) and two (6.9%) of the 29 IDHmut/1p19qcodel tumors exhibiting these features, respectively. Of note, tumors with solid enhancement were significantly less likely to be IDHmut/1p19qcodel (OR 0.2, 95% CI 0.1–0.7, p = 0.016). Enhancement and necrosis were both more common in molecular GBM (enhancement in 16/24, 80.0%; necrosis in 9/24, 37.5%) than IDHwt NEC (enhancement in 2/7, 28.6%; necrosis in 1/7, 14.3%), and both differences were statistically significant (p < 0.001 and p < 0.001, respectively). Additionally, all four IDHwt tumors with evidence of hemorrhage were molecular GBMs, while both IDHwt tumors with calcification were IDHwt NEC.
In terms of an overall algorithmic approach, identification of ≥ 25% mismatch was shown to be the key first step, allowing high confidence of IDHmut/1p19qint status regardless of other features which might otherwise predict a different molecular subtype (such as necrosis). Once mismatch had been considered, the ability of other features (in particular well-defined tumor margins) to predict IDHmut/1p19qint status diminished, but the ability to predict an IDHwt tumor improved. Specifically, after excluding tumors with ≥ 25% mismatch, all tumors with evidence of hemorrhage were IDHwt, the OR for enhancement improved from 5.1 to 5.6, and the OR for necrosis increased from 5.2 to 8.2. One possible approach would be to assess for magnetic susceptibility (calcification and hemorrhage) after T2-FLAIR mismatch and then for the presence of enhancement. In our cohort, this would yield an overall positive predictive value (PPV) of 87.3%, and 60% of tumors would have a positive finding. Alternatively, the enhancement could be replaced by necrosis, improving the positive predictive value to 91.8%, but decreasing the number of tumors with a positive finding to 51%.
Discussion
Our findings reinforce the high-positive predictive value of the T2-FLAIR mismatch sign, but go well beyond just confirming the findings of previous studies. Firstly, we have stratified tumors by the percentage of T2-FLAIR mismatch, adding and specifically assessing tumors with a smaller proportion of mismatch (25–50%). While most tumors with T2-FLAIR mismatch demonstrate > 50% mismatch, smaller percentages of mismatch are not uncommon, and it is unclear from the existing literature how such tumors should be considered. In the original description by Patel et al., T2-FLAIR mismatch was defined as the “presence or absence of complete/near-complete hyperintense signal on T2WI, and relatively hypointense signal on FLAIR except for a hyperintense peripheral rim” [11]. This implies, without explicitly defining, substantially greater than 50% mismatch. One study predominantly used a threshold of > 50% mismatch, though also assessed for 33–50% mismatch, with only one further tumor identified [7]. Our findings suggest that the percentage of mismatch required to consider the feature positive can be dropped to 25% without a drop in PPV (remaining at 100%), but with improved sensitivity. This accounts for the relatively larger proportion of IDHmut/1p19qint tumors demonstrating mismatch in our cohort compared to those reported previously. We have also investigated the accuracy of the T2-FLAIR mismatch sign in enhancing tumors, which is less clear in the literature. There have been suggestions that the PPV may be lower in enhancing tumors [20], but this was not the case in our cohort. Given that all other features were less accurate, we suggest that T2-FLAIR remains a very helpful feature in enhancing tumors, including those with evidence of necrosis.
A lower threshold for T2-FLAIR mismatch is in line with a more recently described feature, named “fluid attenuation in non-contrast-enhancing tumor,” which has been shown to predict IDH mutations in glioblastoma [21]. This feature bears substantial similarities to the T2-FLAIR mismatch sign in LGG and was defined as “any tumor volume with hyperintense signal on T2WI and corresponding hypointense signal on FLAIR” [21]. This definition implies that less than 50% fluid attenuation—and potentially even less than 25%—would nevertheless allow this feature to be considered present. This suggests that grade 2–4 gliomas could be assessed together using a threshold of ≥ 25% mismatch, though this has yet to be formally assessed to our knowledge. Such analysis is particularly relevant in the context of WHO 2021, given that the distinction between tumor grade has become less important, with molecular status playing a greater role [4].
