
Abstract
Inventories of biodiversity are crucial for helping support conservation and management efforts, yet the deep-sea, which is the largest biome on earth remains vastly understudied. Recent advances in molecular detection methods offer alternative techniques for studying inaccessible ecosystems, including those at depth. In this study we utilized environmental DNA metabarcoding, a first for studying deep-sea benthic environments in southern Africa, to assess biological diversity and to test the effects of depth and historical trawling activities on deep-sea communities. Utilising 29 sediment samples (thus focussing on predominantly meiofaunal and epifaunal biodiversity) and targeting a 313 bp region of the mtDNA cytochrome oxidase I gene, we recovered 444 OTUs across a wide array of species and genera. Even though many OTUs could only be assigned to higher taxonomic levels, results showed that biodiversity differed significantly across depth, suggesting that even at relatively small spatial scales (~ 6 km, across a depth gradient of 355 m to 515 m), eDNA derived biodiversity detected variation linked to the depth gradient. Comparison of the OTU database with known species inventories from the sampled area revealed little overlap, highlighting the need for expanding barcoding efforts of deep-sea species to aid future eDNA survey efforts. Overall our results suggest that within a South African context, increased barcoding efforts, in combination with eDNA metabarcoding and physical sampling could capture a greater proportion of benthic deep-sea biodiversity. This provides additional opportunities to underpin conservation and management decision-making in the region, such as evaluating potential sites for future protection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The deep-sea, composed of the water column and seafloor below a depth of 200 m (Ramirez-Llodra et al. 2011), is the largest biome on earth (Glover and Smith 2003; Thurber et al. 2014; Costello and Chaudhary 2017) providing numerous important ecological and economic functions and services (Dallagnolo et al. 2009; Armstrong et al. 2012; Norse et al. 2012; Mengerink et al. 2014; Thurber et al. 2014). Yet deep-sea habitats remain poorly studied, mostly because they are difficult to access and costly to sample, driven largely by their remoteness (Benn et al. 2010; Costello et al. 2010; Thurber et al. 2014). The seafloor is presumed to have high levels of biodiversity (Levin et al. 2001; Armstrong et al. 2012; Thurber et al. 2014; Sinniger et al. 2016; Laroche et al. 2020) of which many species are still undiscovered (Mengerink et al. 2014; Thurber et al. 2014; Sinniger et al. 2016). In spite of the value and the high levels of biodiversity, the deep-sea is threatened by overexploitation of fisheries and other natural resources, pollution as well as more indirect threats from climate change (Paulus 2021). Formal protection of deep-sea environments remains rare, particularly in Areas Beyond National Jurisdiction (Baco et al. 2016; Johnson et al. 2018; Da Ros et al. 2019; Combes et al. 2021). As such, there remains an urgent need to better characterize the deep-sea so that anthropogenic impacts on deep-sea environments can be better understood and managed (Johnson et al. 2018; Da Ros et al. 2019), especially as recovery of impacted areas could take many decades (Vanreusel et al. 2016). Essential to this is knowledge of biodiversity, particularly for establishing baselines against which future changes can be measured (Paulus 2021).
Environmental DNA (eDNA) metabarcoding studies have targeted deep-sea biodiversity in various ecosystems including the seafloor, seamounts, hydrothermal vents and deep-sea coral reefs (e.g. Sinniger et al. 2016; Everett and Park 2018; Laroche et al. 2020; Canals et al. 2021), as well as investigating optimal sampling and analytical methodologies (Brandt et al. 2021; Kawato et al. 2021). In the deep-sea, environmental DNA has been collected from several sources, including sediment, the water column and nodules, focussing on detection of target groups such as cephalopods (Merten et al. 2021), as well as characterising entire communities (see for example Sinniger et al. 2016; Laroche et al. 2020). Different source types are likely to capture different communities; whereas water samples will mostly represent planktonic species, those are likely to be missed when collecting sediments. Numerous studies have shown eDNA results are comparable to more traditional methods of sampling (e.g. using nets, traps or cameras), including for invertebrate (Seymour et al. 2021), and vertebrate communities (e.g. Fujii et al. 2019; Valentini et al. 2014), though some studies show less congruence (e.g. Djurhuss et al. 2018) between methods. Additional challenges around the effectiveness of eDNA metabarcoding as a biodiversity assessment tool is that reference databases lack adequate representation of many taxa, including those from the deep-sea (Marques et al. 2021; Gaither et al. 2022). Despite this limitation, eDNA surveys can help expand biodiversity inventories, thus supporting and allowing for more targeted further research.
South Africa is characterised by high levels of marine biodiversity with over 12,000 species having been described (Griffiths et al. 2010), although only a minority of known species were collected beyond 100 m depth. As such, most of the known biodiversity is from research on coastal and pelagic habitats and from commercially important species (Griffiths et al. 2010). More recently, there has been a more concerted focus on South Africa’s deeper ecosystems (see Currie et al. 2020; Button et al. 2021; Heyns-Veale et al. 2022). Little is known, however, regarding the ecology, including functional diversity and life histories of many deep-sea species, as many of the smaller species are still not accounted for or have yet to be discovered and identified.
