
Abstract
Introduction
Alexander disease is a rare disorder of the central nervous system with characteristic symmetric white matter abnormalities with frontal predominance on magnetic resonance (MR) images. Histopathology shows a lack of myelin in the affected white matter, variably interpreted as hypomyelination or demyelination. To increase our insight into the nature of the pathology leading to the MR imaging findings in Alexander disease, we applied serial MR imaging, spectroscopy, magnetization transfer (MT) imaging (MTI), and diffusion tensor imaging (DTI) in six patients with juvenile Alexander disease.
Methods
The MR imaging protocol comprised T1- and T2-weighted spin echo images and fluid-attenuated inversion recovery images. Fractional anisotropy (FA), apparent diffusion coefficient (ADC), and MT ratio (MTR) maps were generated, and MR spectroscopy concentrations were quantified for several metabolites.
Results
MR imaging showed similar cerebral white matter abnormalities in all patients, with only minor increase on prolonged follow-up, despite sometimes serious clinical progression. MR spectroscopy showed highly elevated levels of myo-inositol, lactate, and choline-containing compounds and decreased total N-acetyl-aspartate and N-acetyl-aspartyl-glutamate levels in the abnormal white matter. High values of ADC were observed, and both FA and MTR were attenuated.
Conclusion
The sequential MR imaging findings in Alexander disease provide strong evidence against active demyelination as sole explanation for the underlying pathology. An alternative explanation for our spectroscopic, DTI, and MTI findings—which would suggest demyelination—could be hyperplasia and hypertrophy of astrocytes, as seen in low grade gliomas.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Alexander disease is a rare disorder of the central nervous system. Dominant, usually de novo, mutations in the gene GFAP, encoding the glial fibrillary acidic protein (GFAP), have been identified in patients with histopathologically proven Alexander disease [1, 2]. Three clinical subgroups can be distinguished: infantile, juvenile, and adult. The most common form is the infantile form, which presents as a progressive leukodystrophy with developmental delay, seizures, macrocephaly, spasticity, and rapid deterioration with early demise. The juvenile and adult variants have a later onset and slower disease course. Macrocephaly is not a consistent finding. Bulbar symptoms are almost invariably present [1, 2].
Characteristic diagnostic magnetic resonance (MR) imaging (MRI) criteria have been proposed, including symmetric white matter abnormalities with frontal predominance, a periventricular rim with high signal on T1-weighted images and low signal on T2-weighted images, abnormalities of the basal ganglia and thalami, brainstem lesions, and contrast enhancement of particular gray and white matter structures [3]. Since the presence of a pathogenic mutation in the GFAP mutation has become the gold standard for the diagnosis of Alexander disease, it has become clear that the MR imaging picture is much more variable than initially thought. Especially in juvenile and adult cases of Alexander disease, the leukoencephalopathy with frontal preponderance may be missing and the brain stem involvement may dominate, often with multifocal tumor-like lesions [4, 5].
Neuropathologically, the most distinctive feature of Alexander disease is the presence of massive numbers of Rosenthal fibers, which accumulate in astrocytes. Rosenthal fibers are scattered throughout the white matter in all areas of the brain, but are most intense in subependymal, subpial, and perivascular locations. Astrocytic density is increased in the same areas. Another distinctive feature is the paucity of myelin, generally most pronounced in the frontal white matter, variably interpreted as hypomyelination or demyelination. The axons appear intact in those areas, suggesting a lack of myelin deposition. However, infiltrating macrophages with perivascular lymphocytic cuffs may also occur, suggesting a component of active demyelination [6, 7]. The nature of the white matter pathology is therefore not clear.
Previously, a method was developed to discriminate between various types of white matter pathology that affect children, such as demyelination, hypomyelination, myelin vacuolation, and cystic degeneration, using quantitative MR parameters [8]. To increase our understanding of the pathophysiological mechanisms leading to the MR findings in juvenile Alexander disease, we conducted MR spectroscopy, magnetization transfer (MT) imaging (MTI), and diffusion tensor imaging (DTI) in six patients with juvenile Alexander disease.