Our data comparing the MRI features of IDHwt tumors with and without additional molecular markers of GBM is another novel aspect of our work. We have shown that features predictive of IDHwt status are predominantly present in molecular GBMs. We have also clarified some uncertainty regarding the overlap in MRI features, which has been under-studied in the literature. Specifically, while there have been some suggestions in the literature that well-defined margins predict an IDHmut/1p19qint tumor [5], this seems to largely relate to an overlap with the T2-FLAIR mismatch sign; well-defined tumor margins have limited (and possibly no) predictive value once T2-FLAIR mismatch is accounted for. On the other hand, the value of other features, such as necrosis, improved once tumors with ≥ 25% mismatch were removed. This suggests that a step-wise, multi-feature algorithmic approach has value—a suggested algorithm is provided in Fig. 3. An alternate algorithm, considering necrosis ahead of magnetic susceptibility, achieved slightly lower accuracy in our cohort, as the one tumor with evidence of calcifications and necrosis was IDHmut/1p19qcodel. A limitation of the assessment of magnetic susceptibility related to the uncertainty in distinguishing between calcifications and hemorrhage in many cases, due to the frequent unavailability of preoperative CT and/or SWI phase images in this dataset. The sensitivity, and potentially specificity, of the algorithm would be expected to increase if these were uniformly available.

A proposed algorithm for the prediction of LGG genotype using visual features
ADC has shown promise for predicting genotype [5, 15], and the lack of inclusion of ADC measurements is a potential limitation of our study. Being a continuous variable, with overlap across the three genotypes, the ADC threshold can be adjusted to optimize either sensitivity or specificity, as is considered most appropriate [2]. We had not incorporated ADC as we had felt that it was unclear from the literature how best to do so, but now our findings provide some guidance. ADC would seem to have the greatest potential in the tumors without any of the key features described. However, ADC values are likely to correlate with T2-FLAIR mismatch to some extent—specifically, ADC values are likely to be high in tumor regions demonstrating T2-FLAIR mismatch. Thus, as with well-defined tumor margins, the predictive value may decrease once T2-FLAIR mismatch has been accounted for, and this warrants further investigation. Another use of ADC could be to supplement the secondary features (calcifications, hemorrhage, enhancement, and necrosis), by confirming that the ADC values lie within the expected range for the predicted genotype.
Despite including a larger number of features than many previous studies, a substantial proportion of tumors did not demonstrate any of the key features we identified. Thus, our goal of using conventional MRI features to predict all three molecular subtypes with high accuracy has not yet been realized. We had hoped that the inclusion of additional features (such as cortical involvement and the morphology of nCET) would allow some differentiation of these equivocal cases, but this was limited by the lower inter-rater agreement. There may nevertheless be some potential in these additional features, but a tighter definition is required. AI techniques which can visually represent the imaging appearances correlating with a particular glioma subtype, such as the technique of principal component analysis as reported by Chang et al. [22], hold promise in this regard. For example, we observed that there was variability in the appearances of tumors demonstrating cortical predominance, for example, in the degree of gray-white matter differentiation, but it was not clear how to objectively distinguish these patterns and whether they indeed reflected differences in the genotype. If AI techniques were able to identify subtle differences in the appearances of such tumors, this could facilitate a clearer definition for visual assessment and thus improve the inter-observer agreement, or at least supplement aspects of visual assessment which are more challenging.
