
Abstract
Purpose
The aim of non-interventional studies (NIS) with medicinal products is to investigate the use of authorized medicinal products in daily routine. In the past, this type of study has been subject to frequent criticism, and many recommendations have been published. The aim of our study was to assess the quality of NIS study protocols.
Methods
Nearly all NIS study protocols submitted to the German Federal Institute for Drugs and Medical Devices (BfArM) within a period of one year could be analyzed. The protocols were evaluated in terms of objectives, methods and included patients, as well as with regard to their compliance with quality recommendations for NIS by federal authorities and pharmaceutical industry associations.
Results
The 136 NIS available for study were scheduled to enroll approximately 330,000 patients (2,500 patients per study) and 43,000 healthcare professionals. Of these NIS, 58 % were performed with medicinal products that had been authorized within the past 5 years; however, 68 % of the investigated active pharmaceutical ingredients were older than 5 years, and 19 % were even older than 19 years. Only 56 % of the protocols provided information on publication policy, and 65 % required the involvement of ethic committees. The adherence to current quality recommendations was average, but the compliance of NIS performed by member companies of the Association of Research-Based Pharmaceutical Companies was significantly higher than that of other sponsors.
Conclusions
Current quality recommendations are still not fully implemented in most NIS protocols. Therefore, the scientific value of many NIS is still questionable, and the criticism that NIS are mainly conducted for marketing reasons could not be refuted by the data analyzed here.
Similar content being viewed by others


Explore related subjects
Find the latest articles, discoveries, and news in related topics.Avoid common mistakes on your manuscript.
Introduction
In the European Union (EU), a marketing authorization is the legal prerequisite for placing a new medicinal product on the market. In order to be granted this authorization, the manufacturer has to prove the efficacy and safety of his product by using controlled, randomized Phase II and III clinical trials [1]. The high internal validity of the results of properly conducted randomized clinical trials (RCTs) is well accepted; consequently, RCTs have become the gold standard for proving the efficacy of medicinal products [1–3]. However, there are certain questions which cannot be answered by randomized trials—or only with enormous and disproportional effort [2, 3]. Due to the fact that clinical trials are performed under strictly controlled conditions and protocols, they can provide only limited information about the usage of a medicinal product in daily routine [3, 4]. This includes, for example, incomplete information about medical prescription practice, dosage adaptations by the physician or patients’ compliance [5]. As a result of the strict inclusion and exclusion criteria, the patients of a clinical trial are usually not a fully representative sample of the actual population, and certain populations are underrepresented [2, 3, 5]. Therefore, the information about the influence of concomitant diseases and medications typically gained in such trials can only be incomprehensive [3, 6]. Since clinical trials are subject to time constraints and contain small study populations, only limited data can be collected on long-term safety and effectiveness, as well as on rare adverse drug reactions (ADR) [1, 2, 4, 5].
In the Clinical Trials Directive 2001/20/EC, the EU has legally defined the non-interventional study (NIS) as opposed to the clinical trial. Article 2c defines the term “non-interventional” as follows: “a study where the medicinal product(s) is (are) prescribed in the usual manner in accordance with the terms of the marketing authorization. The assignment of the patient to a particular therapeutic strategy is not decided in advance by a trial protocol but falls within current practice and the prescription of the medicine is clearly separated from the decision to include the patient in the study. No additional diagnostic or monitoring procedures shall be applied to the patients and epidemiological methods shall be used for the analysis of collected data” [7]. The German legislative authority transferred the NIS definition into national law in 2004 with the 12th AMG (German Medicinal Products Act) amendment [8]. It has become obligatory in subsequent amendments to the AMG to notify the German federal competent authorities prior to commencement of an NIS [9]. The legal specifications for NIS predominantly allow observational studies to be conducted with the aim of gaining scientific knowledge on such subjects as long-term safety rather than efficacy/effectiveness, rare ADR, the usage of a medicinal product in daily routine or the influence of concomitant diseases and medications, among others. While clinical trials focus on efficacy, NIS tend to focus more on effectiveness, which is efficacy under real-life conditions. Interventions differing from clinical routine or randomization of patients’ treatment are not consistent with the legal requirements.
NIS have been criticized frequently over the past years. They have been accused of poor scientific quality, and it is alleged that the pharmaceutical companies as sponsors of these studies are more interested in marketing their medicinal products than in gaining additional scientific knowledge [3, 5, 10–15]. As a consequence of this criticism, there have been different attempts in Germany to raise the quality of the NIS. Starting in 1998, the Federal Institute for Drugs and Medical Devices (BfArM), the German higher federal authority for authorization of most of the medicinal products for human use in Germany, published recommendations for planning and conducting NIS [16]. These recommendations were revised in the course of the AMG amendments in cooperation with the Paul-Ehrlich Institut (PEI), published for public consultation in 2007 and finalized after minor modifications in 2010 [17–19]. This compilation includes information about the definition and possible objectives of NIS, about the observational plan, the analysis plan, quality management, patient’s consent and education, conflicts of interests, duty to notify and recommendation to report, publishing, archiving and finally reimbursement of the participating healthcare professionals (HCPs). The “Association of Research-Based Pharmaceutical Companies” (VFA) and the organization for “Voluntary Self-regulation for the Pharmaceutical Industry” (FSA) have also both published recommendations and procedures for NIS which are binding for their member companies. The VFA recommendations amplify the 2007 recommendations of BfArM and PEI and were also published in 2007. They include additional recommendations for the internal responsibility for the study, patient’s informed consent, involvement of independent ethics committees, publication policies for study results, as well as public study registers and the need to establish standard operating procedures for planning and conducting NIS [18, 20]. The FSA code requires following the BfArM and PEI recommendations and requests additional procedures for conducting NIS. These procedures include the same recommendations as those of the VFA but additionally require obligatory scientific objectives. Furthermore, they contain advice on quality management, content and function of the observational plan, analysis of the results and reimbursement of the participating HCP [21]. In addition to these, there have been many other publications with recommendations for enhancing the quality of NIS, including guidelines and recommendations for assurance of good epidemiological practice [4, 5, 13, 22–26].