Materials and methods
Patients and control subjects
This study was performed with the informed consent of the patients and control subjects or of their parents and with the approval of the institutional ethics review board.
We included three boys and three girls with juvenile Alexander disease. In all six patients, the diagnosis of Alexander disease was confirmed by the demonstration of a heterozygous de novo mutation (i.e., not detected in DNA of the parents) in the GFAP gene (Table 1).
In three patients, all quantitative measures were obtained on at least one occasion. One patient was studied with the same protocol, but without DTI. Two patients underwent conventional MRI only (without DTI and MTI) and MRS, but another MRS technique was used which did not allow quantification. The ages of the patients ranged from 3.3 to 19.9 years at the MR investigations that were used for quantitative measurements.
Quantitative MR imaging and spectroscopy results of the patients were compared with the results of control subjects (25 males, 16 females; age range 2.0 to 26.4 years). Overall, the most rapid changes in values of quantitative MRI parameters and MRS metabolites occur during the first 2 years and close to final values are reached at the age of 2 years, when myelination is almost complete [9–11]. To avoid a significant influence of age-dependent changes, only patients and control subjects of 2 years and older were included.
The control subjects were either healthy volunteers or pediatric subjects with normal results at neurologic examination and normal MRI results. The latter subjects underwent MRI for reasons like headache and epileptic seizures.
MR imaging and proton MR spectroscopy
All examinations were performed on a 1.5-T MR scanner (Siemens Vision, Erlangen, Germany). The conventional imaging protocol included sagittal T1-weighted images using a three-dimensional (3D) magnetization-prepared rapid acquisition gradient-echo sequence (repetition time (TR) 15 ms, echo time (TE) 4 ms, one excitation), transverse T2-weighted spin echo images (TR 3,000 ms, TE 22, 60, and 120 ms, one excitation), and coronal or transverse fluid-attenuated inversion recovery (FLAIR) images (TR 9,000 ms, TE 105 ms, inversion time 2,200 ms, one excitation). All MRIs were reviewed by two authors (MSvdK and FB) in consensus. The MRIs were analyzed for localization, extent, and appearance of the cerebral white matter abnormalities in order to compare these findings to the quantitative MR parameters, as described below.
DTI was performed in three patients (patients 2, 4, and 5) on several occasions with a multislice echo planar imaging (EPI) sequence with optimized gradients according to the method described by Jones et al. [12] using a reference b = 0 s/mm2 and eight noncollinear gradient vectors with b = 1,044 s/mm2. In transverse orientation, 16 slices of 5 mm were acquired, with a 128 × 128 matrix, using TR = 3,600 ms and TE = 123 ms. The DTI analysis included a correction of eddy current-induced distortion and calculation of eigenvalues of the diffusion tensor, resulting in a fractional anisotropy (FA) map and an apparent diffusion coefficient (ADC) map. In patient 2 at age 15 years 7 months, no DTI but only diffusion-weighted imaging was performed, using an EPI sequence with b values of 0, 500, and 1,000 s/mm2 (TR 5,100 ms, TE 137 ms), yielding an automatically generated ADC map (but no FA map).
MTI was performed in four patients (patients 1, 2, 4, and 5) on several occasions with a 3D fast low-angle shot sequence. Two sets of images were obtained with (M S) and without (M 0) MT saturation pulse (7.68 ms Gaussian radio frequency pulse, 1,500 Hz off-resonance), using TR = 23 ms, TE = 4 ms, flip angle 20°, and a 3D slab consisting of 54 transverse slices of 3 mm. MT ratio (MTR) maps were created according to MTR = 1 − M S/M 0.