The oceanography of the west coast of South Africa is dominated by the cold Benguela current (Hutchings et al. 2009), which in conjunction with strong seasonal upwelling results in a highly productive marine environment that supports numerous important fisheries species such as the Cape hakes (Merluccius capensis and M. paradoxus) and kingklip (Genypterus capensis) (Griffiths et al. 2010). This region also hosts unique and vulnerable ecosystems, including canyons, seamounts (Sink et al. 2012) and a productive fishing area adjacent to Childs Bank, targeted by demersal trawl and longline fishing vessels (Fairweather et al. 2006). In South Africa, as elsewhere, trawling is the most commonly utilised method for demersal fishing and conducting research surveys for stock assessment (Sink et al. 2012). Of all human impacts on the seafloor, demersal trawling is identified to have the greatest spatial impact (Benn et al. 2010), with about one fifth of the global sea floor having been trawled at least once (Mengerink et al. 2014, with impacts including habitat destruction, disruption of sediment processes and nutrient cycling, all of which may lead to negatively impacting biodiversity (Pusceddu et al. 2014; Atkinson et al. 2011a,b; Da Ros et al. 2019). Trawling pressures are not necessarily uniform across all taxa; for example Atkinson et al. (2011b) suggest that trawling affects epi-and-infaunal communities in different ways, with epifauna showing more sensitivity to heavy trawling at two sites in the southern Benguela region.
Within an African context, environmental DNA surveys from marine systems are rare, although more generally, the African continent is currently under-represented in studies utilising eDNA for biodiversity surveys (von der Heyden 2023). As such, this study presents the first attempt at utilising metabarcoding of deep-sea sediment eDNA collected at depth from the South African coastline, where we are likely targeting meiofaunal and epifaunal biodiversity. Utilising sediment eDNA in conjunction with metabarcoding of a partial fragment of the cytochrome oxidase I gene, the objectives of this research were to (i) evaluate sediment eDNA metabarcoding for the detection of benthic biodiversity at Childs Bank, (ii) to compare the biodiversity inventory detected from sediment eDNA with existing species lists/taxonomic records generated by traditional sampling methods and (iii) to test the effects of depth and historical trawling activities on sediment biodiversity.
Methods
Study site and sample collection
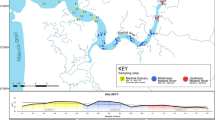
The study site, adjacent to Childs Bank, is located ~ 180 km off Hondeklip Bay on the west coast of South Africa (Fig. 1). This area is considered to be an Ecologically or Biologically Significant Area (EBSA, Harris et al. 2022) and lies adjacent to one of the new Marine Protected Areas (MPAs) from Operation Phakisa (RSA 2019). The area is considered vulnerable to mining and trawling, as well as being identified as an important area for supporting bycatch management and fisheries sustainability (Harris et al. 2014, 2022). Our study was carried out as an extension of an existing long-term benthic trawl experiment, that sought to quantify the impact of trawling on benthic marine biodiversity. The experimental design of the project was implemented in an area ± 90 km2 and included five ‘lanes’ spanning a depth range from 355 to 515 m (Fig. 1). Alternating lanes one, three and five were closed to trawling in 2014, while lanes two and four remained open to trawling, after which annual surveys were conducted in the experimental area to monitor for resulting benthic ecosystem changes. Sediment samples for eDNA research were obtained in 2018, four years after the closure of some of the lanes as described above; sediments were collected opportunistically, given logistical and capacity challenges that prevent targeted sediment eDNA collection at the time of the project. Sediment was sampled using a Van Veen benthic grab from three stations in each of the five lanes. At each station, sediment for eDNA analyses were collected from two replicate grabs, except for at one station in lane 5 (the shallowest lane), where sediment for eDNA analysis was only collected from one grab. From each of the 29 grabs a sediment sample from the top 3-5 cm (~ 350 ml of sediment) was collected into pre-sterilised plastic jars, whilst wearing fresh gloves and taking care to open and close the jar for as short a time as possible. Samples were frozen immediately after collection at − 20 °C until DNA extraction. The depth at which each sample was collected and sediment composition were also recorded for each site.

a Map showing the location of trawling study in relation to the west coast of South Africa, the lines indicated bathymetry of the area and depth is given as metres (m); the inset shows a map of South Africa. b Diagram showing the sampling design and the dimensions of the sampling area. There were three sampling stations per lane, as indicated by black dots. At each sampling station three replicate grab samples were collected which were subsampled for sediment eDNA. Depth from surface is indicated as m
DNA extraction
Sediment samples were subsampled for DNA extraction, as it has been shown that extraction replication improves diversity estimates and coverage of target groups (Lanzén et al. 2017). Three subsamples (technical replicates) were taken from each grab for extraction purposes by dividing each sample into thirds, resulting in 87 samples in total. One extraction replicate was taken from each third, with a total of ~ 1 g of sediment extracted per sample. Negative extraction controls were also carried out for each extraction session. DNA was extracted directly from the sediment using the DNeasy PowerSoil extraction kit (Qiagen, Venlo, The Netherlands) following the manufacturer’s protocol, except for a final elution volume of 25 ul. The final elute was passed through the spin column a second time before storage to ensure that as much DNA as possible was recovered. DNA extractions were stored at − 20 °C and replicates from each sample were pooled before library preparation.