In view of the continuing criticism of NIS, it is unclear whether the numerous recommendations are being followed and whether an improvement in NIS design can be observed. Therefore, the aim of the study reported here was to investigate the quality of NIS by analyzing their observational plans in a nearly complete cross-sectional analysis. Until 2009 the competent authorities (BfArM or PEI) in Germany had only to receive notification of an NIS—and then only if the NIS were performed on behalf of pharmaceutical entrepreneurs. The legal basis for such notifications did not include the obligation to also submit the observational plan. This was changed in the 15th amendment of the German Medicinal Products Act in 2009. Since then, pharmaceutical entrepreneurs are also obliged to submit the observational plan together with the notification of a NIS [27]. This change in legislation opened the door to analyses of observational plans of NIS sponsored by pharmaceutical companies by non-participating third parties and/or without major selection bias.
Methods
Study design
This study evaluated the quality of NIS by analyzing the content and quality of their respective observational plans. The goal was to analyze the observational plans of all NIS submitted to the BfArM within 1 year after the new legal requirements for NIS came into force in Germany. All available observational plans were reviewed according to a standardized review procedure, and the scientific quality evaluation was performed using a predefined scoring system. The authors who performed the investigation are organizationally in a department where they are not involved in—and thus independent of—the BfArM NIS assessment. Even though this study is not an observational study in the classical sense, the reporting of the results is based on the STROBE statement [28].
Setting
All NIS of which the BfArM was notified during the period from September 1, 2009 to August 31, 2010 and for which the BfArM is the competent authority were included in the investigation. NIS that commenced prior to September 1, 2009, report updates and amendments and extensions of ongoing NIS that were started prior to September 1, 2009 as well as NIS for which the PEI is the competent authority were excluded.
Data set selection/study size
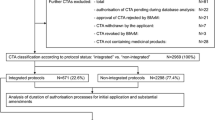
A total of 387 submissions were reviewed and assessed for inclusion and exclusion criteria; 136 NIS of these remained evaluable and were subsequently included in the analysis (Fig. 1).

Data set selection/study size. All documents associated with the topic non-interventional studies (NIS) and collected in the chosen time period were selected from the archive of the Federal Institute for Drugs and Medical Devices (BfArM). The resulting 387 documents were screened. Three documents were excluded since they were not related to an NIS. Of the remaining 384 documents, 243 NIS documentations were excluded for the following reasons: the NIS were not new notifications but additional information on running studies (n = 206); repeated notifications of already notified NIS (n = 21); NIS that had been commenced prior to September 1, 2009 (n = 8); the NIS was not within the competence of the BfArM (n = 8). Of the 141 studies that were not excluded, five NIS were not available for review due to in-house processes
Variables
A standardized data set was assessed for each NIS. Aside from administrative data, the variables in Table 1 were recorded and entered into the study database. Variables were either encoded dichotomously (“yes”, “no”), as respective numbers or as free text. Data not available in the NIS protocol were recorded as missing data (“not available”).
Data sources
The sponsoring companies of each NIS were categorized in accordance with their membership in the VFA [29]. It was also checked whether the NIS had actually been registered in the NIS register of the VFA as planned [30]. The German federal drug information system (AMIS) was used to gain information on the date and status of marketing authorization of the medicinal product, the respective active pharmaceutical ingredients (API) and the originator of the medicinal product(s) with the respective API [31]. For further classification of the API, the Anatomical Therapeutic Chemical (ATC) classification system was used [32]. The summary of product characteristics (SmPC) of the specific medicinal product was used to compare the dosage information and indications given in the observational plan with those given in the SmPC.
Measurements
An evaluation was performed if the diagnostic and therapeutic procedures of the NIS and the documentation requested from the HCPs were consistent with usual medical practice and if no additional diagnostic or monitoring procedures were applied (in accordance with the requirements of European Directive 2001/20/EC and the applicable national legal requirements) [7]. The assessment of compliance of the diagnostic and therapeutic procedures with clinical practice was performed by both authors jointly in a semi-blinded design: the senior expert who performed the major rating was blinded with respect to the company and the specific product, while the second author presented the setting. The assessment was based on the SmPC, German and European expert guidelines and recommendations of German and European scientific societies. A four-point scale was used to rate the adherence to these guidelines: completely consistent with current medical practice (procedures performed completely within the guidelines); partially consistent with current medical practice (additional procedures were only performed if decided by the HCP and could be counted as current medical practice); partially not consistent with current medical practice (additional procedures were only performed if decided by the HCP but procedures could not be counted as current medical practice); not consistent with current medical practice at all (additional procedures required by the protocol). The quality of the observational plans was rated using a specially developed scoring system. The observational plans could reach a score of 25 by fulfilling requirements (score items) of the recommendations for developing and conducting an NIS published by the competent authorities (BfArM and PEI) [17]. A “bonus” score of up to 6 could be reached if the extra VFA and FSA recommendations, i.e, over and above those of the BfArM/PEI, were followed [20, 21]. The score items are depicted in detail along with the results in Table 2. Each score item was weighted equally. In cases where no data on a certain score item were provided in the observational plan, the score item was counted as not fulfilled. A maximum score of 25 (+ 6) was assigned when all requirements of the BfArM/PEI and VFA/FSA recommendations were addressed and fulfilled.
Statistical methods
The statistical analysis was performed using the IBM© SPSS© Statistic software package ver. 20 for Microsoft Windows operating systems (SPSS, Chicago, IL). Categorical variables were evaluated using absolute numbers, frequencies and percentages, while discrete variables were evaluated using numbers, frequencies, mean, minimum, maximum, percentile and median. On the basis of free-text variables, a division into subgroups was performed. A non-parametric Mann–Whitney U test was carried out to compare the recommendation compliance of member companies of the VFA with that of non-member companies.