Over-fitting is a common criticism of AI radiogenomics research and is also a potential consideration for the algorithmic approach we have suggested. Reassuringly, however, the individual features included in the final analysis have all been investigated previously, and our results are in line with previous reports. As such, we believe that similar results can be expected in a broader cohort, though subsequent validation will nevertheless be important. The retrospective nature of our study is a further limitation, as it is for much of the work in this field. We note that the patients in our cohort were from several years ago, and modern scanners could potentially achieve even better results. For example, the routine inclusion of susceptibility-weighted imaging including phase images should improve the detection of, and the distinction between, calcifications and hemorrhage, while modern volumetric post-contrast sequences will aid in the identification of enhancement and necrosis. Similarly, the inclusion of images from a variety of scanners, including external imaging, provides reassurance that the results can be translated to other institutions. We consider the proportions of our molecular subtypes to be concordant for the broader literature—for example, a cohort reported by The Cancer Genome Research Network had 50.0% IDHmut/1p19qint tumors, 19.9% IDHwt, and 30.1% IDHmut/1p19qcodel [23]—though we note a slightly higher proportion of IDHwt tumors and a slightly lower proportion of IDHmut/1p19qcodel tumors in our cohort compared to the above.
Conclusions
The T2-FLAIR mismatch sign is highly predictive of IDHmut/1p19qint status in LGG, including for tumors demonstrating post-contrast enhancement, necrosis, and smaller proportions of mismatch (≥ 25%). Well-defined tumor margins are associated with T2-FLAIR mismatch and are no longer independently predictive of IDHmut/1p19qint status after accounting for T2-FLAIR mismatch. For tumors with < 25% mismatch, secondary predictive features include the presence of enhancement, necrosis, and/or hemorrhage (favoring an IDHwt tumor, especially those with additional molecular features of glioblastoma) and the presence of calcifications (predicting IDHmut/1p19qcodel).
Availability of data and materials
N/A.
Abbreviations
- AI:
-
Artificial intelligence
- GBM:
-
Glioblastoma
- IDH:
-
Isocitrate dehydrogenase
- IDHmut/1p19qcodel :
-
IDH-mutant with 1p/19q-codeletion
- IDHmut/1p19qint :
-
IDH-mutant with intact 1p/19q
- IDHwt :
-
IDH-wildtype
- IDHwt NEC:
-
IDH-wildtype not elsewhere classified
- LGG:
-
Lower grade glioma (WHO grade 2–3)
- nCET:
-
Noncontrast-enhancing tumor
- NGS:
-
Next-generation sequencing
- OR:
-
Odds ratio
- CI:
-
Confidence interval
- PPV:
-
Positive predictive value
- WHO:
-
World Health Organization
References
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW (2016) The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 131(6):803–820. https://doi.org/10.1007/s00401-016-1545-1
Lasocki A, Rosenthal MA, Roberts-Thomson SJ, Neal A, Drummond KJ (2020) Neuro-oncology and radiogenomics: time to integrate? AJNR Am J Neuroradiol 41(11):1982–1988. https://doi.org/10.3174/ajnr.A6769