Library preparation and sequencing
Extracted DNA from each of the selected sites were sent to the Advanced Identification Methods (AIM; https://www.aimethods-lab.com/) lab in Leipzig, Germany for library preparation using a two-step PCR protocol, targeting a partial fragment of mtDNA COI. The primers jgHCO2198 (TAIACYTCIGGRTGICCRAARAAYCA; Geller et al. 2013) and mtCOIintF (GGWACWGGWTGAACWGTWTAYCCYCC; Leray et al. 2013) were utilised to amplify a 313 bp fragment of the mtDNA COI gene and have successfully been utilized for a wide variety of eDNA surveys, including sediment samples (Lacoursière‐Roussel et al. 2018). Primers were synthesis by Metabion (Steinkirchen, Germany). PCRs followed Morinière et al. (2019) with the MyTAQ Plant-PCR (Bioline, Luckenwalde, Germany) and 5 μL (up to 250 ng of DNA). The initial PCR reaction was as follows: 95 °C for 5 min, 3 cycles of [96 °C for 15 s; 48 °C for 30 s; 65 °C for 90 s], 30 cycles of [96 °C for 15 s; 55 °C for 30 s; 65 °C for 90 s] and 76 °C for 10 min. Amplification success and fragment length were then observed using gel electrophoresis. Amplified DNA was cleaned and resuspended in 50 μL molecular water for each sample before proceeding. Illumina Nextera XT (N7xx and N5xx Nextera Index Kit; Illumina Inc., San Diego, USA) indices were attached to the samples in a second PCR reaction applying the same annealing temperature as for the first PCR reaction but with only 7 PCR cycles. Ligation success was confirmed by gel electrophoresis. DNA concentrations were measured using a Qubit fluorometer with Kit Qubit™ 1X dsDNA (Life Technologies, Carlsbad, USA), and samples were combined into 40 μL pools containing equimolar concentrations of 100 ng each. Pools were purified using MagSi-NGSprep Plus (Steinbrenner Laborsysteme GmbH) beads. A final elution volume of 20 μL was used. High-Throughput Sequencing (HTS) was performed on an Illumina MiSeq using v3 (2*300 bp, 600 cycles, maximum of 25mio paired-end reads) chemistry.
Bioinformatic analyses
After sequencing, an initial quality control analysis of the reads was performed using FastQC version 0.11.8. Paired-end merging was performed using usearch v11.0.667 with the parameters –fastq_maxdiffs 99 –fastq_pctid 75 –fastq_trunctail 0. Primers were trimmed using cutadatpt 1.18 with Python 2.7.15. In the next step, sequences above a minimum length of 300 were retained, and with a maximum of 1 expected error. Of those, unique sequences and singletons were filtered for. Quality filtering was performed using vsearch 2.9.1 with the parameters –fastq_minlen 300 –fastq_maxee 1. To save processing power, OTU clustering was performed before detecting chimaeras. Clustering was performed using vsearch 2.9.1 and the parameters –id 0.98 –iddef 029–centroids. OTU cut-off was 97%. Chimeras were detected using vsearch 2.9.1 and the parameters –uchime_denovo –nonchimeras. For full commands for bioinformatic processing see Supplementary Materials Note 1.
Taxonomic assignment of the OTUs generated was performed using a BLAST search, using the GenBank (NCBI) database. All sequences and their identifications were carefully inspected, with OTUs that were classified as non-marine organisms discarded and OTUs that could not be identified further than Domain level also removed. The final list of OTUs was compared to both the Field Guide to the Offshore Marine Invertebrates of South Africa (Atkinson and Sink 2018) and the infaunal species list from the same sampling cruise obtained from physical sampling methods (grab samples). The latter was kindly provided by Dr N. Karenyi from the University of Cape Town, South Africa. All data is available through www.github.com/vonderheydenlab.
Statistical analyses
An accumulation curve for total diversity was generated as well as separate curves for trawled and untrawled sites. The final OTU table was used to create a presence/absence dataset (github.com/vonderHeydenLab/Oosthuizen_Deep-Sea-benthic-eDNA) for each site, which was subsequently used for further analyses. All statistical analyses were performed using the Program R version 4.0.2 (Team R.C. 2020).
Sequence reads per site were rarefied to 3351 reads (lowest read count for sites with at least 3000 sequences reads) prior to formal analyses using the rarefy function in the r-package vegan. Community richness was calculated as the total number of unique OTUs per site and was calculated to quantify the level of biodiversity observed at each site. To describe the spatial change in biodiversity composition between sites a Bray–Curtis similarity resemblance measure was calculated between all site pairs. Using the vegan package in R (Oksanen et al. 2020), a PERMANOVA was used to test the effects of trawling, absolute depth and the interaction between trawling using 999 permutations. As a significant effect of depth between communities was observed (see results below), a Kendall’s exact test was applied, to assess individual OTUs in relation to depth (Hollander and Wolfe 1973). NMDS ordination was calculated using Bray–Curtis dissimilarities and the function metaMDS in the r-package vegan.
Results
Sequencing results and comparison with species lists obtained from physical sampling
A total of 1,844,876 paired-end reads were obtained from sequencing, but for three of the 29 samples no reads were recovered. After paired-end merging, quality filtering, de-replications, and removal of singletons, a total of 24,154 were retained (~ 1000 reads per sample; 19 samples had > 3000 reads/sample), clustered into 1975 OTUs (Fig. 2). After taxonomic assignment and filtering, 444 OTUs were retained and used for further analyses (Fig. 3). Based on a 97% OTU cut-off, of the 444 OTUs, ~ 2.7% could be identified to species, approximately 4.05% to Genus, 10.59% to Family, 24.55% to Order, 36.26% to Class, and 45.49% to Phylum. (Supplementary Table 1). The number of unique taxonomic assignments to OTUs included 12 species, 18 genera, 71 families and 109 orders.

Krona figure showing the proportions of OTUs belonging to the different phyla identified through sediment eDNA metabarcoding adjacent to Childs Bank

Stacked column OTU richness for all families recovered across 19 samples (each with > 3000 reads per sample after rarefaction analyses)
The accumulation curve did not reach an asymptote (Supplementary Fig. 1), showing that there are likely to still be portions of biodiversity not reached by our sampling and filtering approaches. When comparing taxonomic assignments from OTUs with both a published field guide (Atkinson and Sink 2018) and species list specific to the area sampled (N. Karenyi pers comm.), OTU assignments matched the field guide for 1 species, 4 genera, 6 families and 21 orders and matched the species list for no species, 2 genera, 71 families and 8 orders (Table 1).