Results
Sponsors, study location and study duration
The 136 reviewed NIS were initiated by 73 different sponsors, of which. 96 % were pharmaceutical companies that conducted nearly 98 % of the NIS under review. Three studies were submitted by non-pharmaceutical companies, two of them by hospitals and one by a contract research organization (CRO) which did not provide information on the sponsoring client. Approximately one-fourth of the sponsors were VFA member companies and initiated approximately 35 % of the NIS. Geographic information on centers was provided in approximately 90 % of the observational plans: 68 % of the NIS were supposed to be conducted as multicenter NIS with no regional restriction in Germany, 2 % were located in one German Federal Land only and 4 % of the NIS were conducted in a mono-centric setting. Approximately 20 % of the NIS were reported as multinational studies. All observational plans, with two exceptions, contained information on the planned total study duration. One-fourth of the studies had been designed to last more than 24 months, around 35 % to last between 13 and 24 months and 38 % of the protocols provided for a study duration of 12 months or less. The distribution of the individual observational period for each patient is presented in Table 3 (13 % of the protocols did not provide data on this topic). For 48, 22, 6 and 6 % of the NIS, up to three, four to six, seven to nine and more than nine visits per patient were scheduled, respectively, while 18 % of the protocols did not provide any information on the number of visits per patient.
Publication policy
Of the investigated protocols, 56 % provided information on the publication policy. A statement that a publication of the results was at least planned was contained in 43 % of the NIS protocols; an additional 11 % stated that a publication was planned within a certain time period, and 7 % contained statements that the results would be reported to the participating HCP. One-fourth (26 %) of the protocols provided for study registration in public study registries and 50 % in the VFA register for NIS [30]. Review of this register revealed that one of the NIS for which a registration in that register had been announced was recorded with information on the start of the study which was not consistent with the observational plan.
Data protection, informed consent and ethics committees
Patient informed consent was required in 75 % of the NIS, while 7 % explicitly stated that no patient informed consent was required; the remaining 18 % of the NIS did not provide information on this topic. A pseudonymized transfer of patient-related data from the investigator to the sponsor/CRO was scheduled in 73 % of the NIS protocols, while 17 % of the protocols reported a complete data anonymization before data transfer; 10 % of the protocols did not provide information on data protection and data transfer. Interestingly, only 81 % of the NIS reporting a pseudonymized data transfer (73 %) also provided for an informed consent. Of the 44 % of the NIS protocols that specified monitoring of the study in the observational plan, less than half had stipulated an informed consent of the patients on this subject. Approximately two-thirds (68 %) of the NIS protocols contained information on the involvement of an ethics committee, with 65 % requiring the opinion of at least one ethics committee while 2 % explicitly declined the involvement of any ethics committee. Two protocols of internationally performed NIS stated that the opinion of an ethics committee would be requested if demanded by national legislation but did not provide information on whether an opinion was requested in Germany. For 10 % of the NIS reviewed, the favorable opinion of an ethics committee was attached to the study protocols.
Study design
In terms of study design, 85 % were prospective, 2 % were retrospective and 13 % had a prospective design with retrospective parts. The settings were multicentric in 93 % of the NIS and monocentric in 4 %; five protocols did not provide information on this topic. In terms of number of study arms, 95 % of the NIS were performed as a single-arm study, and the remainder were performed as two-arm or even multiple-arm studies. Thirteen percent of the investigated NIS described an additional subgroup analysis. The rationale for choosing a NIS design was not discussed in 65 % of the studies.
Recruitment and sample size
Provisions regarding the mode of patient recruitment were included in 19 % of the NIS protocols, and 41 % provided information on the method of recruiting the participating HCP. Information on the location where the investigation was scheduled to take place was provided in 66 % of the NIS. Most of the NIS were performed in private practices exclusively (38 %) or in private practices and hospitals (23 %). Only 4 % of the NIS were exclusively performed in hospitals, and two NIS were planned to be performed in pharmacies only. Information on the specialization of the participating HCPs was provided in 70 % of the protocols, with the majority focusing on general practitioners and internists [for additional information, see Electronic Supplementary Material (ESM) 1]. All protocols except for one defined the number of participating HCPs, and 88 % provided a sample size for the patient population, with 43 % of the protocols defining the number of patients per HCP. The distribution is presented in Table 3. The 136 reviewed protocols included over 330,000 patients examined by approximately 43,000 HCPs. A justification of the sample size was provided by 58 % of protocols, but a sample size calculation by effect estimation was performed in only 11 % of the NIS; 35 % justified the sample size by the width of a predefined confidence interval and 12 % justified the sample size by simply declaring their sample size as appropriate for answering the study objectives.
Medical indications
With regard to the inclusion process for patients, 95 % of the NIS provided data on the stage of treatment in which the patients should be recruited for the study. In most of the NIS, the patients were supposed to be recruited at the beginning of the treatment with the investigated medicinal product (68 %), in 35 % of the NIS, the patients were to be recruited during therapy, in 14 % after a change in therapy and in one case the intention was to recruit the patients after therapy (multiple answers were possible). Data on inclusion and exclusion criteria for patients were provided by 81 and 68 % % of the NIS, respectively. The most frequently specified inclusion criteria were presence of a certain medical condition (66 %) followed by patient characteristics (e.g. gender, body weight, height; 45 % and a certain medical history of the patient (27 %). The presence of contraindications (39 %) followed by the presence of a certain medical condition (25 %) and concomitant therapies (24 %) were the most commonly specified exclusion criteria. Pregnancy or nursing were specified as exclusion criteria in 17 % of NIS, and patients with known hypersensitivity to one or more ingredients were excluded in 16 % of the NIS. A medical condition in the observational plan was specified in 93 % of NIS protocols which complied with the marketing authorization conditions specified by the SmPC in all instances. The remaining protocols did not provide a medical condition as inclusion criterion. The investigated medical conditions are presented in Fig. 2.

