
Abstract
The level of habitat availability influences genetic divergence among populations and the genetic diversity within populations. In the marine environment, near-shore species are among the most sensitive to habitat changes. Knowledge of how historical environmental change affected habitat availability and genetic variation can be applied to the development of proactive management strategies of exploited species. Here, we modeled the contemporary and historical distribution of Lutjanus jocu in Brazil. We describe patterns of genomic diversity to better understand how climatic cycles might correlate with the species demographic history and current genetic structure. We show that during the Last Glacial Maximum, there were ecological barriers that are absent today, possibly dividing the range of the species into three geographically separated areas of suitable habitat. Consistent with a historical reduction in habitat area, our analysis of demographic changes shows that L. jocu experienced a severe bottleneck followed by a population size expansion. We also found an absence of genetic structure and similar levels of genetic diversity throughout the sampled range of the species. Collectively, our results suggest that habitat availability changes have not obviously influenced contemporary levels of genetic divergence between populations. However, our demographic analyses suggest that the high sensitivity of this species to environmental change should be taken into consideration for management strategies. Furthermore, the general low levels of genetic structure and inference of high gene flow suggest that L. jocu likely constitutes a single stock in Brazilian waters and, therefore, requires coordinated legislation and management across its distribution.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Understanding how genetic variability is partitioned between and within populations of the same species is relevant for conservation management because it defines how many populations should be managed (Ovenden et al. 2015; Petrolo et al. 2021), and their potential differences in population size (Hauser and Carvalho 2008). Such patterns of genetic diversity are strongly influenced by migration and, thus, by how the connectivity between areas of suitable habitat changes over time. Although it is well understood how spatial and temporal changes in habitat suitability have driven diversification in terrestrial species (Laiolo and Tella 2006; Lim et al. 2011; Dennison et al. 2015; Camurugi et al. 2021), relatively less is known about how these processes affect marine species (Nanninga et al. 2014; Gonzalez et al. 2016; Choo et al. 2021).
High levels of genetic divergence have been revealed in less vagile marine species and attributed to unsuitable habitat restricting gene flow between suitable ones (e.g., Riginos and Nachman 2001; Bernardi and Vagelli 2004; Hemmer-Hansen et al. 2007; Nanninga et al. 2014; Catarino et al. 2017; Momigliano et al. 2017). Conversely, it has long been assumed that a lack of genetic structure will result from unrestricted gene flow between populations in species with higher dispersal capacity (Roughgarden et al. 1985; Ward et al. 1994; Doherty et al. 1995; Waples 1998). While this hypothesis has been supported by studies with limited numbers of genetic markers (e.g., Habib and Sulaiman 2016; Lacerda et al. 2016; Souza et al. 2019), recent genomic studies are changing this paradigm (e.g., Lah et al. 2016; Rodrígues-Ezpeleta et al. 2016; Cheng et al. 2021; Fahradi et al. 2022).
Studies of several marine species that have high dispersal capacity have shown that environmental gradients, such as ocean depth, salinity, oceanic currents, and water temperature are associated with patterns of genetic differentiation (Bekkevold et al. 2005; Galarza et al. 2009; Amaral et al. 2012; Ludt and Rocha 2015; e.g., the Black Sea Bass, Roy et al. 2012; the Atlantic Cod, Bradbury et al. 2013; the Atlantic Bluefin Tuna, Albaina et al. 2013; the Spiny Lobster, Truelove et al. 2017; and the Red Cusk-Eel, Córdova-Alarcón et al. 2019). Therefore, more effort is needed to describe present day environmental associations with genetic structure in marine species of high dispersal capacity to ascertain whether there are general patterns and to identify the relative importance of particular life history traits or habitat requirements.
The historical distribution of environmental features has also been shown to contribute to current patterns of genetic variation in marine species (Féral 2002; Hauser and Carvalho 2008; Briggs and Bowen 2012). During the glacial periods of the Pleistocene, lower sea levels changed the extent and distribution of available habitat, particularly for coastal species (Reeder-Myers et al. 2015; Ludt and Rocha 2015; Dolby et al. 2020). For example, during the Last Glacial Maximum (LGM), some 20 kya, sea surface temperature, salinity and sea level were lower than present (sea level 120 ± 5 m lower; Fairbanks 1989; Chappell et al. 1996). This led to demographic contractions and genetic drift in isolated populations of many coastal species (Keller et al. 2005; Alò and Turner 2005; Hauser and Carvalho 2008; England et al. 2010; Pavlova et al. 2017; Brauer and Beheregaray 2020). In addition, glacial cycles also affected oceanic currents, which will directly determine patterns of migration and gene flow in species with a pelagic larval phase (Ottersen et al. 2010; White et al. 2010; Neves et al. 2016).
Demographic changes induced by glacial cycles have been hypothesized to explain the low effective population sizes relative to census size generally observed in marine species (Hauser and Carvalho 2008). Although the LGM has been associated with demographic and genetic bottlenecks in many marine species (e.g., cod, Ólafsdóttir et al. 2014), some species were shown to have experienced a demographic expansion (e.g., Atlantic Bluefin Tuna and Swordfish, Alvarado Bremer et al. 2005; Thornback Ray, Chevolot et al. 2006; Angler, Charrier et al. 2006; Sand goby, Larmuseau et al. 2009; Jenkins et al. 2018). This suggests that the impact of the LGM on demography and genetic variation cannot be broadly generalized across species. Understanding genetic diversity variation due to environmental changes is especially relevant for exploited species because it can affect adaptation capacity.
Species of the Lutjanidae family, known as Snappers, are heavily exploited worldwide, reaching a yearly catch of 125,000 tons in 2018 (Pauly et al. 2020). In some regions of Brazil, the catch of this species represents approximately 40% of all the fishery resources (Rezende et al. 2003; Frédou et al. 2006), making it one of the most economically important species of the country. Nevertheless, several species of this group are already in an overexploited or collapsed state in Brazil (Anderson et al. 2015; Lindeman et al. 2016; Verba et al. 2020), demonstrating the need for establishing sustainable fisheries practices. Among the most exploited species is the large carnivorous Dog snapper Lutjanus jocu, with a yearly global catch close to 2000 tons, of which 99% is in Brazil (Freire et al. 2015).
Lutjanus jocu inhabits reefs, estuaries, and mangroves, and is strongly dependent on coastal and rocky shores (Caló et al. 2009; Moura et al. 2011; Reis-Filho et al. 2019). It is distributed throughout the western coast of the Atlantic Ocean, from north-eastern USA to south-eastern Brazil, including isolated oceanic islands (Feitoza et al. 2003; Lima Viana 2009). Similar to other closely related species, L. jocu has a pelagic larval phase that lasts between 20 and 40 days, which potentially allows long-distance dispersal facilitated by oceanic currents (Zapata and Herrón 2002; Pineda et al. 2007; Bezerra et al. 2021). Accordingly, a study using one mitochondrial gene found no genetic structure along the Brazilian coast and demographic signals consistent with a range expansion (Souza et al. 2019). Yet, mitochondrial studies offer a limiting view of population divergence and diversity because these markers are often under selection, are only maternally inherited, cannot detect subtle and recent divergence due to incomplete lineage sorting, and are sensitive to sample size (Hurst and Jiggins 2005; Fratini et al. 2016; Younger et al. 2017). Genomic data are required to infer finer grained spatial genetic structure and the demographic history of wild marine species (e.g., Petrolo et al. 2021). Understanding the habitat connectivity and demographic history of L. jocu is timely, because it is listed as ‘data deficient’ in the IUCN Red List, has no identified areas dedicated to its conservation (Lindeman et al. 2016), and shows a decreasing population size and body size consistent with overexploitation (Bender et al. 2013).
To examine how past environmental cycles have affected genetic divergence and diversity of L. jocu we applied species distribution modeling to compare habitat availability during the LGM and present day and characterized genetic variation using a data set of 6286 Single Nucleotide Polymorphisms (SNPs). With this data set, we tested the hypotheses that less habitat will be associated with a lower population size and that low genetic structure will be found across areas of largely contiguous habitat. More generally we describe genetic structure and diversity for its application to the sustainable management of a heavily exploited and data deficient fish species.
Materials and methods
Species distribution modeling
To understand how glacial cycles affected the distribution of L. jocu, we have inferred the habitat suitability of the species during the current interglacial period, projected this model to the LGM, and compared the two models. First, we collected recent presence data of L. jocu along the entire coast of Brazil, using the geographic coordinates of the databases Global Biodiversity Information Facility (GBIF, https://www.gbif.org) and SpeciesLink project (https://www.splink.cria.org.br). After the exclusion of duplicates, we considered a total of 130 presence points. Since we do not have information about absences, pseudo-absences were generated by randomly selecting other 130 points within the study area with maximum bathymetry of 100 m (similar to the known limit of depth for this species; Froese and Pauly 2000; Feitoza et al. 2003; Frédou and Ferreira 2005; Olavo et al. 2011).