Community diversity analyses
Community richness analyses suggest that taxa were relatively evenly spread across sites, although the PERMANOVA showed that the biodiversity composition differed significantly across absolute depth (df = 1; F = 1.6; p value = 0.006). There was no significant difference between communities in relation to the main effect of trawling or the trawling by depth interaction effect (Supplementary Table 2; Fig. 4). The correlation analyses further showed that OTUs belonging to Arthropoda, Mollusca and Nematoda increased in frequency with depth; in contrast, other OTUs belonging to Amphipoda and Polymastiida in particular, became less frequent with increasing depth (Supplementary Table 3).

NMDS plot showing the effects of trawling and depth. Only depth was shown to be a significant predictor of community richness
Discussion
This study presents the first attempt at using sediment eDNA metabarcoding to characterise deep-sea benthic communities in South Africa. We recovered novel biodiversity across numerous families and genera that had previously not been recorded from the region, such as the Placozoa, that are generally poorly understood. As expected there was little overlap between the eDNA survey and the known biodiversity of the area sampled, including from specimens identified from the same sampling opportunity. The lack of barcodes (see for example Gaither et al. 2022; von der Heyden 2023), hampered species identification and we reiterate the call for concerted foundational barcoding efforts of marine life, particularly of biodiversity beyond coastal areas. Given that with only a few samples collected during one cruise and recovering over 400 OTUs, we show that metabarcoding of sediment eDNA is a promising complimentary tool for capturing South African biodiversity in areas that are logistically challenging to survey. However, there also exists a need to extend several aspects of the eDNA workflow in southern Africa to ensure that this methodology becomes firmly embedded in biodiversity monitoring of the unique diversity of the region’s deep-sea systems.
Novel biodiversity inventories and little overlap with known biodiversity
We recovered a high level of deep-sea benthic diversity, with over 400 OTUs spread across diverse families. Although a major challenge of our study pertained to low read numbers, which likely does not allow for the detection of low abundance species (Alberdi et al. 2018; Shirazi et al. 2021) and as such will contribute to an under-estimation of actual biodiversity, this study provides exciting insights into deep-sea benthic biodiversity in an understudied system. Notably, there was little overlap with species records from either the physical sampling or know field guides (Table 1) and less than 3% of OTUs could be identified to species level based on global reference databases (Supplementary Materials Table 1). The overlap of taxa identified between the eDNA samples and other sampling methods decreased with the level of identification, with lower taxonomic levels such as species or genus not having a strong overlap in terms of organisms identified between different sampling methods. In this study, both the field guide and the species list contained unknown species that could only be broadly classified to a certain phylum or order (see Atkinson and Sink 2018), and it is possible that some of the OTUs identified may represent some of these unknown species. Confirming this, however, is difficult, since many species in this region lack barcoding information (Singh et al. 2021; von der Heyden 2023) making molecular identification difficult. While the infaunal species list of identified taxa collected at the same time as the eDNA samples identifies 87 taxa to species level, none of them are shared with the 10 species identified in the eDNA samples and of the 409 epifaunal species identified in the field guide, only one was shared with the species identified by the eDNA samples. One explanation could pertain to how we sampled sediment; in our study we used Van Veen grabs to collect sediment samples, which would have exposed the collected sediment to the water column and potentially and accidentally facilitated the sampling non-benthic taxa/eDNA, thereby potentially increasing the detected biodiversity beyond benthic species. In addition, our eDNA survey may well have included meiofauna, whereas biodiversity guides for Childs Bank are based on larger-bodied epi-and-infauna, thus providing additional opportunities for mismatches. Finally, the persistence and transport of eDNA in the deep-sea are not understood and we may have sampled the DNA of an individual that was not captured during the physical sampling, providing additional mismatches between OTUs from the eDNA samples and other species inventories.
Within the context of capturing the broadest possible diversity of a biological community, different sampling methods are frequently used in broad biodiversity studies to ensure that as many taxa as possible are covered in the sampling effort. For example, Thomsen et al. (2016) found small differences between the fish taxa caught in a trawl net and those detected through water eDNA thus expanding the known fish community. Lejzerowicz et al. (2015) also found numerous differences between the taxa detected in physical sampling and by sediment eDNA analysis. Numerous reasons can explain such differences, such as taxa avoiding physical capture equipment such as nets, grabs or cameras (Thomsen et al. 2016) or that some taxa are difficult to identify and could be cryptic species (e.g. Everett and Park 2018). Importantly, ours and other studies highlight the value of using complementary sampling methods to attain better representation of the target communities. For example, in their results on recolonisation of deep-sea vent communities, Cowart et al. (2020) also recommended that physical sampling should be combined with eDNA sampling to give a more complete picture of the recolonisation process. In the South African context, significant advances in the foundational barcode database are required before multiple techniques, such as eDNA metabarcoding, will likely add significantly to the knowledge of deep-sea benthic biodiversity.
Drivers of community diversity
The composition of deep-sea benthic communities is influenced by a variety of environmental factors such as food availability, depth, natural disturbance regimes, sediment type and organic matter (Harris 2014; Rosli et al. 2018; Wang et al. 2019), although measuring environmental variation in the deep-sea is difficult, and there is still much that remains unknown. In this study, depth had a significant effect on biodiversity composition detected with eDNA metabarcoding, with other studies also reporting that depth, in particular, plays an important role in structuring deep-sea communities. For example, in the Yap Trench in the Western Pacific Ocean, the distribution of meiofaunal communities was influenced by depth, sediment grain size, sediment type and factors relating to food availability, where depth had a strong positive correlation to organic matter (Wang et al. 2019). Along the West coast of South Africa, Roel (1987) and Atkinson et al. (2011b) found that depth played a significant role in the structuring demersal fish assemblages with fish assemblages showing a distinct difference between 300 and 400 m along the shelf-break, which is the same depth range of the sites sampled in this study. Depth was also the major factor influencing benthic epifauna along the west coast of South Africa in a study conducted by Lange and Griffiths (2014).