Medical conditions investigated by the NIS under review. NIS investigating malignant tumors were summarized in the category “Oncology” only and excluded from other categories (indicated with an asterisk)
Objectives
All NIS protocols provided information on the primary objective(s), whereas 62 % of the protocols discussed the scientific rationale for the study itself. Nearly one-quarter of the investigated NIS specified a new, currently not investigated objective as the reason for conducting the NIS. Around 10 % of the NIS were conducted either to investigate a larger collective and/or use in everyday life, to confirm results of prior studies or upon recommendation of competent authorities, 7 % were performed to observe new dosages, 4 % were long-term observations and 7 % of the rationales were imprecise and not clear. The objectives defined are presented in Table 4. Objectives were measured by number of the ADR (52 %), score items in the case report forms (CRF) measuring the success of therapy (48 %), results of laboratory tests or results of examinations (33 %), results of special questionnaires (28 %) or dosages (12 %) (multiple answers were possible). Information on the primary measurements was not specified in 12 % of all protocols.
CRF, questionnaires and data management
In 71 % of the NIS protocols, the CRF were filled in only by the HCP, in 27 % by the HCP and the patient and in one NIS only by the patient. Additional information on the content of the CRF is given in ESM 2 and information on the content of special questionnaires is given in ESM 3. Information on data management and quality assurance was provided by 91 % of the NIS. Some kind of monitoring was planned for nearly one-half of the NIS (44 %). Furthermore, 56 % of the protocols provided information on the archiving of the study data. Detailed information on data management, quality assurance and classification systems is given in ESM 4. About 93 % of the NIS specified data on statistical analysis procedures. Such data were often not very precise and contained only information on an analysis of distribution (68 %) or frequencies (63 %), or differentiated whether a descriptive (57 %) or an explorative analysis (22 %) was planned.
Medicinal products and API
Most of the NIS (80 %) investigated only one medicinal product containing one API; 10 % investigated a medicinal product which was a combination product with more than one API; 4 % investigated more than one medicinal product but products with the same API; 5 % investigated different medicinal products with different API. Approximately two-thirds of the NIS (66 %) investigated brand-name medicinal products (originator products), one-third investigated generic medicinal products and in two studies only the API was given in the study protocol.
Nearly one-third of the API were classified as antineoplastic and immunomodulating agents. Additional information on the distribution of the APIs according to ATC code is given in ESM 5. An analysis of the temporal relation between the year of first marketing authorization of the investigated medicinal product and the respective API is provided in Fig. 3.

Year of authorization of the medicinal product and active pharmaceutical ingredient (API) [31]. For combination medicinal products the year the combination was authorized for the first time was chosen as the year of marketing authorization of the API. If several medicinal products containing the same API were investigated, the year the newest medicinal product was granted marketing authorization was chosen. If medicinal products containing different API were investigated, the year the newest medicinal product was granted marketing authorization was chosen as well as that of the corresponding API. For an API without determination of a medicinal product, the year the newest medicinal product with this API was authorized was chosen. For a therapeutic regimen with different API without determination of distinct medicinal products, the year the newest API was authorized was chosen, and the year of authorization of the medicinal product was determined to be the year of authorization of the corresponding API
Compliance with current medical practice and recommendations
For 94 % of the NIS an evaluation of the compliance with current medical practice was performed; such an evaluation could not be performed for the other 6 % due to lack of information in the observational plan. Nearly one-half of the NIS (46 %) could be evaluated as being completely consistent with current medical practice, while approximately 38, 8 and 2 % were evaluated as being partially consistent, partially non-consistent and not consistent with current medical practice, respectively. The scores according to the recommendations of the German competent authorities (BfArM and PEI) are presented in Fig. 4 and Table 2, and the additional scores for the recommendations issued by the VFA and FSA are presented in Fig. 5 and Table 2 [17, 18, 20, 21]. A comparison of the results of member companies of the VFA and other pharmaceutical companies revealed that recommendation compliance by member companies of the VFA was significantly higher than that of other pharmaceutical companies (p < 0.001).


Discussion
In our study we analyzed the quality of 136 NIS protocols submitted to the BfArM over a 1-year period. The protocols analyzed could not be classified as high-quality studies because the studies only partially fulfilled the recommendations for conducting NIS by the BfArM and the VFA/FSA. Furthermore, a remarkably large number of patients was investigated. The API investigated included on the one hand expensive drugs and on the other hand also generic products which have been on the market for more than 5 years.
In contrast to earlier studies, this study investigated a representative sample since it was possible to investigate almost all NIS that had been submitted to the BfArM within the period of 1 year because the new legal regulations require the submission of observational plans for all newly notified NIS to the competent authorities in Germany [27]. As expected, most of these studies were conducted as multicenter studies, but only a small proportion was planned as multinational NIS. The entire study duration of nearly all NIS was shorter than 2 years, and the individual observation period for a single patient was less than or equal to 6 months. Overall, approximately 330,000 patients were scheduled for inclusion in all of the 136 NIS, with an average of approximately 2,500 patients per study, which is in the range of large Phase III clinical trials. All in all, more than 43,000 HCPs participated in the NIS. Although most of the investigated medicinal products were in the therapeutic fields of oncology and neurology, most of the NIS were conducted in the framework of private practices and with general practitioners or internists as investigators.