Quantitative MR spectroscopy was performed in four patients (patients 1, 2, 4, and 5) using a short-echo time stimulated echo acquisition mode (STEAM) sequence (TR/TE/mixing time = 6,000/20/10 ms, 64 accumulations). Single-acquisition reference measurements without water suppression were acquired additionally to enable eddy current correction. Spectra were acquired from one or two volumes of interest (VOIs; 4–6 ml) within parietal and/or frontal white matter (in case of patients, all in affected white matter). The VOIs were positioned on the conventional MR images, taking care to avoid or minimize contamination by cerebrospinal fluid and gray matter. Metabolite concentrations were calculated using LCModel [13] and expressed as millimoles per liter VOI. Concentrations were determined for the metabolites total creatine and phosphocreatine (tCr), total N-acetyl-aspartate and N-acetyl-aspartyl-glutamate (tNAA), choline-containing compounds (Cho), myo-inositol (Ins), and lactate (Lac). MR spectroscopy in the other two patients (patients 3 and 6) had been performed with STEAM sequence as well (TR 2,500 ms), but before regular calibration of the scanner for absolute quantification was performed. The changes in MRS metabolites were observed qualitatively in these two patients.
Regions of interest (ROIs) corresponding to the MR spectroscopy VOIs were transferred to the equivalent ADC, FA, and MTR maps and mean ADC, FA, and MTR in these ROIs were determined. Mean values ± standard deviations were determined for the patients and controls.
Classification
The discriminant functions for four white matter disease groups (hypomyelination, demyelination, myelin vacuolation, and cystic degeneration) from the linear discriminant analysis, given in the appendix of a previously published paper [8], were used to determine into which group the patients who underwent all quantitative investigations (patients 2, 4, and 5) were classified when seven MR parameters (MTR, ADC, tCr, tNAA, Cho, Ins, and Lac) were substituted into these functions.
Results
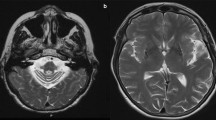
MRIs (ages 3 and 6 years) in patient 1 showed symmetric signal abnormalities in the cerebral white matter, with mild swelling and frontal predominance. Despite serious clinical deterioration and demise soon after the second MRI, the white matter abnormalities showed only minor increase (Fig. 1a, b). In patient 2, the initial MRI at the age of 2 years showed signal abnormalities in the periventricular and deep cerebral white matter without swelling and without frontal predominance. During a follow-up of 18 years, during which the patient clinically remained remarkably stable, the frontal white matter abnormalities showed some increase and became more marked than the parieto-occipital white matter abnormalities, although the cerebral white matter abnormalities remained limited in their extent in all areas. In patient 3, three MRI studies (ages 6–10 years) showed bilateral frontal white matter abnormalities with swelling (Fig. 1c, d). The only change regarding the white matter abnormalities over time was that the FLAIR images revealed a progressive decrease of signal intensity, indicating white matter rarefaction and cystic degeneration. During this time, the patient deteriorated seriously and died shortly after the third MRI. In patient 4, three MRI studies (4–8 years) demonstrated extensive cerebral white matter abnormalities with swelling and frontal preponderance. FLAIR images show evidence of white matter rarefaction and cystic change. Over time, the cerebral white matter signal abnormalities showed some increase in extent. The most marked change was progressive white matter atrophy with secondary dilatation of the lateral ventricles. This patient showed moderate clinical deterioration during this time. In patient 5, extensive areas of abnormal signal in the white matter were found on MRI at 6 years with a mild frontal predominance. Follow-up MRI after 2 years was unchanged. The patient was clinically stable. MRI in patient 6 revealed symmetric frontal white matter abnormalities almost restricted to the frontal and parietal lobes at 4 years. After 1 year, only some increase in white matter swelling was observed. This patient experienced serious clinical deterioration and died.

Transverse T2-weighted images (3,000/22, 60 and 120, one excitation) in two patients. MRIs at ages 3 (a) and 6 years (b) in patient 1 showed symmetric signal abnormalities in the cerebral white matter, with mild swelling and a frontal predominance. The white matter abnormalities showed only minor increase on follow-up. In patient 3, two MRI studies at age 8 (c) and 10 years (d) showed little change in the bilateral frontal white matter abnormalities with swelling
A total of 17 MRS measurements were obtained in the abnormal white matter of the four patients, six in the (most affected) frontal white matter and 11 in the parietal white matter. A proton MR spectrum of patient 5 is depicted in Fig. 2 for frontal white matter. The quantified metabolite concentrations of patients 1, 2, 4, and 5 are shown in Table 2. Main findings were a marked reduction of the concentration of tNAA in both frontal and parietal regions (progressive over time), a strong elevation of the concentrations of Ins (increasing over time in most patients) in both regions, and accumulation of Lac in all patients. Furthermore, Cho concentrations were strongly elevated in most measurements. The level of tCr was elevated when Ins was increased. Similar spectroscopic changes were observed qualitatively in patients 3 and 6, in whom we did not have the possibility of quantification of the metabolite concentrations.