Trawling is known to impact benthic communities in the deep-sea in a number of different ways such as by causing disruption of sediment, habitat destruction, disruption of nutrient cycling and loss of biodiversity (Bluhm 2001; Pusceddu et al. 2014; Hiddink et al. 2017; Da Ros et al. 2019), with Good et al. (2022) utilising eDNA metabarcoding to show impacts of trawling on deep-sea communities in the Mediterranean Sea. Within the context of our study, the broader trawl impact project investigated changes in the benthic community (infauna and epifauna) over the entire four year period, with results showing significant shifts in the community composition over this time (L. Atkinson, pers. comm.). Our eDNA metabarcoding did not detect any significant difference in biodiversity composition between trawled and untrawled sites in the sampled area and although unexpected, there are several explanations that can provide insight into these results. The first is that, on a national level the experimental area did not reflect sufficiently intense trawl effort at the outset (Currie et al. 2020), limiting the detectable contrast in change between open and closed lanes. Since fishing pressure was then comparatively low at all sites, there may not have been sufficient contrast in the effect to detect after four years. Second, trawling may not have as great an impact on communities of certain taxonomic groups, especially infauna, here targeted via sediment eDNA. For example, Kaiser et al. (2006) demonstrated that the impacts of trawling and rates of recovery differ among habitat types, trawling gear types and different taxonomic groups. In addition, Atkinson et al. (2011a) found that infaunal and epifaunal communities responded differently to trawling pressures and Fleddum et al. (2013) using biological traits analysis on benthic communities in southern Africa also reported that epifaunal groups were generally more impacted by trawling than infaunal groups. For our study, most OTUs could only be identified to higher taxonomic levels which meant that it was not possible to carry out more detailed community assemblage analyses.
Overall, our study provides a first glimpse at the potential of sediment eDNA metabarcoding for detecting biodiversity of South Africa’s deep-seas and shows that much of the eukaryotic diversity is as yet unaccounted for. Given the general lack of overlap between species detected through physical sampling and sediment eDNA, we encourage a complimentary approach to better capture more of the diversity of the deeper areas of the ocean. In particular, within the context of the recent extension of the MPA and EBSA networks, that include numerous offshore features, eDNA based approaches could significantly support future conservation and management decision-making. Biodiversity surveys based on a wide range of eDNA sources (such as sediments and water) will undoubtedly open up new and exciting opportunities to dive into the world of the deep unexplored depths of southern Africa’s oceans.
Data availability
All data are available through https://github.com/vonderHeydenLab/Oosthuizen_Deep-Sea-benthic-eDNA.
References
Alberdi A, Aizpurua O, Gilbert MTP, Bohmann K (2018) Scrutinizing key steps for reliable metabarcoding of environmental samples. Methods Ecol Evol 9:134–147. https://doi.org/10.1111/2041-210X.12849
Armstrong CW, Foley NS, Tinch R, van den Hove S (2012) Services from the deep: Steps towards valuation of deep-sea goods and services. Ecosyst Serv 2:2–13. https://doi.org/10.1016/j.ecoser.2012.07.001
Atkinson L, Sink K (2018) Field guide to the offshore marine invertebrates of South Africa. Malachite Marketing and Media, Pretoria
Atkinson LJ, Field JG, Hutchings L (2011a) Effects of demersal trawling along the west coast of Southern Africa: multivariate analysis of benthic assemblages. Mar Ecol Prog Ser 430:241–256. https://doi.org/10.3354/meps08956
Atkinson LJ, Leslie RW, Field JG, Jarre A (2011b) Changes in demersal fish assemblages on the west coast of South Africa, 1986–2009. A J Mar Sci 33:157–170. https://doi.org/10.2989/1814232X.2011.572378
Baco AR, Etter RJ, Ribeiro PA, von der Heyden S, Beerli P, Kinlan BP (2016) A synthesis of genetic connectivity in deep-sea fauna and implications for marine reserve design. Mol Ecol 25:3276–3298. https://doi.org/10.1111/mec.13689
Benn AR, Weaver PP, Billet DSM, van den Hove S, Murdock AP, Doneghan GB, Le Bas T (2010) Human activities on the deep seafloor in the North East Atlantic: an assessment of spatial extent. PLoS ONE 5:e12730. https://doi.org/10.1371/journal.pone.0012730
Bluhm H (2001) Re-establishment of an abyssal megabenthic community after experimental physical disturbance of the seafloor. Deep Sea Res 48:3841–3868. https://doi.org/10.1016/S0967-0645(01)00070-4
Brandt MI, Pradillon F, Trouche B, Henry N, Liautard-Haag C, Cambon-Bonavita MA, Cueff-Gauchard V et al (2021) Evaluating sediment and water sampling methods for the estimation of deep-sea biodiversity using environmental DNA. Sci Rep 11:1–14. https://doi.org/10.1038/s41598-021-86396-8
Button RE, Parker D, Coetzee V, Samaai T, Palmer RM, Sink K, Kerwath SE (2021) ROV assessment of mesophotic fish and associated habitats across the continental shelf of the Amathole region. Sci Rep 11:1–11. https://doi.org/10.1038/s41598-021-97369-2