We evaluated the NIS with regard to their compliance with the recommendations of the BfArM, VFA and FSA. In the observed period of time, the scores of the NIS evaluated fell neither into the top nor into the lowest quintile, and most could be found in the middle quintile (Fig. 4). However, the recommendation compliance of the NIS performed by companies which were members of the VFA was significantly higher than that of those performed by other pharmaceutical companies. Interestingly, not even a VFA member company provided sufficient information in the observational plan to fulfill the six additional score items of the VFA/FSA recommendations, but at least two NIS sponsored by VFA member companies fulfilled five of the six additional score items. Overall, three-quarters of the NIS provided information in the protocol on compliance with only one or two additional score items (Fig. 5). The most frequently neglected—or at least not mentioned—VFA/FSA requirements were the score items “result reporting to the participating HCPs” and “definition and use of standard operation procedures for NIS”; each was missing from approximately 90 % of NIS. Upon closer inspection, the findings on many score items were disappointing. Only slightly more than one-third of the NIS provided a rationale for choosing a non-interventional design, and a little more than one-half of these provided information on archiving study data or on the publication policy. Furthermore, only about one-third of the NIS planned a registration of the study in a public register. The study designs also exhibited some shortcomings: only 11 % of the NIS protocols described a sample size calculation, only 5 % analyzed control groups and only 7 % provided data on confounder analysis.
Overall, the results of our study support the results of previous studies indicating a more or less unsatisfactory publication policy in NIS protocols, as already reported by Windeler and colleagues and de Mey in 2000 [33, 34]. In view of this poor publication policy it has to be seriously considered whether involved ethics committees should attach greater importance to this topic and should refuse a favorable opinion for protocols with such inadequate publication policies. This consideration also applies to other shortcomings of the study protocols.
In general, these findings might also be relevant to the competent authorities, but due to the legal situation the BfArM is currently not in the position to grant approval for NIS but is only notified of the fact that they are being conducted. Thus, freedom of research and quality of research may be in competition with each other to some extent . The internal assessment at the BfArM mainly ensures that the notified NIS are indeed non-interventional as opposed to being clinical trials which would require approval. As long as the study is truly non-interventional, the participating patients are by definition not exposed to a greater risk.
Despite the generally less favorable outcome regarding many score items, some results were more satisfying. For example, over two-thirds of the NIS planned the involvement of an ethics committee, and over 80 % provided data on study duration, observational period for each patient, number of visits of the patients, number of patients, number of HCPs and reported the survey instruments. It also has been recognized that over 90 % of the NIS provided data on quality assurance, data management and statistical analysis. Nevertheless, it would have been preferable if the information provided on these topics had been more detailed.
With respect to the primary objective of these NIS, it once again is obvious that the most frequently analyzed objectives (>70 %) were effectiveness/efficacy as well as safety and tolerability of the investigated medicinal products. This is in line with results published by Hasford and Lamprecht in 1998 [6] and confirmed by Dietrich in 2009 [10]. The authors of both studies discussed that NIS are usually not an appropriate scientific instrument for analyzing effectiveness/efficacy. Against this background it is worth mentioning that the 1998 BfArM recommendations on NIS listed “enhancing of findings regarding effectiveness” as a possible objective for an NIS, but this was omitted in the draft version in 2007 [16, 18].
Although NIS seem to be an appropriate instrument for detecting new ADR and for calculating ADR incidences, the role of controlled study designs has also been discussed frequently in the past. Hasford and Lamprecht [6] as well as Dietrich [10] stipulated that a control group should be included to adjust ADR rates. Even though over 70 % of the NIS investigated ADR as the primary or secondary objective, only 5 % of them included a control group. Other “typical” NIS objectives such as “everyday life experience” and “treatment compliance” were only reported in 41 and 13 %, respectively [6, 13, 18, 21].
It is remarkable that nearly one-third of the NIS analyzed the treatment with antineoplastic and immunomodulating agents which are generally expensive products. Similar results were published by Dietrich [10] and Müller 2009 [34] and supported by Ruppert and colleagues in 2012 [35], who stated that “oncology” was the medical field to be observed most frequently in NIS. The high cost of the treatments has to be borne by the respective insurance companies. It was also remarkable that 10 % of the NIS focused on only two biosimilar medicinal products, namely erythropoietin and interferon. These findings support the often-mentioned suspicion that NIS are conducted for marketing reasons and in order to generate a greater turnover.
More than one-half of the analyzed API were well-known API (on the market for more than 10 years), but in contrast more than one-half of the analyzed medicinal products were “newly introduced” products, i.e. they had been on the market for less than 5 years. In addition to this, 32 % of the medicinal products were non-originator products.
A closer look at the relationship between the year of marketing authorization of the medicinal product and the respective API yields a few surprising facts. Thus, 17 % of the NIS were conducted with new generic products up to 2.5 years after their market entry, but with API from 1998 or even earlier. Furthermore, 9 % of the NIS were conducted with originator medicinal products which had been on the market for approximately 10 years (1997–1999), and 10 % of the NIS were conducted with API which received their marketing authorization as early as 1978, the first year after Germany started requiring marketing authorizations [36]. Taking these findings together, it appears questionable whether it was really intended to gain new scientific data on API which have been on the market for longer than 10 years. This is all the more so as it is only allowed to conduct NIS within the authorized medical indication [8], and the simplified marketing authorization process for generic medicinal products is based on the assumption of a well-known safety and efficacy profile of such API. These doubts are supported by the observation that over two-thirds of the NIS that analyzed generic medicinal products focused on effectiveness/efficacy as at least one study objective.
When analyzing the number of scheduled patients it became obvious that nearly two-thirds of the 330,000 patients were included in only 10 % of the NIS, with each of the latter including more than 10,000 patients. A subgroup analysis revealed that nearly one-third of the group of patients was analyzed by only five NIS investigating X-ray contrast agents. Four of the respective contrast API received their marketing authorization between 1982 and 1995; only one API was on the market for less than 5 years. Against this background we cannot prove the suspicion false that a considerable number of the NIS were conducted primarily for marketing reasons.