Second, to identify the most important environmental variables that affect the distribution of L. jocu, we used an approach based on Bayesian Additive Regression Trees (BART), implemented in the embarcadero package (Carlson 2020) in R (R Core Team 2021). BART was shown to perform better than more traditional methods when using small datasets as it implements a Bayesian approach that allows a better quantification of associated uncertainties (Baquero et al. 2021; Konowalik and Nosol 2021). These models estimate the probability of the suitability (chance of occurrence) of a species presence based on decision “trees” that divide predictor variables with nested binary rule sets. The rules for generating these trees in BART are defined by posterior probabilities (Carlson 2020).
To fit BARTs, we tested the following environmental variables for the present time, extracted from the Marine Spatial Ecology database (MARSPEC, Braconnot et al. 2007; Sbrocco and Barber 2013; Sbrocco 2014): mean bathymetry (m, 30 arc-second); slope (degrees, 30 arc-second); aspect (East–West and North–South, the azimuthal direction of the steepest slope, a horizontal orientation of the seabed; radians, 30 arc-second); distance from the coast (km); minimum, maximum, mean and range of Sea Surface Temperature (SST; ºC, 2.5 arc-minute); and minimum, maximum, mean and range of Sea Surface Salinity (SSS; psu, 1 arc-degree) (Fig. S1). These variables were selected because they are likely to have an effect on marine fish distribution, dispersal, reproduction, and survival (Godoy et al. 2002; Hopkins and Cech 2003; Wiley et al. 2003; Lara and González 2005; Pittman and Brown 2011; Barneche et al. 2018; Wellington et al. 2021). Variables selection procedure was done using the automatic variable.step function of the embarcadero package (Carlson 2020). This function estimates for 50 times a complete model with all predictors and a small predefined set of trees (n = 20), eliminating the least informative variables in all 50 runs. Each time the least informative variable is removed, the function runs the models again (n = 50 more times), recording the Root Mean Square Error (RMSE). These steps are repeated automatically until there are only three covariates left and the model with the lowest average RMSE is selected.
Once the most important variables for the present time were selected, the same variables were extracted for the LGM (also from MARSPEC) and habitat suitability was projected for the current and the LGM scenarios. Given that the occurrence of this species has a relatively small number of points and that these are not randomly distributed across environmental variables, we estimated favorability instead of suitability using the probability of occurrence. Environmental favorability reflects the variation of the occurrence probability and is less affected by the presence/absence ratio (Real et al. 2006; Acevedo and Real 2012). It can vary between 0 (unsuited for the species) and 1 (ideal for occurrence of the species). Here, we considered that favorability values higher than 0.70 are highly suitable for the species, values between 0.40 and 0.70 have intermediate suitability, and values below 0.40 have low suitability.
Genetic analyses
To assess if the habitat suitability variation is consistent with genetic patterns, we collected samples along most of the species distribution in Brazil and used SNP data to calculate genetic diversity, infer population structure, and investigate changes in effective population size through time.
Specimen collection and genomic sequencing
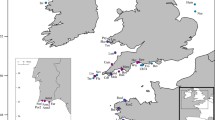
To assess population differentiation, we sampled tissues from 82 individuals of L. jocu collected from nine sites in Brazil (Fig. 1), representing ~ 60% of the whole distribution range of the species (Froese and Pauly 2000). These localities encompass several possible barriers to gene flow, such as two opposing oceanic currents (the Brazil Current and the North Brazil Current; Stramma and England 1999), and about 400 km of deeper habitat separating the continental shelf from an oceanic island (Fernando de Noronha Archipelago). Muscle tissues were collected during landings and from fish markets when the specific fishing locality was known, or provided by researchers from different institutions, and were preserved in 70% ethanol.

Sampling of Lutjanus jocu. a Sampling localities along the Brazilian coast: MA Maranhão, CE Ceará, RN Rio Grande do Norte, FN Fernando de Noronha Archipelago, PE Pernambuco, AL Alagoas, BAN Bahia North, BAS Bahia South, ES Espírito Santo; main oceanic currents are represented by arrows: NECC North Equatorial Countercurrent, NBC North Brazil Current, SEC South Equatorial Current, and BC Brazil Current; Depth is represented by shades of blue. b An adult individual of L. jocu caught near BAS (Photo: União das Associações Brasileiras de Pesca Subaquática) and c An adult near FN (Photo: Natália Roos)
DNA extraction, library preparation, sequencing and SNPs genotyping were carried out by DArT™ (Diversity Array Technology), following Georges et al. (2018). In short, this protocol is similar to ddRAD-sequencing approaches, and uses two restriction enzymes (Pst I and Sph I) to digest genomic DNA at homologous sites across samples. Adaptors with individual barcodes are ligated to DNA fragments and the resulting library amplified for 77 PCR cycles. The cleaned libraries are then sequenced on Illumina Hiseq 2500 at a minimum of 25x coverage per individual. Technical replicates were carried out by the company for 30% of the samples. Each SNP is classified according to three indexes: “Reproducibility”, which varies from 0 to 1 and represents the percentage of times that the same SNPs were found for the technical replicates; “Call Rate”, which varies between 0 and 1 and represents the percentage of individuals that were scored for that particular SNP; and “Polymorphism Information Content”, which varies between 0 and 1 and indicates how polymorphic each SNP is.
Data filtering
The initial raw dataset consisted of 41,462 SNPs and 82 individuals. Because missing data can lead to an underestimation of population structure, we excluded 14 individuals collected in eight different sites with more than 20% of missing data, likely due to low quality of the samples. For the remaining 68 individuals, the raw genotypic data were imported to R in genlight format using gl.read.dart function, and the data was filtered using the dartR package (Gruber et al. 2018).
We filtered the SNPs with the following criteria and order: (1) to guarantee high-quality sequences, we kept SNPs with > 97% Reproducibility (function gl.filter.repAvg); (2) to exclude uninformative loci, we only maintained polymorphic SNPs (function gl.filter.monomorphs); (3) to avoid linked SNPs, i.e., those known to be physically linked, we retained one SNP per read (per locus), selecting the SNP with the higher frequency (function gl.filter.secondaries); and (4) SNPs with a minimum Call Rate of 1, 0.95, 0.80 and 0.60, which reflect 0, 5, 20, and 40% of missing data, respectively, to test for biases caused by missing data (function gl.filter.callrate). After filtering, the number of SNPs retained was 6286 SNPs with 0% missing data, 9204 SNPs with 5% of missing data, 12,020 SNPs with 20%, and 14,253 SNPs with 40% missing data (Fig. S2). The dataset with 0% of missing data was used for all analyses, except when indicated.
Population genetic analysis
To estimate levels of genetic diversity in each individual and in each sampling locality, we used the package dartR in R and the Software DNAsp (Rozas et al. 2017). To estimate expected and observed heterozygosity for each sampling locality, the genlight file was used, and the function gl.report.heterozygosity from the package dartR was applied. To calculate π-SNP for each individual and for each sampling locality, DNAsp was used. To convert our data (genlight format) to a suitable format (fasta format), we used the function gl2fasta from the dartR package in R, coding all heterozygous positions as ambiguity codes (Gruber et al. 2018). Our fasta file was imported to DNAsp and split into two pseudohaplotypes, using the function Unfold fasta file for diploid individuals with ambiguity codes. The exported unfolded file was again imported to DNAsp, and the samples from each sampling locality were aggregated into sets using the Define sequence sets function. The function DNA Polymorphism from DNAsp was used on each sequence set to calculate π-SNP for each sampling locality, and for each individual to calculate individual diversity.