Louis DN, Aldape K, Brat DJ, Capper D, Ellison DW, Hawkins C, Paulus W, Perry A, Reifenberger G, Figarella-Branger D, Wesseling P, Batchelor TT, Cairncross JG, Pfister SM, Rutkowski S, Weller M, Wick W, von Deimling A (2017) Announcing cIMPACT-NOW: the consortium to inform molecular and practical approaches to CNS tumor taxonomy. Acta Neuropathol 133(1):1–3. https://doi.org/10.1007/s00401-016-1646-x
Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, Hawkins C, Ng HK, Pfister SM, Reifenberger G, Soffietti R, von Deimling A, Ellison DW (2021) The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol 23(8):1231–1251. https://doi.org/10.1093/neuonc/noab106
Bhandari AP, Liong R, Koppen J, Murthy SV, Lasocki A (2021) Noninvasive determination of IDH and 1p19q status of lower-grade gliomas using MRI radiomics: a systematic review. AJNR Am J Neuroradiol 42(1):94–101. https://doi.org/10.3174/ajnr.A6875
Matsui Y, Maruyama T, Nitta M, Saito T, Tsuzuki S, Tamura M, Kusuda K, Fukuya Y, Asano H, Kawamata T, Masamune K, Muragaki Y (2020) Prediction of lower-grade glioma molecular subtypes using deep learning. J Neurooncol 146(2):321–327. https://doi.org/10.1007/s11060-019-03376-9
Lasocki A, Gaillard F, Tacey M, Drummond K, Stuckey S (2018) Morphologic patterns of noncontrast-enhancing tumor in glioblastoma correlate with IDH1 mutation status and patient survival. Journal of clinical neuroscience: official journal of the Neurosurgical Society of Australasia 47:168–173. https://doi.org/10.1016/j.jocn.2017.09.007
Saito T, Muragaki Y, Maruyama T, Komori T, Tamura M, Nitta M, Tsuzuki S, Kawamata T (2016) Calcification on CT is a simple and valuable preoperative indicator of 1p/19q loss of heterozygosity in supratentorial brain tumors that are suspected grade II and III gliomas. Brain Tumor Pathol 33(3):175–182. https://doi.org/10.1007/s10014-016-0249-5
Batchala PP, Muttikkal TJE, Donahue JH, Patrie JT, Schiff D, Fadul CE, Mrachek EK, Lopes MB, Jain R, Patel SH (2019) Neuroimaging-based classification algorithm for predicting 1p/19q-codeletion status in IDH-mutant lower grade gliomas. AJNR Am J Neuroradiol 40(3):426–432. https://doi.org/10.3174/ajnr.A5957
Broen MPG, Smits M, Wijnenga MMJ, Dubbink HJ, Anten M, Schijns O, Beckervordersandforth J, Postma AA, van den Bent MJ (2018) The T2-FLAIR mismatch sign as an imaging marker for non-enhancing IDH-mutant, 1p/19q-intact lower grade glioma: a validation study. Neuro Oncol. https://doi.org/10.1093/neuonc/noy048
Patel SH, Poisson LM, Brat DJ, Zhou Y, Cooper L, Snuderl M, Thomas C, Franceschi AM, Griffith B, Flanders AE, Golfinos JG, Chi AS, Jain R (2017) T2-FLAIR mismatch, an imaging biomarker for IDH and 1p/19q status in lower-grade gliomas: a TCGA/TCIA project. Clinical cancer research : an official journal of the American Association for Cancer Research 23(20):6078–6085. https://doi.org/10.1158/1078-0432.Ccr-17-0560
Park SI, Suh CH, Guenette JP, Huang RY, Kim HS (2021) The T2-FLAIR mismatch sign as a predictor of IDH-mutant, 1p/19q-noncodeleted lower-grade gliomas: a systematic review and diagnostic meta-analysis. Eur Radiol 31(7):5289–5299. https://doi.org/10.1007/s00330-020-07467-4
Lasocki A, Gaillard F (2019) Non-contrast-enhancing tumor: a new frontier in glioblastoma research. AJNR Am J Neuroradiol 40(5):758–765. https://doi.org/10.3174/ajnr.A6025
Kanazawa T, Fujiwara H, Takahashi H, Nishiyama Y, Hirose Y, Tanaka S, Yoshida K, Sasaki H (2019) Imaging scoring systems for preoperative molecular diagnoses of lower-grade gliomas. Neurosurg Rev 42(2):433–441. https://doi.org/10.1007/s10143-018-0981-x