Although the medical indication and the dosage information given for all NIS in the observational plan were compliant with the respective SmPCs, overall 10 % of the NIS were only partially or even non-consistent with current medical practice. In these cases, either the performed documentation or the clinical investigations were beyond the current usual medical practice, and the NIS were misused as a substitute for clinical trials. It has to be considered that on the one hand this is not in line with the legal requirements and on the other hand the respective healthcare insurance has to cover the costs for the extended diagnostics.
Not of relevance for costs and scientific quality but from an ethical point of view it remains remarkable that nearly 20 % of the NIS apply a pseudonymized data transfer from the investigator to the sponsor without written informed consent from the participating patients. This privacy issue may interfere with the patient–doctor relationship of trust and may further damage the reputation of NIS in the public opinion.
Our results are in contrast to those results of two studies published by Hahn and colleagues in 2008 and 2010 who reported that the VFA recommendations on NIS were implemented by the member companies [37, 38]. However, the results of these two studies cannot be compared directly to our results because the studies performed by Hahn and colleagues analyzed the recommendation adherence in planning and conducting of NIS by self-assessment by the pharmaceutical companies, while our study focused on the quality of single NIS protocols independently of the pharmaceutical companies.
Overall, the results of our study can be seen as a continuation of the criticism on NIS. While Hasford and Lamprecht identified poor conduct of NIS over the last two decades of the past century [6, 39], Dietrich renewed the criticism in 2009 [10]. This ongoing criticism stimulated the authorities as well as industry associations and others to publish detailed recommendations [4, 13, 16–18, 20–26] for NIS. Nevertheless, the results of our study have confirmed once again that despite these updated recommendations, the quality of NIS protocols remains in need of improvement.
Limitations
In this study, we performed a cross-sectional analysis of nearly all NIS study protocols submitted to the BfArM within a period of 1 year to determine the current state of NIS. However, our study does have some limitations.. During the data set selection and research phases, five NIS which should have been analyzed according to the inclusion criteria were not eligible for analysis due to in-house processes at the BfArM. Another limiting factor is that only NIS conducted and/or sponsored by pharmaceutical companies are subject to the obligation of submitting NIS protocols to the BfArM and could therefore be included in our study. There is still a larger number of NIS initiated by non-commercial sponsors which was not available for analysis.
Our results only show the findings based on variables for which information was provided in the observational plans. Thus, it might be possible that a certain NIS applied more variables or provided other measures, such as publication policies not mentioned in the observational plan. This is especially relevant for the scoring system in terms of being able to rate the compliance with BfArM/PEI and VFA/FSA recommendations. An NIS could only be scored if data on a score item was presented in the observational plan. It could happen that some studies fulfilled the score items of the recommendation (e.g. the definition of standard operating procedures) but could not be scored due to the missing description thereof in the observational plan. Nevertheless, it is considered good scientific standard as well as good clinical practice to include all relevant information on planning, conducting, reporting and archiving of a study in the respective protocol. Therefore, a lack of such a description in the protocol is considered to be a quality issue. Furthermore, it cannot be excluded that some protocols were changed after notification to the BfArM, possibly based on suggestions by the involved ethics committees.
With regard to the scoring system applied it should also be mentioned that all score items were rated equally. The discriminatory power of some score items was therefore poor in some cases because they were fulfilled either by nearly all or by hardly any of the NIS.
The assessment of the compliance of the diagnostic and therapeutic procedures with clinical practice was performed only in a semi-blinded design.
Conclusions and outlook
The results of this study once again demonstrate that the NIS conducted during the 1-year study period were only of a medium quality and that the recommendations which were published to enhance the quality of NIS have been only partially followed. On the positive side it has to be mentioned that NIS conducted by VFA member companies were of a relatively better—but still not of the highest—quality. More or less most of the NIS still analyze objectives less suitable for the study design type, and our results do not eliminate the suspicion that some of the NIS were conducted for marketing reasons.
It is disappointing that after all these years and publications and after criticism and published recommendations the potential of the NIS to be an important scientific instrument has still not been sufficiently exploited. As stated in the Introduction, the NIS could be a scientific instrument for complementing the results of RCTs. The NIS could contribute important data on the use of medicinal products in everyday life, such as on medical prescription practices, dosage adaptations or patients’ compliance and, with a suitable study design, on safety aspects. Instead, researchers and pharmaceutical companies are still trying to obtain data on objectives that have been determined as unsuitable for this type of study design in the past.
Based on the results of our analysis, we could not negate the current suspicion that some NIS are conducted for marketing reasons and not to gain scientific data; in fact, in some cases, such as with generic medicinal products, we were able to substantiate this suspicion. Against this background the future of NIS has to be discussed in a critical manner. The preservation of NIS as a scientific instrument is only reasonable if the pharmaceutical companies finally start to integrate all items of the recommendations in the planning of their NIS and stop using the NIS as marketing or pseudoscientific instruments, especially if ten thousands of patients are involved in the NIS. It may be helpful to consider increasing the competency of the authorities and/or ethics committees, but this would require further legal modifications.
Recently, the term “post-authorization safety studies” (PASS) was incorporated into European law, and these studies are now closer to NIS [40, 41]. It would be desirable, and it can only be hoped that in the course of enhancing and improving the quality of the PASS, the quality of the NIS will also be improved and enhanced.