To infer population structure, we selected complementary methods that differ in their assumptions. First, the distribution of genetic variation was visualized using Principal Coordinate Analysis (PCoA) based on the Pearson Correlation Coefficient using the function gl.pcoa from dartR package (Gruber et al. 2018). We used the 4 datasets with the different thresholds of missing data for the PCoA, to investigate if different levels of missing data could affect the results. Second, using the 0% missing data dataset, population structure was further evaluated using the STRUCTURE software (Pritchard et al. 2000). STRUCTURE clusters individuals into K ancestral populations that maximize Hardy–Weinberg and linkage equilibria (Pritchard et al. 2000). For this, the data were converted from genlight to STRUCTURE format in R using the gl2structure function from the dartR package. We used the Admixture model, to account for possible genetic connectivity between differentiated clusters, and considered the allele frequencies of each population to be dependent because population divergence, if existent, was likely to still involve gene flow. We considered models with 1–4 ancestral clusters, representing the four coastal regions that are ecologically distinct: the north coast (MA, CE and RN), oceanic island (FN), northeast coast (PE, AL, BAN) and central coast (BAS and ES). We considered 10,000 iterations of burn-in, and a run length of 100,000 MCMC steps. We ran 10 independent replicates of each model to assess convergence of the alpha parameter, which was assessed visually based on the plots provided by the software. The most likely number of ancestral clusters (K) was inferred based on (1) higher values of log-likelihood visualized in CLUMPAK (Kopelman et al. 2015), (2) the stabilization of the curve of the mean log-likelihood, (3) the standard deviation of log-likelihood for each K, and (4) the geographic proximity of individuals assigned to the same cluster. When models assuming different number of clusters show similar log-likelihoods, we favored the lowest number of K, following the recommendation of Pritchard et al. (2009).
To assess whether gene flow is influenced by the geographic distance among sampling localities, we tested for isolation by distance. For each pairwise comparison among sampling localities, we calculated FST and their linear geographic distance (km). We used a Mantel test (gl.ibd function from dartR package in R) with 999 permutations to test the significance of any association between genetic and geographic distances.
Demographic history
To infer the historical changes of the effective population size of L. jocu from the study area, we applied two methods: Tajima’s D and δaδi. First, we estimated Tajima’s D as a measure of departure from demographic stability based on the mutation-drift equilibrium, and the significance was assessed based on the confidence limits of D (two-tailed test) and a calculation of a p value, assuming that D follows a beta distribution (Tajima 1989). In DNAsp the same unfolded fasta file used to calculate π-SNP was used and Tajima’s D was estimated for the whole study area using the function Tajima’s test. Additionally, to investigate differences between demographic histories on the different sampling localities, samples from each locality were aggregated using the function Define sequence sets in DNAsp, and Tajima’s D was estimated for each set.
Second, to further investigate changes in population size based on deviations of the site frequency spectrum (SFS), we used the program δaδi (Gutenkunst et al. 2010) that implements a diffusion approximation method to explicitly compare alternative demographic models for the species on the study area. We converted the genlight file with 0% missing data (68 individuals, 6286 SNPs) into the SNP data format supported by δaδi with the R package radiator (Gosselin 2020) and calculated the observed one-dimensional folded SFS. We simulated four demographic models with an increasing number of parameters: (1) a neutral model with constant population size (no parameters); (2) a two-epoch model with an instantaneous change in population size to NuF at time T (2 parameters); (3) a bottlegrowth model with an instantaneous size change to NuB at Tt followed by an exponential size change to the present population size NuF (3 parameters); and (4) a three-epoch model with two instantaneous size changes (NuB, NuF) at times TB and TF (4 parameters). We used the δaδi pipeline (Portik et al. 2017) to optimize these models. This tool implements a customizable number of optimization rounds and the number of replicates per round, where the parameters with the highest likelihood score of any given round are passed as starting input to the next round. For the neutral model (model 1), we did not perform multiple rounds of optimization, as no parameters have to be fitted. For the three other models, we performed optimizations with the following settings: three rounds of optimization with 10, 20, 100 replicates in each round, with maximum iterations of 5, 10, 50 per replicate in each round. For optimization, we used the default Nelder–Mead method (Nelder and Mead 1965). We reran this approach five times to assess convergence of parameter estimates and likelihood of the models. To visualize the model fit we plotted the optimized SFS of each model against the empirical SFS, also plotting the residuals. Lastly, we compared model fit by choosing the model with the lowest Akaike information criterion (AIC) score (Akaike 1974), which takes into account both model likelihood and the number of parameters estimated.
Results
Species distribution modeling
Based on the lowest RMSE, the three out of thirteen environmental variables selected to model the L. jocu distribution on the study area were: (i) distance from the coast, (ii) range of Sea Surface Temperature (SST) and (iii) minimum Sea Surface Salinity (SSS). Using these variables, the distribution of the present data was successfully calibrated and favorability was estimated (Fig. S3, Fig. 2). For the LGM projection, favorability was always lower than 0.70, with an intermediate favorability (between 0.56 and 0.70) mostly concentrated in the southern region of the study area, in BAS (Fig. 2a). The region in the center of the study area, where the continental shelf is narrower, presented a lower favorability (between 0.42 and 0.56). Two areas with the lowest favorability (less than 0.40) appear between the intermediate regions during the LGM (Fig. 2a). The present day model projection indicates that the habitat suitability is high along the coast, with favorability values consistently over 0.70, and continuous (Fig. 2b). Overall, both the extent of habitat suitability and the connectivity along the coast of Brazil has increased from the LGM to the present, with the strongest increase of habitat suitability being observed in the isolated oceanic archipelago of FN, and in the region between the states of AL and RN.

Habitat favorability for Lutjanus jocu in Brazil during the last glacial cycle estimated using BART. a Favorability model for the Last Glacial Maximum and b Favorability model for present day. Gray brackets indicate regions with lower favorability during LGM (maximum around 0.40)
Population genetic analysis
Genetic diversity measures of L. jocu per sampling location were similar in general. The π-SNP varied between 0.198 in AL, at the center of our sampling area, and 0.254 in ES (the southernmost site; Table 1). The observed levels of heterozygosity per sampling locality varied between 0.177 (MA) and 0.246 (ES; Table S1 in the Supplement), and the individual levels of genetic diversity ranged from 0.167 (one individual from RN) and 0.350, (one from PE; Fig. S4).
Using the standard data set with 0% of missing data, the first four dimensions of the PCoA explained a total of 8.1% of the genetic variation observed across the study area (Fig. 3). PC1, PC2 and PC4 indicate some dissimilarity of ES samples (the southernmost sample locality) in comparison with the others, but this is explained by a low percentage of the variance. Less stringent filtering datasets resulted in the same pattern (Fig. S5). In the STRUCTURE analyses assuming K = 1, the parameter alpha did not converge even when increasing length to 1,000,000 of burn-in and 1,000,000 of run length. For the remaining Ks tested (2–4), alpha converged with the initial burn-in and length run (10,000 and 100,000; respectively). The assessment of likelihood showed that both K = 1 and K = 2 resulted in similar scores: average for K = 1 is − 372,998.7 (standard deviation = 19.11) and, for K = 2, − 368,582.4 (standard deviation = 5081.80; Fig. S6 and Table S2). When assuming K = 2, we find one ancestral cluster spread across most sampling localities, and a less frequent cluster represented by individuals in ES and PE sites, which are more than 1400 km apart, and also individuals from FN and BAN, with a lower percentage of contribution (Fig. S7). For K = 3, we find a third cluster spread in all the individuals, except the two samples from ES and one from PE. Given the combination of no convergence of the alpha parameter in STRUCTURE, low difference in likelihood scores between K = 1 and K = 2, the large standard deviations for K = 2, and the lack of geographic cohesion of clusters, we consider that there is no significant population structure in our dataset. This conclusion is also in agreement with the results visualized in the PCoA (Fig. 3).

Clustering analysis of Lutjanus jocu using 0% missing data (6286 SNPs). a PC1 and PC2, b PC3 and PC4, and c histogram showing percentage of explanations for each axis. MA Maranhão, CE Ceará, RN Rio Grande do Norte, FN Fernando de Noronha Archipelago, PE Pernambuco, AL Alagoas, BAN Bahia North, BAS Bahia South, ES Espírito Santo
Most FST values were below 0.006, with the only exceptions being between ES and other sites, in which maximum FST was 0.04 (ES x RN), between BAN and PE (0.01) and between BAN and FN (0.01; Table S3). Isolation by distance results showed a positive but non-significant correlation between genetic and geographic distances (Mantel r = 0.366, p value = 0.061) (Fig. S8).
Demographic history
Tajima’s D for the whole study area was positive (0.4712) but not significant (p > 0.05). When calculating for each sampling locality, Tajima’s D was positive for eight of the nine sampling localities (Table 1) and negative for ES, yet none of these deviations from the neutral expectation was significant (p > 0.05).
In the demographic modeling estimated using δaδi, the neutral model with constant population size had the highest AIC score (Fig. 4a, Table 2), and was therefore rejected in favor of every model incorporating a change in population size. The two-epoch model showed an intermediate AIC score and no convergence in the estimated parameters across replicates, reflecting a poor representation of the empirical data. The bottlegrowth and three-epoch models showed the lowest AIC scores with little differences and the parameter estimates converged across replicates and across models (Fig. 4a, Table 2), showing that the observed data fit equally well to both models. The residuals reflect the same trend as the AIC values, with the simpler models showing a deficit of singletons and excess of low-frequency SNPs and the two most complex models showing virtually no residuals (Fig. S9). Estimates of effective population size reflect an initial bottleneck down to 6.49 and 4.01% of the ancestral population size, followed by a subsequent expansion of 240.98 and 489.67% of the ancestral populations, respectively for the three-epoch and the bottlegrowth models (Fig. 4, Table 2).