VOI localization for MRS (STEAM TR/TE/TM = 6,000/20/10 ms, 64 accumulations) on the transverse T2-weighted image (a) (3,000/22, 60 and 120, one excitation), and equivalent transverse ADC (b), FA (c), and MTR (d) maps with the corresponding spectrum (e) in patient 5: high ADC, decreased FA and MTR, and a decrease in tNAA and increases in Cho, Ins, and Lac are evident. Note focal areas of lowered ADC values in the parietal white matter
A total of eight MTI and five DTI measurements were obtained in the white matter of the patients. Examples of MTR, ADC, and FA maps that were obtained in patient 5 are shown in Fig. 2. MTR, ADC, and FA values in ROIs corresponding to the spectroscopy VOIs are shown in Table 2. In the white matter of all Alexander patients, high values of ADC were observed, and both FA and MTR were attenuated. Although overall ADC is increased in affected white matter, we observed some regional heterogeneity. In patients 4 and 5, a thin periventricular rim of lowered ADC values was observed in the parietal white matter (Fig. 2b).
Substituting the seven quantitative MR parameters MTR, ADC, tCr, tNAA, Cho, Ins, and Lac as measured in the frontal or parietal regions for patients 2, 4, and 5 in the discriminant functions for the four white matter disease groups of the linear discriminant analysis [8] resulted in classification of these patients into the demyelination group in all patients at each time point.
Discussion
The nature of the pathology underlying the white matter abnormalities in Alexander disease is not clear. Histopathology shows a lack of myelin in the affected white matter, variably interpreted as hypomyelination or demyelination [6, 7]. In this study, we applied serial MRI to visualize structural abnormalities, proton MR spectroscopy to assess metabolic abnormalities, and MTI and DTI to determine MTR, ADC, and FA values in the white matter of several patients with juvenile Alexander disease in order to obtain better insight into the nature of the pathology.
MR spectroscopy showed highly elevated Ins, Lac, and Cho levels and decreased tNAA levels in our patients. In the context of a white matter disorder, such findings are generally interpreted as suggestive of active demyelination [14–16]. Multiple studies in demyelinating diseases have reported that MR spectroscopy of the affected white matter is characterized by decreased NAA and elevated Cho, Ins, and Lac [14–16]. These metabolic changes are interpreted as evidence of axonal damage or loss, enhanced membrane turnover or accumulation of myelin breakdown products or both, astrogliosis, and infiltrating macrophages, respectively [14–16]. In addition, we found high ADC and decreased MTR an FA values in the abnormal white matter.
We applied discriminant functions determined in a previously published paper [8], in which pathophysiological mechanisms underlying white matter diseases were classified into four categories using quantitative MR parameters: demyelination, hypomyelination, myelin vacuolation, and cystic degeneration. With this method, the white matter pathology in the present patients was predicted to be demyelination rather than hypomyelination.
Conventional MRI showed cerebral white matter abnormalities with frontal preponderance in our patients, the extent of which was remarkably stable on follow-up, even at prolonged follow-up, irrespective of the clinical progression of the disease, which was serious in some cases. This finding is in line with previous MRI studies on Alexander disease [17, 18] and, in fact, not compatible with active demyelination as sole cause of the clinical deterioration and the white matter abnormalities on MRI. In diseases characterized by active demyelination, for instance metachromatic leukodystrophy and X-linked adrenoleukodystrophy, in which the aforementioned MR spectroscopic abnormalities are typically found [15–18], MRI reveals that the extent of the white matter abnormalities increases over time, sometimes rapidly. Therefore, sequential MRI in juvenile Alexander disease provides strong evidence against active demyelination as sole pathophysiological mechanism, although, of course, we cannot exclude the possibility of low-grade myelin loss as part of the pathology.