References
Pockock SJ, Elbourne DR (2000) Randomized trials or observational tribulations? N Engl J Med 342:1907–1909
Black N (1996) Why we need observational studies to evaluate the effectiveness of health care. BMJ 312:1215–1218
Concato J, Shah N, Horwitz RI (2000) Randomized, controlled trials, observational studies, and the hierarchy of research design. N Engl J Med 342:1887–1892
Eberhardt R, Kori-Lindner C, Stammer H (2006) Nicht interventionelle Untersuchungen (qualifizierte Anwendungsbeobachtungen) [author’s translation: Non-interventional studies (qualified observational studies)]. Pharm Ind 68(5):542–550
de Mey C (2000) Sinn und Unsinn von Anwendungsbeobachtungen (Sense and nonsense in post-authorization surveillance). Med Klin 95:56–62
Hasford J, Lamprecht T (1998) Company observational post-marketing studies: drug risk assessment and drug research in special populations—a study-based analysis. Eur J Clin Pharamacol 53:369–371
European parliament and the Council (2001) Directive 2001/20/EC of the European parliament and of the council of 4 April 2001 on the approximation of the laws, regulations and administrative provisions of the Member States relating to the implementation of good clinical practice in the conduct of clinical trials on medicinal products for human use. Official Journal of the European Communities L121:34–44. Available at: http://ec.europa.eu/health/files/eudralex/vol-1/dir_2001_20/dir_2001_20_en.pdf. Accessed 12 Aug 2012
Section 4 sub-section 23 sentence 3 of the German Medicinal Products Act Version 2004 (AMG, Gesetz über den Verkehr mit Arzneimitteln, Arzneimittelgesetz, in der Fassung des Zwölften Gesetzes zur Änderung des Arzneimittelgesetzes vom 30.07.2004 BGBl. I S. 2031)
Section 67 sub-section 6 of the German Medicinal Products Act Version 1986 (AMG, Gesetz über den Verkehr mit Arzneimitteln, Arzneimittelgesetz, in der Fassung des Zweiten Gesetzes zur Änderung des Arzneimittelgesetzes vom 16.08.1986 BGBl. I S. 1296)
Dietrich ES (2009) Die meisten deutschen Anwendungsbeobachtungen sind zur Generierung wissenschaftlich valider Erkenntnisse nicht geeignet (Most post-marketing surveillance studies in Germany are not useful to generate valid scientific evidence). PharmacoEconomics 7:3–14
Bausch J (2010) Small gifts sustain sales. Dtsch Arztebl Int 107(22):390–391
Korzilius H (2007) Anwendungsbeobachtungen zwischen Marketing und Wissenschaft (Author’s translation: observational studies between marketing and science). Dtsch Arztebl 104(9):A528
Sickmüller B, Breitkopf S et al (2009) „Points to consider“zu Anwendungsbeobachtungen (author’s translation: „points to consider“with regard to observational studies). Pharm Ind 71(5):764–769
Dahlkamp J, Ludwig U (2005) Ärzte Betrug; das große Geschäft mit wertlosen Studien (author’s translation: fraud of physicians; big business with worthless studies) Spiegel Online http://www.spiegel.de/wissenschaft/mensch/0,1518,336730,00.html. Accessed 12 Aug 2012
Grill M (2007) Pharmaindustrie: Die Schein-Forscher (author’s translation: pseudo researchers). Available at: http://www.stern.de/wirtschaft/news/maerkte/pharmaindustrie-die-schein-forscher-581173.html. Accessed 12 Aug 2012
Federal Institute for Drugs and Medical Devices (1998) Bekanntmachung über die Zulassung und Registrierung von Arzneimitteln: Empfehlung zur Planung und Durchführung von Anwendungsbeobachtungen (Announcement regarding the authorization and registration of medicinal products: recommendations for planning and conducting of non-interventional studies). Bundesanzeiger Nr. 229:16884 (4 December 1998)
Federal Institute for Drugs and Medical Devices, Paul-Ehrlich-Institut (2010) Empfehlungen des Bundesinstituts für Arzneimittel und Medizinprodukte und des Paul-Ehrlich-Instituts zur Planung, Durchführung und Auswertung von Anwendungsbeobachtungen (Recommendations of the Federal Institute for Drugs and Medical Devices and of the Paul-Ehrlich-Institut for planning, conducting and analysis of non-interventional studies). Bundesinstitut für Arzneimittel und Medizinprodukte. Available at: http://www.bfarm.de/DE/Arzneimittel/1_vorDerZul/klinPr/nichtInterventPruef/nichtInterventPruef-node.html. Accessed 12 Aug 2012
Federal Institute for Drugs and Medical Devices, Paul-Ehrlich-Institut (2007) Gemeinsame Empfehlungen des Bundesinstituts für Arzneimittel und Medizinprodukte und des Paul-Ehrlich-Instituts zur Planung, Durchführung und Auswertung von Anwendungsbeobachtungen Entwurfsfassung vom 9 Mai 2007 zur Kommentierung durch die Fachöffentlichkeit (Joint recommendations of the Federal Institute for Drugs and Medical Devices and of the Paul-Ehrlich-Institut for planning, conducting and analysis of non-interventional studies draft version from 9 May 2007 to comment by the professional public). Arbeitskreis klinische Studien e.V. Available at: http://www.akstudien.de/upload/docs/Bfarm_PEI_AWB.pdf. Accessed 12 Aug 2012
Paul-Ehrlich-Institut (PEI) The German higher federal authority for vaccines and biomedicines. Available at: http://www.pei.de/. Accessed 12 Aug 2012
The association of research-based pharmaceutical companies (2007) VFA-Empfehlungen zur Verbesserung der Qualität und Transparenz von nicht-interventionellen Studien (vfa-recommendations for improvement of the quality and of non-interventional studies). Verband Forschender Arzneinmittelhersteller e.V. Available at:http://www.vfa.de/de/arzneimittel-forschung/datenbanken-zu-arzneimitteln/nisdb/nis-empfehlungen. Accessed 12 Aug 2012