Demographic history of Lutjanus jocu. a Parameters estimated for the bottlegrowth model and b for the three epochs model. nuB ratio of population size immediately after change to the ancient population size, nuF ratio of the contemporary to ancient population size, T time in the past in units of 2Ne*generations, TB length of bottleneck, TF time since bottleneck recovery
Discussion
Knowledge of the evolutionary processes shaping the distribution of genetic variation is fundamental to developing sustainable management practices and predicting responses to environmental changes (Ward 2006; Reiss et al. 2009; Young et al. 2015; Benestan et al. 2021). Coastal marine species are generally sensitive to glacial cycles because changes in sea level alter the areas of habitat suitability. However, the extent to which these cycles impacted the size of current populations and the genetic variation of marine species appears to vary considerably and general patterns are not yet apparent for species in tropical waters.
Here, we modeled temporal changes in habitat suitability along the Brazilian coast for the commercially exploited marine fish L. jocu, which inhabits intermediate depths (20–90 m; Frédou and Ferreira 2005; Olavo et al. 2011), and is, therefore, expected to have experienced decreases in habitat as the sea level retracts during glacial periods. We then characterized current patterns of genetic diversity for L. jocu and looked for evidence of genetic structure associated with historical discontinuities in habitat and genetic bottlenecks associated with past reductions in habitat. More generally, the knowledge of how genetic variation of L. jocu is distributed across the study area is needed to establish whether fishing pressure is being applied to one or to several genetically distinct populations, and if the fishery is impacting genetically depauperate populations (Petrolo et al. 2021). In addition, levels of genetic variation can help managers gauge the likely resilience to environmental change because genome-wide levels of genetic variation reflect adaptive potential and the risks imposed by inbreeding (Harrisson et al. 2014).
Low genetic structure in a species with high vagility
The distribution of genetic variation revealed by our PCA and STRUCTURE analyses shows no evidence for strong genetic divergence across the entire study area. Both analyses did, however, show that individuals from the ES site (far south) and the PE site (center) were genetically different. Notably, these two sites also have higher nucleotide diversity and observed heterozygosity than the remaining localities, suggesting that this genetic difference could reflect local demographic effects, such as higher effective population size or migration rates, rather than independent ancestry (Lawson et al. 2018). Future studies with a denser population level sampling in this region can test these hypotheses. We find no significant isolation by distance (p value = 0.061; Fig. S8), suggesting that gene flow is not restricted by geographic distance at the scale of our sampling. Although our sampling covers most of the species’ distribution in Brazil (Fig. 3), we note that we did not include samples from the extremes of the species range in the country, such as the states of São Paulo, in the south, and Pará, in the north.
The lack of evidences of strong genetic structure or isolation by distance in L. jocu is consistent with other species with similar life history traits, such as an extensive pelagic larval phase, long lifespan and high dispersal capability (Palumbi 1994, 2003; Pineda et al. 2007; Haye et al. 2014). Therefore, it is likely that the larvae are capable of long-distance movement with oceanic currents, thereby maintaining the genetic connectivity. In addition, L. jocu forms large spawning aggregations throughout its range (Claro and Lindeman 2003; Heyman and Kjerfve 2008; Biggs and Nemeth 2014), including in Brazil (França and Olavo 2015; Bezerra et al. 2021; França et al. 2021). Wide-spread movement of adults to spawning aggregations will also contribute to genetic homogenization. Lack of genetic structure has been found in other co-occurring Lutjanidae species with similar dispersal rates, such as L. purpureus (Gomes et al. 2012), L. analis (Souza et al. 2019), and Ocyurus chrysurus (Vasconcellos et al. 2008), indicating that this might be a general pattern for species of this group.
The low levels of genetic structure are consistent with the high habitat connectivity shown in the contemporary distribution model (Fig. 2b) that potentially facilitate dispersal and gene flow. In contrast, the LGM distribution model shows an area of low habitat suitability in the region of CE and BAN potentially acting as a historical barrier to gene flow (Fig. 2a). Such low level of population divergence across a historical barrier to dispersal can be explained either because habitats that were less suitable for adults did not constrain the exchange of pelagic larvae carried by oceanic currents, or because more recent periods with higher habitat connectivity were sufficient to homogenize previously differentiated populations (Taylor et al. 2006).
Historical changes in habitat suitability are concordant with demographic change
Our demographic modeling strongly supports the hypothesis of a past genetic bottleneck, a result that is consistent with the distribution modeling showing a reduction in available habitat during the LGM. The two best supported demographic models (Three epoch and bottlegrowth models; Fig. 4, Table 2) suggest an initial strong bottleneck to around 5% of the ancestral population size, consistent with the positive values of Tajima’s D for nuclear markers. The decrease was followed by a recent two- to fourfold increase in effective population size relative to the ancestral population size. Our finding of a recent population size expansion is consistent with another study of L. jocu covering nearly the same study area, based on a single mitochondrial gene, that reported negative values of Tajima’s D (Souza et al. 2019). Contrasting demographic signals from maternally inherited mitochondrial data compared with bi-parentally inherited nuclear markers can be explained by their differences in effective population size, which in turn translates to different recovery times following a demographic change (Gattepaille et al. 2013). For example, in the Atka Mackerel, microsatellite markers provided no indication for population size variation, while mitochondrial DNA analysis showed significantly negative Tajima’s D, indicating a bottleneck, founder effect or selection (Canino et al. 2010).
The most recent increase in effective population size, following the bottleneck, is consistent with the distribution models that indicate an expansion of suitable habitat from the LGM to the present. During the LGM, when sea level was lower, most of the Brazilian continental shelf was exposed, reducing the habitat of near-shore species. Contraction of available habitat affects L. jocu’s feeding grounds, such as the shallow reefs on the continental shelf where most of its prey occurs. Sea-level reduction also leads to a contraction of estuaries, which are essential for the development of juvenile L. jocu prior to moving to reef areas (Moura et al. 2011). The loss of estuarine habitat during the LGM was possibly stronger on the Brazilian coast in contrast to other areas of distribution of the species, such as the coast of the USA, because Brazilian estuaries are narrower and more sensitive to sea level variation (Lessa et al. 2018).
Implications for management
Our results indicate little population structure (Fig. 3), lack of isolation by distance (Fig. S7), and similar levels of genetic diversity throughout L. jocu’s range in Brazil (Fig. S8). The combination of high gene flow and sufficient levels of protection could result in localized exploited areas being supplemented by dispersal from elsewhere (Bar-David et al. 2007; Goñi et al. 2010; Lawton et al. 2011; Sutherland et al. 2012). Conversely, the existence of highly connected populations means that high levels of overexploitation within regions of the species range impacts the entire population, including no-take protected areas (Jones et al. 2007; Agardy et al. 2011; Moffitt et al. 2011).
The extensive exploitation of L. jocu (Freire et al. 2015) in conjunction with our data showing high levels of connectivity point to the need of a coordinated approach to ensuring a sustainable fishery. Furthermore, our demographic analysis demonstrates the sensitivity of L. jocu to past environmental changes. Consequently we suggest that management also be vigilant for additional reductions in population size in this commercially important and heavily exploited species that are a result of habitat alteration from ongoing global warming (Woodroffe 2007; Hauser and Carvalho 2008; Albouy et al. 2013; James et al. 2013).
Data availability
Raw data are available on https://github.com/juliaverba/Lutjanus_jocu/blob/main/Report_DLu18-3697_SNP_singlerow_1.csv and code is available upon request to the corresponding author.