Canals O, Mendibil I, Santos M, Irigoien X, Rodríguez-Ezpeleta N (2021) Vertical stratification of environmental DNA in the open ocean captures ecological patterns and behaviour of deep-sea fishes. Limnol Oceanogr 6:339–347. https://doi.org/10.1002/lol2.10213
Combes M, Vaz S, Grehan A, Morato T, Arnaud-Haond S, Dominguez-Carrió C, Fox A et al (2021) Systematic conservation planning at an ocean basin scale: Identifying a viable network of deep-sea protected areas in the North Atlantic and the Mediterranean. Front Mar Sci 8:611358. https://doi.org/10.3389/fmars.2021.611358
Costello MJ, Chaudhary C (2017) Marine biodiversity, biogeography, deep-sea gradients, and conservation. Curr Biol 27:R511–R527. https://doi.org/10.1016/j.cub.2017.04.060
Costello MJ, Coll M, Danovaro R, Halpin P, Ojaveer H, Miloslavich P (2010) A census of marine biodiversity knowledge, resources, and future challenges. PLoS ONE 5:e12110. https://doi.org/10.1371/journal.pone.0012110
Cowart DA, Matabos M, Brandt MI, Marticorena J, Sarrazin J (2020) Exploring environmental DNA (eDNA) to assess biodiversity of hard substratum faunal communities on the lucky strike vent field (Mid-Atlantic Ridge) and investigate recolonization dynamics after an induced disturbance. Front Mar Sci 6:783. https://doi.org/10.3389/fmars.2019.00783
Currie JC, Atkinson LJ, Sink KJ, Attwood CG (2020) Long-term change of demersal fish assemblages on the inshore Agulhas bank between 1904 and 2015. Front Mar Sci 7:355. https://doi.org/10.3389/fmars.2020.00355
Dallagnolo R, Perez JAA, Pezzuto PR, Wahrlich R (2009) The deep-sea shrimp fishery off Brazil (Decapoda: Aristeidae): development and present status. Lat Am J Aquat Res 37:327–345
Djurhuus A, Pitz K, Sawaya NA, Rojas-Márquez J, Michaud B, Montes E, Muller-Karger F et al (2018) Evaluation of marine zooplankton community structure through environmental DNA metabarcoding. Limnol Oceanog 16:209–221. https://doi.org/10.1002/lom3.10237
Everett MV, Park LK (2018) Exploring deep-water coral communities using environmental DNA. Deep-Sea Res 150:229–241. https://doi.org/10.1016/j.dsr2.2017.09.008
Fairweather TP, Booth AJ, Sauer WHH, Leslie RW (2006) Spatial description of hake-directed fishing activity off the west coast of South Africa. A J Mar Sci 28:13–24. https://doi.org/10.2989/18142320609504129
Fleddum A, Atkinson LJ, Field JG, Shin P (2013) Changes in biological traits of macro-benthic communities subjected to different intensities of demersal trawling along the west coast of Southern Africa. J Mar Biol Assoc UK 93:2027–2038. https://doi.org/10.1017/S0025315413000647
Fujii K, Doi H, Matsuoka S, Nagano M, Yamanaka SH, H, (2019) Environmental DNA metabarcoding for fish community analysis in backwater lakes: a comparison of capture methods. PLoS ONE 14:e0210357. https://doi.org/10.1371/journal.pone.0210357
Gaither MR, DiBattista JD, Leray M, von der Heyden S (2022) Metabarcoding the marine environment: from single species to biogeographic patterns. Environ DNA 4:3–8. https://doi.org/10.1002/edn3.270
Geller J, Meyer C, Parker M, Hawk H (2013) Redesign of PCR primers for mitochondrial cytochrome oxidase subunit I for marine invertebrates and application in all-taxa biotic surveys. Mol Ecol Resour 13:851–861. https://doi.org/10.1111/1755-0998.12138
Glover AG, Smith CR (2003) The deep-sea floor ecosystems: current status and prospects of anthropogenic change by the year 2025. Environ Conserv 30:219–241. https://doi.org/10.1017/S0376892903000225
Good E, Holman LE, Pusceddu A, Russo T, Rius M, Iacono CL (2022) Detection of community-wide impacts of bottom trawl fishing on deep-sea assemblages using environmental DNA barcoding. Mar Poll Bull 183:114062. https://doi.org/10.1016/j.marpolbul.2022.114062
Griffiths CL, Robinson TB, Lange L, Mead A (2010) Marine biodiversity in South Africa: an evaluation of current states of knowledge. PLoS ONE 5:e12008. https://doi.org/10.1371/journal.pone.0012008
Harris PT (2014) Shelf and deep-sea sedimentary environments and physical benthic disturbance regimes: a review and synthesis. Mar Geol 353:169–184. https://doi.org/10.1016/j.margeo.2014.03.023
Harris LR, Holness SD, Finke G, Amunyela M, Braby R, Coelho N, Gee K et al (2022) Practical marine spatial management of ecologically or biologically significant marine areas: emerging lessons from evidence-based planning and implementation in a developing-world context. Front Mar Sci 9:831678. https://doi.org/10.3389/fmars.2022.831678
Harris J, Livingstone T, Phadima J, Sink K, Fasheun T, Boyd A, Mfeka X (2014) Phakisa Initiative: fast-tracking establishment of an effective and representative network of Marine Protected Areas for South Africa. In: Symposium of contemporary conservation practice, Howick, KwaZulu-Natal
Heyns-Veale ER, Bernard ATF, Midgley JM, Herbert DG (2022) The distribution of offshore benthic molluscs provides new insight into South Africa’s marine biogeography. Ocean Coast Manag 217:106001. https://doi.org/10.1016/j.ocecoaman.2021.106001
Hiddink JG, Jennings S, Sciberras M, Szosteck C, Hughes KM, Ellis N, Rijnsdorp AD et al (2017) Global analysis of depletion and recovery of seabed biota after bottom trawling disturbance. Proc Nat Acad Nat Sci USA 114:8301–8306. https://doi.org/10.1073/pnas.1618858114