Section 12 Voluntary Self-regulation for the Pharmaceutical Industry (2004) FSA code of conduct on the collaboration with healthcare professionals (FSA code of conduct healthcare professionals). BAnz Nr. 76:8732 (22 April 2004), amendment BAnz Nr. 62:2220 (29 March 2006), amendment BAnz Nr. 68:1636 (7 May 2008), amendment BAnz Nr. 22:499 (10 February 2010). Freiwillige Selbstkontrolle für die Arzneimittelindustrie e.V. Available at: http://www.fs-arzneimittelindustrie.de/verhaltenskodex/fachkreise/. Accessed 12 Aug 2012
Working group epidemiology methods of the German association for epidemiology (DAE) (2008) Leitlinien und Empfehlungen zur Sicherung von guter epidemiologischer Praxis (GEP) (Guidelines and recommendations for protection of good epidemiological practice (GEP)). Deutsche Arbeitsgemeinschaft für Epidemiologie. Available at: http://dgepi.de/fileadmin/pdf/leitlinien/GEP.pdf. Accessed 12 Aug 2012
de la Haye R, Herbold M (2000) Anwendungsbeobachtungen: Leitfaden für die praktische Durchführung (author’s translation: observational studies: guideline on practical implementation). Editio Cantor Verlag, Aulendorf
Theobald K, Capan M, Herbold M, Schinzel S, Hundt F (2009) Quality assurance in non-interventional studies. Ger Med Sci 7:Doc29
Victor N, Schäfer H, Nowak H et al (1991) Arzneimittelforschung nach der Zulassung (author’s translation: pharmaceutical research post authorization). Springer, Berlin Heidelberg New York London Paris Tokyo Hong Kong Barcelona Budapest
Wink K (2003) Anwendungsbeobachtungen in der ärztliche Praxis (author’s translation: observational studies in the dialy routine). Bundesverband der Pharmazeutischen Industrie e.V. (BPI), Berlin
Section 67 sub-section 6 sentence 2 of the German Medicinal Products Act Version 2009 (AMG, Gesetz über den Verkehr mit Arzneimitteln, Arzneimittelgesetz, in der Fassung von Artikel 1 Nr. 58 des Gesetzes zur Änderung arzneimittelrechtlicher und anderer Vorschriften vom 17.07.2009 BGBl. I S. 1990)
Vandenbroucke JP, von Elm E, Altman DG, Gøtzsche PC, Mulrow CD, Pocock SJ, Poole C, Schlesselmann JJ, Egger M (2007) Strengthening the Reporting of Observational Studies in Epidemiology (STROBE). Epidemiology 18(6):805–835
The Association of Research-Based Pharmaceutical Companies: member companies. Verband forschender Arzneimittelhersteller e.V. Available at: http://www.vfa.de/de/verband-mitglieder/mitgliedsunternehmen. Accessed 12 Aug 2012
The Association of Research-Based Pharmaceutical Companies: Register nicht-interventioneller Studien (Register of non-interventional studies). Verband forschender Arzneimittelhersteller e.V. Available at: http://www.vfa.de/de/arzneimittel-forschung/datenbanken-zu-arzneimitteln/nisdb. Last accessed 07 April 2012
Federal Institute for Drugs and Medical Devices, Paul-Ehrlich-Institut, Federal Office of Consumer Protection and Food Safety: AMIS—federal drug information system (AMIS-Arzneimittelinformationssystem des Bundes). Available at: http://www.pharmnet-bund.de/dynamic/en/am-info-system/. Accessed 12 Aug 2012
WHO Collaborating Centre for Drug Statistics Methodology (2012): ATC/DDD Index 2012. WHO Collaborating Centre for Drug Statistics Methodology, Oslo. Available at: http://www.whocc.no/atc_ddd_index/. Accessed 12 Aug 2012
Windeler J (2000) Verharren in der Schmuddelecke (author’s translation: remainig in the dirty corner). Dtsch Arztebl 97(42):A2756–2757
Müller CH (2009) Notwendig: mehr Transparenz und Wissenschafltichkeit (author’s translation: necessity: more transparency and scientificity). Dtsch Arztebl 106(42):A2042–2044
Ruppert T, Hahn M, Hundt F (2012) Remuneration for non-interventional studies – results of a survey in the pharmaceutical industry in Germany. Ger Med Sci 10:Doc04
German Medicinal Products Act Version 1976 (AMG vom 24.08.1976 BGBl. I S. 2445, 2448)
Hahn M, Bethke TD, Hecht A, Henn D, Ruppert T, Hundt F (2008) Qualitätssichernde Maßnahmen in nicht-interventionellen Studien: Ergebnisse einer Umfrage unter den Mitgliedsunternehmen des Verbandes Forschenden Arzneimittelhersteller (Quality assurance measures in non-interventional studies: Results of a survey among the members of the Association of Research-Based Pharmaceutical Companies). Ger Med Sci 6:Doc12
Hahn M, Ruppert T, Bethke TD, Hundt F (2010) Results of a survey on applied quality standards on non-interventional studies among members of the German Association of Researched-based Pharmaceutical Companies. Ger Med Sci 8:Doc29
Wardell WW et al (1979) Postmarketing surveillance of new drugs: II. case studies. Clin Pharmacol 19:164–184
European parliament and the Council (2001) Directive 2001/83/EC of the European parliament and of the council of 6 November 2001 on the Community cide realting to medicinal products for human use. Official Journal of the European Communities L311:67–128. Available at: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2001:311:0067:0128:EN:PDF. Accessed 12 Aug 2012
European parliament and the Council (2010) Directive 2010/84/EC of the European parliament and of the council of 15 December 2010 amending, as regards pharmacovigilance, Directive 2001/83/EC on the Community code realting to medicinal products for human use. Official Journal of the European Communities L348:74–99. Available at: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2010:348:0074:0099:EN:PDF. Accessed 12 Aug 2012
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
von Jeinsen, B.K.J.G., Sudhop, T. A 1-year cross-sectional analysis of non-interventional post-marketing study protocols submitted to the German Federal Institute for Drugs and Medical Devices (BfArM). Eur J Clin Pharmacol 69, 1453–1466 (2013). https://doi.org/10.1007/s00228-013-1482-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-013-1482-z