References
Acevedo P, Real R (2012) Favourability: concept, distinctive characteristics and potential usefulness. Naturwissenschaften 99:515–522. https://doi.org/10.1007/s00114-012-0926-0
Agardy T, di Sciara GN, Christie P (2011) Mind the gap: addressing the shortcomings of marine protected areas through large scale marine spatial planning. Mar Policy 35:226–232. https://doi.org/10.1016/j.marpol.2010.10.006
Akaike H (1974) A new look at the statistical model identification. IEEE Trans Autom Control 19:716–723. https://doi.org/10.1109/TAC.1974.1100705
Albaina A, Iriondo M, Velado I, Laconcha U, Zarraonaindia I, Arrizabalaga H, Pardo MA, Lutcavage M, Grant WS, Estonba A (2013) Single nucleotide polymorphism discovery in albacore and Atlantic bluefin tuna provides insights into worldwide population structure. Anim Genet 44:678–692. https://doi.org/10.1111/age.12051
Albouy C, Guilhaumon F, Leprieur F, Lasram FBR, Somot S, Aznar R, Velez L, Le Loc’h F, Mouillot D (2013) Projected climate change and the changing biogeography of coastal Mediterranean fishes. J Biogeogr 40:534–547. https://doi.org/10.1111/jbi.12013
Alò D, Turner TF (2005) Effects of habitat fragmentation on effective population size in the endangered Rio Grande Silvery Minnow. Conserv Biol 19:1138–1148. https://doi.org/10.1111/j.1523-1739.2005.00081.x
Alvarado Bremer JR, Viñas J, Mejuto J, Ely B, Pla C (2005) Comparative phylogeography of Atlantic bluefin tuna and swordfish: the combined effects of vicariance, secondary contact, introgression, and population expansion on the regional phylogenies of two highly migratory pelagic fishes. Mol Phylogenet Evol 36:169–187. https://doi.org/10.1016/j.ympev.2004.12.011
Amaral AR, Beheregaray LB, Bilgmann K, Boutov D, Freitas L, Robertson KM, Sequeira M, Stockin KA, Coelho MM, Möller LM (2012) Seascape genetics of a globally distributed, highly mobile marine mammal: the short-beaked common dolphin (Genus Delphinus). PLoS One 7:e31482. https://doi.org/10.1371/journal.pone.0031482
Anderson W, Claro R, Cowan J, Lindeman K, Padovani-Ferreira B, Rocha LA (2015) Lutjanus campechanus (errata version published in 2017). The IUCN Red List of Threatened Species 2015: e.T194365A115334224. https://doi.org/10.2305/IUCN.UK.2015-4.RLTS.T194365A2322724.en. Downloaded on 28 October 2021. IUCN
Baquero RA, Barbosa AM, Ayllón D, Guerra C, Sánchez E, Araújo MB, Nicola GG (2021) Potential distributions of invasive vertebrates in the Iberian Peninsula under projected changes in climate extreme events. Divers Distrib 27:2262–2276. https://doi.org/10.1111/ddi.13401
Bar-David S, Segev O, Peleg N, Hill N, Templeton AR, Schultz CB, Blaustein L (2007) Long-distance movements by fire salamanders (Salamandra infraimmaculata) and implications for habitat fragmentation. Isr J Ecol Evol 53:143–159. https://doi.org/10.1080/15659801.2007.10639579
Barneche DR, Burgess SC, Marshall DJ (2018) Global environmental drivers of marine fish egg size. Glob Ecol Biogeogr 27:890–898. https://doi.org/10.1111/geb.12748
Bekkevold D, Aandré C, Dahlgren TG, Clausen LAW, Torstensen E, Mosegaard H, Carvalho GR, Christensen TB, Norlinder E, Ruzzante DE (2005) Environmental correlates of population differentiation in Atlantic Herring. Evolution 59:2656–2668. https://doi.org/10.1111/j.0014-3820.2005.tb00977.x
Bender MG, Floeter SR, Hanazaki N (2013) Do traditional fishers recognise reef fish species declines? Shifting environmental baselines in Eastern Brazil. Fish Manag Ecol 20:58–67
Benestan LM, Rougemont Q, Senay C, Normandeau E, Parent E, Rideout R, Bernatchez L, Lambert Y, Audet C, Parent GJ (2021) Population genomics and history of speciation reveal fishery management gaps in two related redfish species (Sebastes mentella and Sebastes fasciatus). Evol Appl 14:588–606. https://doi.org/10.1111/eva.13143
Bernardi G, Vagelli A (2004) Population structure in Banggai cardinalfish, Pterapogon kauderni, a coral reef species lacking a pelagic larval phase. Mar Biol 145(4):803–810
Bezerra IM, Hostim-Silva M, Teixeira JLS, Hackradt CW, Félix-Hackradt FC, Schiavetti A (2021) Spatial and temporal patterns of spawning aggregations of fish from the Epinephelidae and Lutjanidae families: An analysis by the local ecological knowledge of fishermen in the Tropical Southwestern Atlantic. Fish Res 239:105937. https://doi.org/10.1016/j.marenvres.2018.10.004
Biggs C, Nemeth RS (2014) Timing, size, and duration of a Dog (Lutjanus jocu) and Cubera Snapper (Lutjanus cyanopterus) spawning aggregation in the US Virgin Islands. Proc. Annu. Gulf Caribb. Fish. Inst
Braconnot P, Otto-Bliesner B, Harrison S, Joussaume S, Peterchmitt J-Y, Abe-Ouchi A, Crucifix M, Driesschaert E, Fichefet T, Hewitt CD, Kageyama M, Kitoh A, Loutre M-F, Marti O, Merkel U, Ramstein G, Valdes P, Weber L, Yu Y, Zhao Y (2007) Results of PMIP2 coupled simulations of the mid-holocene and last glacial maximum: part 2: feedbacks with emphasis on the location of the ITCZ and mid- and high latitudes heat budget. Clim past 3:279–296. https://doi.org/10.5194/cp-3-279-2007
Bradbury IR, Hubert S, Higgins B, Bowman S, Borza T, Paterson IG, Snelgrove PVR, Morris CJ, Gregory RS, Hardie D, Hutchings JA, Ruzzante DE, Taggart CT, Bentzen P (2013) Genomic islands of divergence and their consequences for the resolution of spatial structure in an exploited marine fish. Evol Appl 6:450–461. https://doi.org/10.1111/eva.12026
Brauer CJ, Beheregaray LB (2020) Recent and rapid anthropogenic habitat fragmentation increases extinction risk for freshwater biodiversity. Evol Appl 13:2857–2869. https://doi.org/10.1111/eva.13128
Briggs JC, Bowen BW (2012) A realignment of marine biogeographic provinces with particular reference to fish distributions. J Biogeogr 39:12–30. https://doi.org/10.1111/j.1365-2699.2011.02613.x
Caló CFF, Schiavetti A, Cetra M (2009) Local ecological and taxonomic knowledge of snapper fish (Teleostei: Actinopterygii) held by fishermen in Ilhéus, Bahia, Brazil. Neotrop Ichthyol 7:403–414. https://doi.org/10.1590/S1679-62252009000300007
Camurugi F, Gehara M, Fonseca EM, Zamudio KR, Haddad CF, Colli GR, Thomé MT, Prado CP, Napoli MF, Garda AA (2021) Isolation by environment and recurrent gene flow shaped the evolutionary history of a continentally distributed neotropical treefrog. J Biogeograph 48(4):760–772
Canino MF, Spies IB, Lowe SA, Grant WS (2010) Highly discordant nuclear and mitochondrial DNA diversities in Atka mackerel. Mar Coast Fish 2(1):375–387
Carlson CJ (2020) Embarcadero: species distribution modelling with Bayesian additive regression trees in r. Methods Ecol Evol. https://doi.org/10.1111/2041-210X.13389
Catarino D, Stanković D, Menezes G, Stefanni S (2017) Insights into the genetic structure of the rabbitfish Chimaera monstrosa (Holocephali) across the Atlantic-Mediterranean transition zone. J Fish Biol 91(4):1109–1122
Chappell J, Omura A, Esat T, McCulloch M, Pandolfi J, Ota Y, Pillans B (1996) Reconciliaion of late quaternary sea levels derived from coral terraces at Huon Peninsula with deep sea oxygen isotope records. Earth Planet Sci Lett 141:227–236. https://doi.org/10.1016/0012-821X(96)00062-3
Charrier G, Chenel T, Durand JD, Girard M, Quiniou L, Laroche J (2006) Discrepancies in phylogeographical patterns of two European anglerfishes (Lophius budegassa and Lophius piscatorius). Mol Phylogenet Evol 38:742–754. https://doi.org/10.1016/j.ympev.2005.08.002
Cheng SH, Gold M, Rodriguez N, Barber PH (2021) Genome-wide SNPs reveal complex fine scale population structure in the California market squid fishery (Doryteuthis opalescens). Conserv Genet 22(1):97–110
Chevolot M, Hoarau G, Rijnsdorp AD, Stam WT, Olsen JL (2006) Phylogeography and population structure of thornback rays (Raja clavata L., Rajidae). Mol Ecol 15:3693–3705. https://doi.org/10.1111/j.1365-294X.2006.03043.x