The question is what the alternative explanation could be for our spectroscopic findings. It should be noted that MR spectra supposedly suggestive of demyelination are also observed in low-grade gliomas [19]. The decreased density or dysfunction of axons in these tumors is reflected in low NAA. The increased membrane density and turnover is reflected in increased Cho. Ins is mainly present in astrocytes and with that increased in tumors of increased glial density. Elevated Lac in gliomas is explained by the reduced oxygenation by impaired blood flow within the tumor. The question is whether gliomas and Alexander disease may have something in common.
Alexander disease is a disease of astrocytes. Histopathology of the affected white matter demonstrates proliferation and hypertrophy of astrocytes that accumulate Rosenthal fibers [7]. The disease is not so different from a circumscribed or diffusely infiltrating low-grade glioma. Some patients with Alexander disease actually display tumor-like lesions, especially in the brain stem, but sometimes also elsewhere [20]. The pathologic similarities between Alexander disease and gliomas may be so striking that diagnostic problems arise, especially in focal brain stem tumors, also because some low-grade gliomas, for example in a pilocytic astrocytoma, may contain Rosenthal fibers. However, despite some intriguing similarities, pseudotumoral forms of Alexander disease are “self-limiting” and ultimately regressive leading to atrophy, unlike true neoplastic processes.
The MR spectroscopic findings in Alexander disease allow an alternative interpretation. The marked Ins elevation can be ascribed to the hyperplasia and hypertrophy of astrocytes. The elevation of Cho can be ascribed to increased membrane density of the affected white matter. The elevated lactate in the affected white matter, observed in all our patients, could be related to metabolic disruption of disordered astrocytes giving rise to nonoxidative glycolysis [19].The low NAA can be explained by the decreased density of nerve fibers in the swollen white matter full of astrocytes.
MTR and DTI provide information about tissue microstructure. The MTR in the white matter of our patients was decreased, indicating a reduced capacity of the macromolecule-bound protons in brain tissue to exchange magnetization with the surrounding protons in free water, probably reflecting lack of myelin and damage of axonal membranes. Within the abnormal white matter, ADC, a measure for water diffusivity, irrespective of direction, was increased in patients, and FA, a measure for the degree of diffusion anisotropy, was decreased. Both observations in white matter indicate damage to the tissue matrix. So far, the findings are as seen in other leukoencephalopathies [8]. Interestingly, however, we observed decreased ADC values in a periventricular rim in the parietal region in two patients. This may be explained by a high density of astrocytes stuffed with Rosenthal fibers, which are preferentially located in the periventricular white matter, which would hamper the diffusion. Similarly, it is known that diffusion is restricted in gliomas with increased cellularity [21].
A limitation of our study is the rather small number of patients and MR studies, which is related to the rarity of the diseases. This precludes analysis of changes over time for MTR, ADC, and FA values and MRS metabolite concentrations. It should also be noted that we only investigated patients with the juvenile form of the disease. The results of our study may not be applicable to all forms of the disease. In the infantile form, myelin build-up may be predominantly affected, leading to hypomyelination, whereas in adult forms, the cerebral white matter is often normal.
Conclusion
In conclusion, the results of our study suggest that abnormalities of the white matter in juvenile Alexander disease are not related to active demyelination, although we cannot exclude minor low-grade demyelination as part of the pathology. Hyperplasia and hypertrophy of astrocytes with Rosenthal deposition probably explain the MR spectroscopic findings.