Hollander M, Wolfe DA (1973) Nonparametric statistical methods. John Wiley & Sons, New York, pp 185–194
Hutchings L, van der Lingen CD, Shannon LJ, Crawford RJM, Verheye HMS, Bartholomae CH, van der Plas AK et al (2009) The Benguela current: an ecosystem of four components. Prog Oceanogr 8:5–32. https://doi.org/10.1016/j.pocean.2009.07.046
Johnson D, Ferreira MA, Kenchington E (2018) Climate change is likely to severely limit the effectiveness of deep-sea ABMTs in the North Atlantic. Mar Policy 87:111–112. https://doi.org/10.1016/j.marpol.2017.09.034
Kaiser MJ, Clarke KR, Hinz H, Austen MC, Somerfield PJ, Karakassis I (2006) Global analysis of response and recovery of benthic biota to fishing. Mar Ecol Prog Ser 311:1–14. https://doi.org/10.3354/meps311001
Kawato M, Yoshida T, Miya M, Tsuchida S, Nagano Y, Nomura M, Yabuki A et al (2021) Optimization of environmental DNA extraction and amplification methods for metabarcoding of deep-sea fish. MethodsX 8:101238. https://doi.org/10.1016/j.mex.2021.101238
Lacoursière-Roussel A, Howland K, Normandeau E, Grey EK, Archambault P, Deiner K, Lodge DM et al (2018) eDNA metabarcoding as a new surveillance approach for coastal arctic biodiversity. Ecol Evol 8:7763–7777. https://doi.org/10.1002/ece3.4213
Lange L, Griffiths CL (2014) Large-scale spatial patterns within soft-bottom epibenthic invertebrate assemblages along the west coast of South Africa, based on the Nansen trawl survey. A J Mar Sci 36:111–124. https://doi.org/10.2989/1814232X.2014.894943
Lanzén A, Lekang K, Jonassen I, Thompson EM, Troedsson C (2017) DNA extraction replicates improve diversity and compositional dissimilarity in metabarcoding of eukaryotes in marine sediments. PLoS ONE 12:e0179443. https://doi.org/10.1371/journal.pone.0179443
Laroche O, Kersten O, Smith CR, Goetze E (2020) Environmental DNA surveys detect distinct metazoan communities across abyssal plains and seamounts in the western Clarion Clipperton zone. Mol Ecol 29:4588–4604. https://doi.org/10.1111/mec.15484
Lejzerowicz F, Esling P, Pillet L, Wilding TA, Black KD, Pawlowski J (2015) High-throughput sequencing and morphology perform equally well for benthic monitoring of marine ecosystems. Sci Rep 5:13932. https://doi.org/10.1038/srep13932
Leray M, Yang JY, Meyer CP, Mills SC, Agudelo N, Ranwez V, Boehm JT et al (2013) A new versatile primer set for targeting a short fragment of the mitochondrial COI region for metabarcoding metazoan diversity: application for characterising coral reef fish gut contents. Front Zool 10:34. https://doi.org/10.1186/1742-9994-10-34
Levin LA, Etter RJ, Rex MA, Gooday AJ, Smith CR, Pineda J, Stuart CT et al (2001) Environmental influences on regional deep-sea species diversity. Annu Rev Ecol Syst 32:51–93. https://doi.org/10.1146/annurev.ecolsys.32.081501.114002
Marques V, Milhau T, Albouy C, Dejean T, Manel S, Mouillot D, Juhel J-B (2021) GAPeDNA: assessing and mapping global species gaps in genetic databases for eDNA metabarcoding. Divers Distrib 27:1880–1892. https://doi.org/10.1111/ddi.13142
Mengerink KJ, Van Dover CL, Ardron J, Baker M, Escobar-Briones E, Gjerde K, Koslow JA et al (2014) A call for deep-ocean stewardship. Science 344:696–698. https://doi.org/10.1126/science.1251458
Merten V, Bayer T, Reusch TBH, Puebla O, Fuss J, Stefanschitz J, Lischka A, Hauss H, Neitzel P, Piatkowski U, Czudaj S, Christiansen B, Denda A, Hoving H-J (2021) An integrative assessment combining deep-sea net sampling, in situ observations and environmental DNA analysis identifies Cabo Verde as a cephalopod biodiversity hotspot in the Atlantic Ocean. Front Mar Sci 8:760108. https://doi.org/10.3389/fmars.2021.760108
Morinière J, Balke M, Doczkal D, Geiger MF, Hardulak LA, Haszprunar G et al (2019) A DNA barcode library for 5,200 German flies and midges (Insecta: Diptera) and its implications for metabarcoding-based biomonitoring. Mol Ecol Res 19:900–928. https://doi.org/10.1111/1755-0998.13022
Norse EA, Brooke S, Cheung WWL, Clark MR, Ekeland I, Froese R, Gjerde KM et al (2012) Sustainability of deep-sea fisheries. Mar Policy 36:307–320. https://doi.org/10.1016/j.marpol.2011.06.008
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, et al. (2020) vegan: Community Ecology Package. R package version 2.5–7
Paulus E (2021) Shedding light on deep-sea biodiversity–a highly vulnerable habitat in the face of anthropogenic change. Front Mar Sci 8:28. https://doi.org/10.3389/fmars.2021.667048
Pusceddu A, Bianchelli S, Martín J, Puig P, Palanques A, Masqué P, Danovaro R (2014) Chronic and intensive bottom trawling impairs deep-sea biodiversity and ecosystem functioning. Proc Nat Acad Nat Sci USA 111:8861–8866. https://doi.org/10.1073/pnas.1405454111
Ramirez-Llodra E, Tyler PA, Baker MC, Bergstad OA, Clark MR, Escobar E, Levin LA et al (2011) Man and the last great wilderness: human impact on the deep sea. PLoS ONE 6:e22588. https://doi.org/10.1371/journal.pone.0022588
Roel BA (1987) Demersal communities off the west coast of South Africa. SA J Mar Sci 5:575–584. https://doi.org/10.2989/025776187784522135