Choo LQ, Bal TM, Goetze E, Peijnenburg KT (2021) Oceanic dispersal barriers in a holoplanktonic gastropod. J Evol Biol 34(1):224
Claro R, Lindeman K (2003) Spawning aggregation sites of snapper and grouper species (Lutjanidae and Serranidae) on the Insular Shelf of Cuba. Gulf Caribb 14:91–106. https://doi.org/10.18785/gcr.1402.07
Córdova-Alarcón VR, Araneda C, Jilberto F, Magnolfi P, Toledo MI, Lam N (2019) Genetic diversity and population structure of Genypterus chilensis, a commercial benthic marine species of the South Pacific. Front Mar Sci 6:748. https://doi.org/10.3389/fmars.2019.00748
de Souza AS, Dias Júnior EA, Perez MF, de Cioffi MB, Bertollo LAC, Garcia-Machado E, Vallinoto MNS, Pedro Manoel G, Molina WF (2019) Phylogeography and historical demography of two sympatric Atlantic snappers: Lutjanus analis and L. jocu. Front Mar Sci 6:545. https://doi.org/10.3389/fmars.2019.00545
Dennison S, McAlpin S, Chapple DG, Stow AJ (2015) Genetic divergence among regions containing the vulnerable great desert skink (Liopholis kintorei) in the Australian arid zone. PLoS One 10(6):e0128874
Doherty PJ, Planes S, Mather P (1995) Gene flow and larval duration in seven species of fish from the great barrier reef. Ecology 76:2373–2391. https://doi.org/10.2307/2265814
Dolby GA, Bedolla AM, Bennett SEK, Jacobs DK (2020) Global physical controls on estuarine habitat distribution during sea level change: consequences for genetic diversification through time. Glob Planet Change 187:103128. https://doi.org/10.1016/j.gloplacha.2020.103128
Duckett PE, Stow AJ (2013) Higher genetic diversity is associated with stable water refugia for a gecko with a wide distribution in arid Australia. Divers Distrib 19:1072–1083
England PR, Luikart G, Waples RS (2010) Early detection of population fragmentation using linkage disequilibrium estimation of effective population size. Conserv Genet 11:2425–2430. https://doi.org/10.1007/s10592-010-0112-x
Fairbanks RG (1989) A 17,000-year glacio-eustatic sea level record: influence of glacial melting rates on the Younger Dryas event and deep-ocean circulation. Nature 342:637–642. https://doi.org/10.1038/342637a0
Farhadi A, Pichlmueller F, Yellapu B, Lavery S, Jeffs A (2022) Genome-wide SNPs reveal fine-scale genetic structure in ornate spiny lobster Panulirus ornatus throughout Indo-West Pacific Ocean. ICES J Mar Sci 79(6):1931–1941
Feitoza B, Rocha L, Luiz O, Floeter S, Gasparini J (2003) Reef fishes of St. Paul’s Rocks: new records and notes on biology and zoogeography. Aqua 7:61–82
Féral J-P (2002) How useful are the genetic markers in attempts to understand and manage marine biodiversity? J Exp Mar Biol Ecol 268:121–145
França AR, Olavo G (2015) Indirect signals of spawning aggregations of three commercial reef fish species on the continental shelf of Bahia, east coast of Brazil. Braz J Oceanogr 63:289–301. https://doi.org/10.1590/S1679-87592015087506303
França A, Olavo G, Rezende S, Ferreira B (2021) Spatio-temporal distribution of mutton snapper and dog snapper spawning aggregations in the South-west Atlantic. Aquat Conserv: Mar Freshw Ecosyst. https://doi.org/10.1002/aqc.3536
Fratini S, Ragionieri L, Deli T, Harrer A, Marino IA, Cannicci S, Zane L, Schubart CD (2016) Unravelling population genetic structure with mitochondrial DNA in a notional panmictic coastal crab species: sample size makes the difference. BMC Evol Biol 16(1):1–5
Frédou T, Ferreira BP (2005) Bathymetric trends of northeastern Brazilian snappers (Pisces, Lutjanidae): implications for the reef fishery dynamic. Braz Arch Biol Technol 48:787–800. https://doi.org/10.1590/S1516-89132005000600015
Frédou T, Ferreira BP, Letourneur Y (2006) A univariate and multivariate study of reef fisheries off northeastern Brazil. ICES J Mar Sci 63:883–896. https://doi.org/10.1016/j.icesjms.2005.11.019
Freire K, Aragão JA, Araújo A, da Silva A, Bispo M, Velasco G, Carneiro M, Gonçalves F, Keunecke K, Mendonça J, Moro P, Motta FS, Olavo G, Pezzuto P, Santana R, Aguiar dos Santos R, Trindade-Santos I, Vasconcelos J, Vianna M, Divovich E (2015) Reconstruction of catch statistics for Brazilian marine waters (1950–2010). Fish Res Rep 23:3–30
Froese R, Pauly D (2000) FishBase. World Wide Web electronic publication. www.fishbase.org, version (08/2021)
Galarza JA, Carreras-Carbonell J, Macpherson E, Pascual M, Roques S, Turner GF, Rico C (2009) The influence of oceanographic fronts and early-life-history traits on connectivity among littoral fish species. PNAS 106:1473–1478. https://doi.org/10.1073/pnas.0806804106
Gattepaille LM, Jakobsson M, Blum MG (2013) Inferring population size changes with sequence and SNP data: lessons from human bottlenecks. Heredity 110:409–419. https://doi.org/10.1038/hdy.2012.120
Georges A, Gruber B, Pauly GB, White D, Adams M, Young MJ, Kilian A, Zhang X, Shaffer HB, Unmack PJ (2018) Genomewide SNP markers breathe new life into phylogeography and species delimitation for the problematic short-necked turtles (Chelidae: Emydura) of eastern Australia. Mol Ecol 27:5195–5213. https://doi.org/10.1111/mec.14925
Godoy EAS, Almeida TCM, Zalmon IR (2002) Fish assemblages and environmental variables on an artificial reef north of Rio de Janeiro, Brazil. ICES J Mar Sci 59:S138–S143. https://doi.org/10.1006/jmsc.2002.1190
Gomes G, Sampaio I, Schneider H (2012) Population structure of Lutjanus purpureus (Lutjanidae - Perciformes) on the Brazilian coast: further existence evidence of a single species of red snapper in the western Atlantic. An Acad Bras Ciênc 84:979–999. https://doi.org/10.1590/S0001-37652012000400013
Goñi R, Hilborn R, Díaz D, Mallol S, Adlerstein S (2010) Net contribution of spillover from a marine reserve to fishery catches. MEPS 400:233–243. https://doi.org/10.3354/meps08419
Gosselin T (2020) Radiator: RADseq data exploration, manipulation and visualization using R. https://thierrygosselin.github.io/radiator/.doi:10.5281/zenodo.3687060.10.5281/zenodo.3687060
Gonzalez BE, Knutsen H, Jorde PE (2016) Habitat discontinuities separate genetically divergent populations of a rocky shore marine fish. PloS One 11(10):e0163052
Gruber B, Unmack PJ, Berry OF, Georges A (2018) dartR: an r package to facilitate analysis of SNP data generated from reduced representation genome sequencing. Mol Ecol Res 18:691–699. https://doi.org/10.1111/1755-0998.12745
Gutenkunst R, Hernandez R, Williamson S, Bustamante C (2010) Diffusion approximations for demographic inference: DaDi. Nat Prec. https://doi.org/10.1038/npre.2010.4594.1
Habib A, Sulaiman Z (2016) High genetic connectivity of narrow-barred Spanish mackerel (Scomberomorus commerson) from the South China, Bali and Java Seas. Zool Ecol 26(2):93–99
Harrisson KA, Pavlova A, Telonis-Scott M, Sunnucks P (2014) Using genomics to characterize evolutionary potential for conservation of wild populations. Evol Appl 7:1008–1025. https://doi.org/10.1111/eva.12149
Hauser L, Carvalho GR (2008) Paradigm shifts in marine fisheries genetics: ugly hypotheses slain by beautiful facts. Fish Fish 9:333–362. https://doi.org/10.1111/j.1467-2979.2008.00299.x
Haye PA, Segovia NI, Muñoz-Herrera NC, Gálvez FE, Martínez A, Meynard A, Pardo-Gandarillas MC, Poulin E, Faugeron S (2014) Phylogeographic Structure in benthic marine invertebrates of the Southeast Pacific Coast of Chile with differing dispersal potential. PLoS One 9:e88613. https://doi.org/10.1371/journal.pone.0088613
Hemmer-Hansen JA, Nielsen EE, Grønkjaer P, Loeschcke V (2007) Evolutionary mechanisms shaping the genetic population structure of marine fishes; lessons from the European flounder (Platichthys flesus L.). Mol Ecol 16(15):3104–18
Heyman WD, Kjerfve B (2008) Characterization of transient multi-species reef fish spawning aggregations at Gladden Spit, Belize. Bull Mar Sci 83:531–551