Maynard J, Okuchi S, Wastling S, Busaidi AA, Almossawi O, Mbatha W, Brandner S, Jaunmuktane Z, Koc AM, Mancini L, Jäger R, Thust S (2020) World Health Organization grade II/III glioma molecular status: prediction by MRI morphologic features and apparent diffusion coefficient. Radiology 296(1):111–121. https://doi.org/10.1148/radiol.2020191832
Lasocki A, Tsui A, Tacey MA, Drummond KJ, Field KM, Gaillard F (2015) MRI grading versus histology: predicting survival of World Health Organization grade II–IV astrocytomas. Am J Neuroradiol 36(1):77–83. https://doi.org/10.3174/ajnr.A4077
Johnson DR, Diehn FE, Giannini C, Jenkins RB, Jenkins SM, Parney IF, Kaufmann TJ (2017) Genetically defined oligodendroglioma is characterized by indistinct tumor borders at MRI. AJNR Am J Neuroradiol 38(4):678–684. https://doi.org/10.3174/ajnr.A5070
Neal A, Kwan P, O’Brien TJ, Buckland ME, Gonzales M, Morokoff A (2018) IDH1 and IDH2 mutations in postoperative diffuse glioma-associated epilepsy. Epilepsy & behavior : E&B 78:30–36. https://doi.org/10.1016/j.yebeh.2017.10.027
Cheung VKY, Buckland ME, Wei G, Lee M, Sy J (2021) Next generation sequencing impacts the classification and management of primary brain tumours. Pathology 53(6):780–782. https://doi.org/10.1016/j.pathol.2020.12.008
Jain R, Johnson DR, Patel SH, Castillo M, Smits M, van den Bent MJ, Chi AS, Cahill DP (2020) “Real world” use of a highly reliable imaging sign: “T2-FLAIR mismatch” for identification of IDH mutant astrocytomas. Neuro Oncol 22(7):936–943. https://doi.org/10.1093/neuonc/noaa041
Patel SH, Batchala PP, Muttikkal TJE, Ferrante SS, Patrie JT, Fadul CE, Schiff D, Lopes MB, Jain R (2021) Fluid attenuation in non-contrast-enhancing tumor (nCET): an MRI marker for isocitrate dehydrogenase (IDH) mutation in glioblastoma. J Neurooncol 152(3):523–531. https://doi.org/10.1007/s11060-021-03720-y
Chang P, Grinband J, Weinberg BD, Bardis M, Khy M, Cadena G, Su MY, Cha S, Filippi CG, Bota D, Baldi P, Poisson LM, Jain R, Chow D (2018) Deep-learning convolutional neural networks accurately classify genetic mutations in gliomas. AJNR Am J Neuroradiol 39(7):1201–1207. https://doi.org/10.3174/ajnr.A5667
Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA, Rheinbay E, Miller CR, Vitucci M, Morozova O, Robertson AG, Noushmehr H, Laird PW, Cherniack AD, Akbani R, Huse JT, Ciriello G, Poisson LM, Barnholtz-Sloan JS, Berger MS, Brennan C, Colen RR, Colman H, Flanders AE, Giannini C, Grifford M, Iavarone A, Jain R, Joseph I, Kim J, Kasaian K, Mikkelsen T, Murray BA, O’Neill BP, Pachter L, Parsons DW, Sougnez C, Sulman EP, Vandenberg SR, Van Meir EG, von Deimling A, Zhang H, Crain D, Lau K, Mallery D, Morris S, Paulauskis J, Penny R, Shelton T, Sherman M, Yena P, Black A, Bowen J, Dicostanzo K, Gastier-Foster J, Leraas KM, Lichtenberg TM, Pierson CR, Ramirez NC, Taylor C, Weaver S, Wise L, Zmuda E, Davidsen T, Demchok JA, Eley G, Ferguson ML, Hutter CM, Mills Shaw KR, Ozenberger BA, Sheth M, Sofia HJ, Tarnuzzer R, Wang Z, Yang L, Zenklusen JC, Ayala B, Baboud J, Chudamani S, Jensen MA, Liu J, Pihl T, Raman R, Wan Y, Wu Y, Ally A, Auman JT, Balasundaram M, Balu S, Baylin SB, Beroukhim R, Bootwalla MS, Bowlby R, Bristow CA, Brooks D, Butterfield Y, Carlsen R, Carter S, Chin L, Chu A, Chuah E, Cibulskis K, Clarke