Ros Da, Dell’Anna A, Morato T, Sweetman AK, Carreiro-Silva M, Smith CJ, Papadopoulou N, Corinaldesi C, Bianchelli S, Gambi C et al (2019) The deep sea: the new frontier for ecological restoration. Mar Policy 108:103642. https://doi.org/10.1016/j.marpol.2019.103642
Rosli N, Leduc D, Rowden AA, Probert PK (2018) Review of recent trends in ecological studies of deep-sea meiofauna, with focus on patterns and processes at small to regional spatial scales. Mar Biodivers 48:13–34. https://doi.org/10.1007/s12526-017-0801-5
RSA (Republic of South Africa) (2019) Department of Environmental Affairs, Government Notices 757–776 declaring 20 marine protected areas in terms of Section 22A of the National Environmental Management: Protected Areas Act (Act No. 57 of 2003). Government Gazette, South Africa 647 (42478)
Seymour M, Edwards FK, Cosby BJ, Bista I, Scarlett PM, Brailsford FL, Glanville HC et al (2021) Environmental DNA provides higher resolution assessment of riverine biodiversity and ecosystem function via spatio-temporal nestedness and turnover partitioning. Commun Biol 4:512–512. https://doi.org/10.1038/s42003-021-02031-2
Shirazi S, Meyer RS, Shapiro B (2021) Revisiting the effect of PCR replication and sequencing depth on biodiversity metrics in environmental DNA metabarcoding. Ecol Evol 11(22):15766–15779
Singh SP, Groeneveld JC, Huggett J, Naidoo D, Cedras R, Willows-Munro S (2021) Metabarcoding of marine zooplankton in South Africa. A J Mar Sci 43:147–159. https://doi.org/10.2989/1814232X.2021.1919759
Sink K, Holness S, Harris L, Majiedt P, Atkinson L, Robinson T, Kirkman S, et al. (2012) National biodiversity assessment 2011: technical report. Vol. 4: marine and coastal component. Pretoria: South African national biodiversity institute
Sinniger F, Pawlowski J, Harii S, Gooday AJ, Yamamoto H, Chevaldonné P, Cedhagen T et al (2016) Worldwide analysis of sedimentary DNA reveals major gaps in taxonomic knowledge of deep-sea benthos. Front Mar Sci 3:92. https://doi.org/10.3389/fmars.2016.00092
Thomsen PF, Møller PR, Sigsgaard EE, Knudsen SW, Jørgensen OA, Willerslev E (2016) Environmental DNA from seawater samples correlate with trawl catches of subarctic, deepwater fishes. PLoS ONE 11:e0165252. https://doi.org/10.1371/journal.pone.0165252
Thurber AR, Sweetman AK, Narayanaswamy BE, Jones DOB, Ingels J, Hansman RL (2014) Ecosystem function and services provided by the deep sea. Biogeosciences 11:3941–3963. https://doi.org/10.5194/bg-11-3941-2014
Valentini A, Taberlet P, Miaud C, Civade R, Herder J, Thomsen PF, Bellemain E et al (2014) Next-generation monitoring of aquatic biodiversity using environmental DNA metabarcoding. Mol Ecol 25:929–942. https://doi.org/10.1111/mec.13428
Vanreusel A, Hilario A, Ribeiro PA, Menot L, Arbizu PM (2016) Threatened by mining, polymetallic nodules are required to preserve abyssal epifauna. Sci Rep 6:26808. https://doi.org/10.1038/srep26808
von der Heyden S (2023) Environmental DNA surveys of African biodiversity: state of knowledge, challenges and opportunities. Environ DNA 5:12-17. https://doi.org/10.1002/edn3.363
Wang X, Liu X, Xu J (2019) Distribution patterns of meiofauna assemblages and their relationship with environmental factors of deep-sea adjacent to the Yap Trench, Western Pacific Ocean. Front Mar Sci 6:735. https://doi.org/10.3389/fmars.2019.00735
Acknowledgements
The project partners of the Benthic Trawl Experiment are acknowledged for their role in endorsing this research as part of the project sampling. The captain and crew of the Department of Forestry, Fisheries and Environment research vessel, R/V Ellen Khuzwayo, Associate Professor Colin Attwood and Karen Tunley are gratefully acknowledged for collecting the sediment samples for this research. A sincere thank you Grant van der Heever for producing the map in Figure 1 and to Dr Natasha Karenyi for sharing species lists. We also acknowledge all funding: DO was funded through a National research Foundation grant-holder linked MSc bursary. This work was supported through the NRF, South Africa grant awarded to SvdH (Grant number: 105842), and an NRF/FBIP grant awarded to LA (Grant number 138572).
Funding
Open access funding provided by Stellenbosch University. DO was funded through a National Research Foundation grant-holder linked MSc bursary. This work was supported through the NRF, South Africa grant awarded to SvdH (Grant number: 105842), and an NRF/FBIP grant awarded to LA (Grant number 138572).
Author information
Authors and Affiliations
Contributions
DO: project conceptualisation, laboratory work, data analysis, drafting and editing of manuscript; MA: data analysis, drafting and editing of manuscript, student supervision; LJA: project conceptualisation, editing of manuscript; project funding; SvdH: project conceptualisation, drafting and editing of manuscript; project funding, student supervision.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
Not required for this work.
Additional information
Responsible Editor: H.J. Hoving.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Oosthuizen, D., Seymour, M., Atkinson, L.J. et al. Extending deep-sea benthic biodiversity inventories with environmental DNA metabarcoding. Mar Biol 170, 60 (2023). https://doi.org/10.1007/s00227-023-04205-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00227-023-04205-4