Hopkins TE, Cech JJ (2003) The influence of environmental variables on the distribution and abundance of three elasmobranchs in Tomales Bay, California. Environ Biol Fish 66:279–291. https://doi.org/10.1023/A:1023907121605
Hurst GD, Jiggins FM (2005) Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: the effects of inherited symbionts. Proc Royal Soc B: Biol Sci 272(1572):1525–1534
James NC, van Niekerk L, Whitfield AK, Potts WM, Götz A, Paterson AW (2013) Effects of climate change on South African estuaries and associated fish species. Clim Res 57:233–248. https://doi.org/10.3354/cr01178
Jenkins TL, Castilho R, Stevens JR (2018) Meta-analysis of northeast Atlantic marine taxa shows contrasting phylogeographic patterns following post-LGM expansions. PeerJ 6:e5684. https://doi.org/10.7717/peerj.5684
Jones GP, Srinivasan M, Almany GR (2007) Population connectivity and conservation of marine biodiversity. Oceanography 20:100–111
Keller I, Excoffier L, Largiadèr CR (2005) Estimation of effective population size and detection of a recent population decline coinciding with habitat fragmentation in a ground beetle. J Evol Biol 18:90–100. https://doi.org/10.1111/j.1420-9101.2004.00794.x
Konowalik K, Nosol A (2021) Evaluation metrics and validation of presence-only species distribution models based on distributional maps with varying coverage. Sci Rep 11:1482. https://doi.org/10.1038/s41598-020-80062-1
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Res 15:1179–1191. https://doi.org/10.1111/1755-0998.12387
Lacerda AL, Kersanach R, Cortinhas MC, Prata PF, Dumont LF, Proietti MC, Maggioni R, D’Incao F (2016) High connectivity among blue crab (Callinectes sapidus) populations in the Western South Atlantic. PloS One 11(4):e0153124
Lah L, Trense D, Benke H, Berggren P, Gunnlaugsson Þ, Lockyer C, Öztürk A, Öztürk B, Pawliczka I, Roos A, Siebert U (2016) Spatially explicit analysis of genome-wide SNPs detects subtle population structure in a mobile marine mammal, the harbor porpoise. PloS One 11(10):e0162792
Laiolo P, Tella JL (2006) Landscape bioacoustics allow detection of the effects of habitat patchiness on population structure. Ecology 87(5):1203–1214
Lara EN, González EA (2005) The relationship between reef fish community structure and environmental variables in the southern Mexican Caribbean. J Fish Biol 53:209–221
Larmuseau MHD, Van Houdt JKJ, Guelinckx J, Hellemans B, Volckaert FAM (2009) Distributional and demographic consequences of Pleistocene climate fluctuations for a marine demersal fish in the north-eastern Atlantic. J Biogeogr 36:1138–1151. https://doi.org/10.1111/j.1365-2699.2008.02072.x
Lawson DJ, van Dorp L, Falush D (2018) A tutorial on how not to over-interpret STRUCTURE and ADMIXTURE bar plots. Nat Commun 9:3258. https://doi.org/10.1038/s41467-018-05257-7
Lawton RJ, Messmer V, Pratchett MS, Bay LK (2011) High gene flow across large geographic scales reduces extinction risk for a highly specialised coral feeding butterflyfish. Mol Ecol 20:3584–3598. https://doi.org/10.1111/j.1365-294X.2011.05207.x
Lessa GC, Santos FM, Souza Filho PW, Corrêa-Gomes LC (2018) Brazilian estuaries: a geomorphologic and oceanographic perspective. Brazilian estuaries. Springer, Cham, pp 1–37
Lim HC, Rahman MA, Lim SL, Moyle RG, Sheldon FH (2011) Revisiting Wallace’s haunt: coalescent simulations and comparative niche modeling reveal historical mechanisms that promoted avian population divergence in the Malay Archipelago. Evol Int J Org Evol 65(2):321–34
Lima Viana D (ed) (2009) O arquipélago de São Pedro e São Paulo: 10 anos de estação científica. SECIRM, Brasília, DF
Lindeman K, Anderson W, Carpenter KE, Claro R, Cowan J, Padovani-Ferreira B, Rocha LA, Sedberry G, Zapp-Sluis M (2016) Lutjanus cyanopterus. The IUCN Red List of Threatened Species e.T12417A506633. https://doi.org/10.2305/IUCN.UK.2016-1.RLTS.T12417A506633.en. Downloaded on 28 October 2021
Ludt WB, Rocha LA (2015) Shifting seas: the impacts of Pleistocene sea-level fluctuations on the evolution of tropical marine taxa. J Biogeogr 42:25–38. https://doi.org/10.1111/jbi.12416
Moffitt EA, Wilson White J, Botsford LW (2011) The utility and limitations of size and spacing guidelines for designing marine protected area (MPA) networks. Biol Conserv 144:306–318. https://doi.org/10.1016/j.biocon.2010.09.008
Momigliano P, Harcourt R, Robbins W, Jaiteh J, Mahardika GN, Sembiring A, Stow A (2017) Genetic connectivity and signatures of selection in grey reef sharks (Carcharinus amblyrhychos). Heredity 119:142
Moura RL, Francini-Filho RB, Chaves EM, Minte-Vera CV, Lindeman KC (2011) Use of riverine through reef habitat systems by dog snapper (Lutjanus jocu) in eastern Brazil. Estuar Coast Shelf Sci 95:274–278. https://doi.org/10.1016/j.ecss.2011.08.010
Nanninga GB, Saenz-Agudelo P, Manica A, Berumen ML (2014) Environmental gradients predict the genetic population structure of a coral reef fish in the red sea. Mol Ecol 23(3):591–602
Nelder JA, Mead R (1965) A simplex method for function minimization. Computer J 7:308–313. https://doi.org/10.1093/comjnl/7.4.308
Neves JMM, Lima SMQ, Mendes LF, Torres RA, Pereira RJ, Mott T (2016) Population structure of the Rockpool Blenny Entomacrodus vomerinus shows source-sink dynamics among ecoregions in the tropical Southwestern Atlantic. PLoS One 11:e0157472. https://doi.org/10.1371/journal.pone.0157472
Ólafsdóttir GÁ, Westfall KM, Edvardsson R, Pálsson S (2014) Historical DNA reveals the demographic history of Atlantic cod (Gadus morhua) in medieval and early modern Iceland. Proc Royal Soc B 281:20132976. https://doi.org/10.1098/rspb.2013.2976
Olavo G, Costa P, Martins A, Ferreira B (2011) Shelf-edge reefs as priority areas for conservation of reef fish diversity in the tropical Atlantic. Aquat Conserv 21:199–209. https://doi.org/10.1002/aqc.1174
Ottersen G, Kim S, Huse G, Polovina JJ, Stenseth NC (2010) Major pathways by which climate may force marine fish populations. J Mar Syst 79:343–360. https://doi.org/10.1016/j.jmarsys.2008.12.013
Ovenden JR, Berry O, Welch DJ, Buckworth RC, Dichmont CM (2015) Ocean’s eleven: a critical evaluation of the role of population, evolutionary and molecular genetics in the management of wild fisheries. Fish Fish 16(1):125–159
Palumbi SR (1994) Genetic divergence, reproductive isolation, and marine speciation. Annu Rev Ecol Evol Syst 25:547–572. https://doi.org/10.1146/annurev.es.25.110194.002555
Palumbi SR (2003) Population genetics, demographic connectivity, and the design of marine reserves. Ecol Appl 13:146–158. https://doi.org/10.1890/1051-761(2003)013[0146:PGDCAT]2.0.CO;2
Pauly D, Zeller D, Palomares MLD (eds) (2020) Sea around Us: concepts, design and data (seaaroundus.org)
Pavlova A, Beheregaray LB, Coleman R, Gilligan D, Harrisson KA, Ingram BA, Kearns J, Lamb AM, Lintermans M, Lyon J, Nguyen TTT, Sasaki M, Tonkin Z, Yen JDL, Sunnucks P (2017) Severe consequences of habitat fragmentation on genetic diversity of an endangered Australian freshwater fish: a call for assisted gene flow. Evol Appl 10:531–550. https://doi.org/10.1111/eva.12484
Petrolo E, Boomer J, O’Hare J, Bilgmann K, Stow A (2021) Stock structure and effective population size of the commercially exploited gummy shark Mustelus antarcticus. Mar Ecol Prog Ser 678:109–124. https://doi.org/10.3354/meps13859
Pineda J, Hare JA, Sponaugle S (2007) Larval transport and dispersal in the coastal ocean and consequences for population connectivity. Oceanography 20:22–39
Pittman SJ, Brown KA (2011) Multi-scale approach for predicting fish species distributions across coral reef seascapes. PLoS One 6:e20583. https://doi.org/10.1371/journal.pone.0020583
Portik DM, Leaché AD, Rivera D, Barej MF, Burger M, Hirschfeld M, Rödel M-O, Blackburn DC, Fujita MK (2017) Evaluating mechanisms of diversification in a Guineo-Congolian tropical forest frog using demographic model selection. Mol Ecol 26:5245–5263. https://doi.org/10.1111/mec.14266