A, Coetzee SG, Dhalla N, Fennell T, Fisher S, Gabriel S, Getz G, Gibbs R, Guin R, Hadjipanayis A, Hayes DN, Hinoue T, Hoadley K, Holt RA, Hoyle AP, Jefferys SR, Jones S, Jones CD, Kucherlapati R, Lai PH, Lander E, Lee S, Lichtenstein L, Ma Y, Maglinte DT, Mahadeshwar HS, Marra MA, Mayo M, Meng S, Meyerson ML, Mieczkowski PA, Moore RA, Mose LE, Mungall AJ, Pantazi A, Parfenov M, Park PJ, Parker JS, Perou CM, Protopopov A, Ren X, Roach J, Sabedot TS, Schein J, Schumacher SE, Seidman JG, Seth S, Shen H, Simons JV, Sipahimalani P, Soloway MG, Song X, Sun H, Tabak B, Tam A, Tan D, Tang J, Thiessen N, Triche T Jr, Van Den Berg DJ, Veluvolu U, Waring S, Weisenberger DJ, Wilkerson MD, Wong T, Wu J, Xi L, Xu AW, Yang L, Zack TI, Zhang J, Aksoy BA, Arachchi H, Benz C, Bernard B, Carlin D, Cho J, DiCara D, Frazer S, Fuller GN, Gao J, Gehlenborg N, Haussler D, Heiman DI, Iype L, Jacobsen A, Ju Z, Katzman S, Kim H, Knijnenburg T, Kreisberg RB, Lawrence MS, Lee W, Leinonen K, Lin P, Ling S, Liu W, Liu Y, Liu Y, Lu Y, Mills G, Ng S, Noble MS, Paull E, Rao A, Reynolds S, Saksena G, Sanborn Z, Sander C, Schultz N, Senbabaoglu Y, Shen R, Shmulevich I, Sinha R, Stuart J, Sumer SO, Sun Y, Tasman N, Taylor BS, Voet D, Weinhold N, Weinstein JN, Yang D, Yoshihara K, Zheng S, Zhang W, Zou L, Abel T, Sadeghi S, Cohen ML, Eschbacher J, Hattab EM, Raghunathan A, Schniederjan MJ, Aziz D, Barnett G, Barrett W, Bigner DD, Boice L, Brewer C, Calatozzolo C, Campos B, Carlotti CG Jr, Chan TA, Cuppini L, Curley E, Cuzzubbo S, Devine K, DiMeco F, Duell R, Elder JB, Fehrenbach A, Finocchiaro G, Friedman W, Fulop J, Gardner J, Hermes B, Herold-Mende C, Jungk C, Kendler A, Lehman NL, Lipp E, Liu O, Mandt R, McGraw M, McLendon R, McPherson C, Neder L, Nguyen P, Noss A, Nunziata R, Ostrom QT, Palmer C, Perin A, Pollo B, Potapov A, Potapova O, Rathmell WK, Rotin D, Scarpace L, Schilero C, Senecal K, Shimmel K, Shurkhay V, Sifri S, Singh R, Sloan AE, Smolenski K, Staugaitis SM, Steele R, Thorne L, Tirapelli DP, Unterberg A, Vallurupalli M, Wang Y, Warnick R, Williams F, Wolinsky Y, Bell S, Rosenberg M, Stewart C, Huang F, Grimsby JL, Radenbaugh AJ, Zhang J (2015) Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med 372(26):2481–2498. https://doi.org/10.1056/NEJMoa1402121
Acknowledgements
This study was presented at the 2022 European Congress of Radiology, with an abstract published in the ECR Book of Abstracts.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions This study was supported by a RANZCR research grant in 2018. Dr Arian Lasocki was supported by a Peter MacCallum Cancer Foundation Discovery Partner Fellowship.
Author information
Authors and Affiliations
Ethics declarations
Ethics approval
Institutional Human Research Ethics Committee approval was obtained (HREC number QA2017093). The study was performed in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki and its later amendments.
Consent to participate
Patient consent was not required.
Consent for publication
N/A
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lasocki, A., Buckland, M.E., Drummond, K.J. et al. Conventional MRI features can predict the molecular subtype of adult grade 2–3 intracranial diffuse gliomas. Neuroradiology 64, 2295–2305 (2022). https://doi.org/10.1007/s00234-022-02975-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00234-022-02975-0