References
Brenner M, Johnson AB (2001) Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat Genet 27:117–120. doi:10.1038/87020
Li R, Messing A, Goldman JE, Brenner M (2002) GFAP mutations in Alexander disease. Int J Dev Neurosci 20:259–268. doi:10.1016/S0736-5748(02)00019-9
Van der Knaap MS, Naidu S, Breiter SN et al (2001) Alexander disease: diagnosis with MR imaging. AJNR Am J Neuroradiol 22:541–552
Van der Knaap MS, Salomons GS, Li R et al (2005) Unusual variants of Alexander’s disease. Ann Neurol 57:327–338. doi:10.1002/ana.20381
Barkovich AJ, Messing A (2006) Alexander disease, not just a leukodystrophy anymore. Neurology 66:468–469. doi:10.1212/01.wnl.0000200905.43191.4d
Klein EA, Anzil AP (1994) Prominent white matter cavitation in an infant with Alexander’s disease. Clin Neuropathol 13:31–38
Graham DI, Lantos PL (2002) Greenfield’s neuropathology. Arnold Publisher, London, pp 491–494
Van der Voorn JP, Pouwels PJ, Hart AA et al (2006) Childhood white matter disorders: quantitative MR imaging and spectroscopy. Radiology 241:510–517. doi:10.1148/radiol.2412051345
Van Buchem MA, Steens SCA, Vrooman HA et al (2001) Global estimation of myelination in the developing brain on the basis of magnetization transfer imaging: a preliminary study. AJNR Am J Neuroradiol 22:762–766
Engelbrecht V, Scherer A, Rassek M et al (2001) Diffusion-weighted MR imaging in children: findings in the normal brain and in the brain with white matter disease. Radiology 222:410–418. doi:10.1148/radiol.2222010492
Pouwels PJ, Brockmann K, Kruse B et al (1999) Regional age dependence of human brain metabolites from infancy to adulthood as detected by quantitative localized proton MRS. Pediatr Res 46:474–482. doi:10.1203/00006450-199910000-00019
Jones DK, Horsfield MA, Simmons A (1999) Optimal strategies for measuring diffusion in anisotropic systems by magnetic resonance imaging. Magn Reson Med 42:515–525. doi:10.1002/(SICI)1522-2594(199909)42:3<515::AID-MRM14>3.0.CO;2-Q
Provencher SW (1993) Estimation of metabolite concentrations from localized in vivo proton MR spectra. Magn Reson Med 30:672–679. doi:10.1002/mrm.1910300604
De Stefano N, Filippi M (2007) MR spectroscopy in multiple sclerosis. J Neuroimaging 17:31S–35S
Pouwels PJ, Kruse B, Korenke GC et al (1998) Quantitative proton magnetic resonance spectroscopy of childhood adrenoleukodystrophy. Neuropediatrics 29(5):254–264. doi:10.1055/s-2007-973571
Kruse B, Hanefeld F, Christen HJ et al (1993) Alterations of brain metabolites in metachromatic leukodystrophy as detected by localized proton magnetic resonance spectroscopy in vivo. J Neurol 241(2):68–74. doi:10.1007/BF00869766
Bobele GB, Garnica A, Schaefer GB et al (1990) Neuroimaging findings in Alexander’s disease. J Child Neurol 5:253–258
Imamura A, Orii KE, Mizuno S et al (2002) MR imaging and H-MR spectroscopy in a case of juvenile Alexander disease. Brain Dev 24(7):723–726. doi:10.1016/S0387-7604(02)00080-3
Imamura K (2003) Proton MR spectroscopy of the brain with a focus on chemical issues. Magn Reson Med Sci 3:117–132. doi:10.2463/mrms.2.117
Hirayama T, Fukae J, Noda K et al (2008) Adult-onset Alexander disease with palatal myoclonus and intraventricular tumour. Eur J Neurol 15(2):e16–e17
Bode MK, Ruohonen J, Nieminen MT et al (2006) Potential of diffusion imaging in brain tumors: a review. Acta Radiol 47(6):585–594. doi:10.1080/02841850600580598
Acknowledgments
The study was supported by the Dutch Organization for Scientific Research (NWO, Grant 903-42-097) and the Optimix Foundation for Scientific Research.
Conflict of interest statement
We declare that we have no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
van der Voorn, J.P., Pouwels, P.J.W., Salomons, G.S. et al. Unraveling pathology in juvenile Alexander disease: serial quantitative MR imaging and spectroscopy of white matter. Neuroradiology 51, 669–675 (2009). https://doi.org/10.1007/s00234-009-0540-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00234-009-0540-9