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1093/genetics/155.2.945
Pritchard JK, Wen X, Falush D (2009) Documentation for STRUCTURE software: Version 2.3. 39
R Core Team (2021). R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria
Real R, Barbosa AM, Vargas JM (2006) Obtaining environmental favourability functions from logistic regression. Environ Ecol Stat 13:237–245. https://doi.org/10.1007/s10651-005-0003-3
Reeder-Myers L, Erlandson JM, Muhs DR, Rick TC (2015) Sea level, paleogeography, and archaeology on California’s Northern Channel Islands. Quatern Res 83:263–272. https://doi.org/10.1016/j.yqres.2015.01.002
Reis-Filho JA, Schmid K, Harvey ES, Giarrizzo T (2019) Coastal fish assemblages reflect marine habitat connectivity and ontogenetic shifts in an estuary-bay-continental shelf gradient. Mar Environ Res 148:57–66. https://doi.org/10.1016/j.marenvres.2019.05.004
Reiss H, Hoarau G, Dickey-Collas M, Wolff WJ (2009) Genetic population structure of marine fish: mismatch between biological and fisheries management units. Fish Fish 10:361–395. https://doi.org/10.1111/j.1467-2979.2008.00324.x
Rezende S, Ferreira B, Fredou T (2003) A pesca de lutjanídeos no nordeste do Brasil: histórico das pescarias, características das espécies e relevância para o manejo. Bol Tec Cient Cepene 11:1–17
Riginos C, Nachman MW (2001) Population subdivision in marine environments: the contributions of biogeography, geographical distance and discontinuous habitat to genetic differentiation in a blennioid fish, Axoclinus nigricaudus. Mol Ecol 10(6):1439–1453
Rodríguez-Ezpeleta N, Bradbury IR, Mendibil I, Álvarez P, Cotano U, Irigoien X (2016) Population structure of Atlantic mackerel inferred from RAD-seq-derived SNP markers: effects of sequence clustering parameters and hierarchical SNP selection. Mol Ecol Res 16(4):991–1001
Roughgarden J, Iwasa Y, Baxter C (1985) Demographic theory for an open marine population with space-limited recruitment. Ecology 66:54–67
Roy EM, Quattro JM, Greig TW (2012) Genetic management of black sea bass: influence of biogeographic barriers on population structure. Mar Coast Fish 4:391–402. https://doi.org/10.1080/19425120.2012.675983
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sánchez-Gracia A (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 34:3299–3302. https://doi.org/10.1093/molbev/msx248
Sbrocco EJ (2014) Paleo-MARSPEC: gridded ocean climate layers for the mid-holocene and last glacial maximum. Ecology 95:1710–1710. https://doi.org/10.1890/14-0443.1
Sbrocco EJ, Barber PH (2013) MARSPEC: ocean climate layers for marine spatial ecology. Ecology 94:979–979. https://doi.org/10.1890/12-1358.1
Stramma L, England M (1999) On the water masses and mean circulation of the South Atlantic Ocean. J Geophys Res: Oceans 104(C9):20863–20883
Sutherland C, Elston DA, Lambin X (2012) Multi-scale processes in metapopulations: contributions of stage structure, rescue effect, and correlated extinctions. Ecology 93:2465–2473. https://doi.org/10.1890/12-0172.1
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Taylor EB, Boughman JW, Groenenboom M, Sniatynski M, Schluter D, Gow JL (2006) Speciation in reverse: morphological and genetic evidence of the collapse of a three-spined stickleback (Gasterosteus aculeatus) species pair. Mol Ecol 15:343–355. https://doi.org/10.1111/j.1365-294X.2005.02794.x
Truelove NK, Kough AS, Behringer DC, Paris CB, Box SJ, Preziosi RF, Butler MJ (2017) Biophysical connectivity explains population genetic structure in a highly dispersive marine species. Coral Reefs 36:233–244. https://doi.org/10.1007/s00338-016-1516-y
Vasconcellos AV, Vianna P, Paiva PC, Schama R, Solé-Cava A (2008) Genetic and morphometric differences between yellowtail snapper (Ocyurus chrysurus, Lutjanidae) populations of the tropical West Atlantic. Genet Mol Biol 31:308–316. https://doi.org/10.1590/S1415-47572008000200026
Verba JT, Pennino MG, Coll M, Lopes PFM (2020) Assessing drivers of tropical and subtropical marine fish collapses of Brazilian exclusive economic zone. Sci Total Environ 702:134940. https://doi.org/10.1016/j.scitotenv.2019.134940
Waples RS (1998) Separating the wheat from the chaff: patterns of genetic differentiation in high gene flow species. J Hered 89:438–450. https://doi.org/10.1093/jhered/89.5.438
Ward RD (2006) The importance of identifying spatial population structure in restocking and stock enhancement programmes. Fish Res 80:9–18. https://doi.org/10.1016/j.fishres.2006.03.009
Ward RD, Woodwark M, Skibinski DOF (1994) A comparison of genetic diversity levels in marine, freshwater, and anadromous fishes. J Fish Biol 44:213–232. https://doi.org/10.1111/j.1095-8649.1994.tb01200.x
Wellington CM, Harvey ES, Wakefield CB, Abdo D, Newman SJ (2021) Latitude, depth and environmental variables influence deepwater fish assemblages off Western Australia. J Exp Mar Biol Ecol 539:151539. https://doi.org/10.1016/j.jembe.2021.151539
White C, Selkoe KA, Watson J, Siegel DA, Zacherl DC, Toonen RJ (2010) Ocean currents help explain population genetic structure. Proc Royal Soc B 277:1685–1694. https://doi.org/10.1098/rspb.2009.2214
Wiley EO, McNyset KM, Peterson AT, Robins CR, Stewart AM (2003) Niche modeling and geographic range predictions in the marine environment using a machine-learning algorithm. Oceanography 16:8
Woodroffe CD (2007) Critical thresholds and the vulnerability of australian tropical coastal ecosystems to the impacts of climate change. J Coast Res. http://www.jstor.org/stable/26481633
Young EF, Belchier M, Hauser L, Horsburgh GJ, Meredith MP, Murphy EJ, Pascoal S, Rock J, Tysklind N, Carvalho GR (2015) Oceanography and life history predict contrasting genetic population structure in two Antarctic fish species. Evol Appl 8:486–509. https://doi.org/10.1111/eva.12259
Younger JL, Clucas GV, Kao D, Rogers AD, Gharbi K, Hart T, Miller KJ (2017) The challenges of detecting subtle population structure and its importance for the conservation of emperor penguins. Mol Ecol 26(15):3883–3897
Zapata FA, Herrón PA (2002) Pelagic larval duration and geographic distribution of tropical eastern Pacific snappers (Pisces: Lutjanidae). MEPS 230:295–300. https://doi.org/10.3354/meps230295
Acknowledgements
The authors thank Luis Costa (UFMA), Paulo Affonso (UESB), Jamille Bitencourt (UESB), Natália Roos (INMA), and Guilherme Longo (UFRN), who contributed with samples for this study. The authors thank the reviewers for their valuable comments. JTV was funded by a PhD scholarship–Coordenação de Aperfeiçoamento de Pessoal de Nível Superior-Brasil (CAPES). PFML (301515/2019-0), SMQL (313644/2018-7) and BPF (309216/2018-4) thank CNPq for productivity grants.
Funding
Open Access funding enabled and organized by Projekt DEAL. This study was financed by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior-Brasil (CAPES)–Finance Code 001, by MCTI/CNPq/Universal 424790/2016-5, and by National Geographic Society–Research and Exploration (CP-077ER-17).
Author information
Authors and Affiliations
Contributions
JTV, AS, PFML and SMQL designed the study. JTV collected the data, with contribution of BPF. JTV and BB analyzed the data, with the contribution of MGP. JTV, AS and RP discussed the results. JTV and RP wrote the manuscript, with the contribution of AS. All authors critically reviewed and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare that are relevant to the content of this article.
Additional information
Responsible Editor: C. Eizaguirre.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tovar Verba, J., Stow, A., Bein, B. et al. Low population genetic structure is consistent with high habitat connectivity in a commercially important fish species (Lutjanus jocu). Mar Biol 170, 5 (2023). https://doi.org/10.1007/s00227-022-04149-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00227-022-04149-1