
Abstract
A considerable number of research works focus on the positive influence of cellulose on the properties of polymer-based composites and their wide range of application possibilities. The present work is focused on the synthesis of novel bio-based polyurethane (bio-PU) composites filled with powdered cellulose (microcellulose, MC) in an amount of 5 wt.%. Bio-PU composites were synthesized via a non-solvent prepolymer method. First, the prepolymer was synthesized from diisocyanate mixture based on hexamethylene diisocyanate and bio-based polyisocyanate Tolonate™ X Flo 100 and α,ω-oligo(ethylene-butylene adipate)diol which contained cellulose. Then, resulted prepolymer was extended by bio-based 1,4 butanediol (bio-BDO). Bio-PU composites were obtained with the different [NCO]/[OH] molar ratios: 0.95, 1.0, 105 and 1.1. Special attention was paid to the influence of MC on the phase separation between soft and hard segments of bio-PU by studying the chemical structure, morphology and thermal and mechanical properties of the prepared cellulose-based composites.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The versatile chemical structure and properties of polyurethanes (PUs) and their composites have led to these materials finding a wide range of practical and different applications, such as in the automotive, medical devices or furniture industries (Głowińska and Datta 2016; Trache et al. 2016). Polyurethane can be obtained by using bio-monomers, resulting in environmentally friendly polymers with high contents of green carbon and desirable properties (Głowińska and Datta 2015a; Pattamaprom et al. 2020; Głowińska et al. 2021; Kasprzyk et al. 2021). The mentioned features are advantages of bio-based polymers in comparison to traditional polymers, with non-renewable carbon in their chemical structure.
It is well known that some types of components utilized in the synthesis of PUs, such as isocyanates, polyols, chain extenders, catalysts or other chemical additives, like active fillers, have a crucial impact on the properties of PUs (including their composites) and their future application. The molar ratio of isocyanate to hydroxyl groups [NCO]/[OH], and thus the phase separation between the hard segments (HS) and soft segments (SS) of PUs, also influence the structure and final properties of the mentioned materials (Desai et al. 2000; Huang and Zhang 2002).
The properties of polymer-based composites not only depend on the matrix and filler type but are also related to the adhesion of the polymer matrix to the filler surface (Auad et al. 2008). Adhesion is an important factor, which should be considered when preparing the composites, because it affects the matrix–filler interaction. For instance, hydroxyl groups of cellulose-based filler can react with chemicals containing isocyanate groups by creating a urethane linkage. This chemical linkage provides strong covalent bonds between the filler and the matrix (Kabir et al. 2012). That is why prior to the preparation of cellulose-based composites, it is important to take into account the effect on the interaction of the filler and polyurethane matrix and on the phase separation in polyurethane materials.
The type and structure of the filler have impact on the resulting materials. Depending on the morphology of cellulose, it might cause different changes in the polyurethane materials. For example, Marcovich et al. (2006) studied polyurethane composite films reinforced with cellulose crystals. Researchers demonstrated that cellulose crystals are chemically bonded to the polyurethane, due to the chemical reaction between the –OH groups from the cellulose and the isocyanate groups present in prepolymers, which was confirmed by infrared spectroscopy. Increases in the glass transition temperature (Tg) and tensile modulus of the PU at filler loadings (0.5–5 wt.%) were observed and related to an increase in the cross-linking density of the materials.
In another work, Kasprzyk et al. (2020) obtained poly(ether-urethane)-based composites containing microcrystalline cellulose (MC) in amounts of 1, 3, 5 and 7 wt.% characterized by good thermo-mechanical and thermal properties. The authors found that cellulose microcrystalline addition to the poly(ether-urethane)-based matrix influenced hydrogen bond formation. Selected properties of the studied materials decrease with increasing content of bio-filler, such as elongation at break or tensile strength (Kasprzyk et al. 2020).
Lee et al. (2017) investigated a series of polyurethane–urea (PUU) elastomeric nanocomposites which contained low amounts (0.5, 1.0 and 2.0 wt.%) of bio-based nanofillers and individualized cellulose nanofibers (TEMPO-oxidized cellulose-individualized nanofibers, TOCNs). The authors confirmed covalent cross-linking with the TOCNs and PUUs as the result of a reaction of carboxyl and/or hydroxyl groups of TOCNs and the polyols with isocyanate groups of MDI. Based on the results, it was found that 2 wt.% of TOCNs caused an increase in tensile strength and strain to failure and a 35% decrease in the coefficient of thermal expansion in comparison to the PUU without TOCNs (Lee et al. 2017).
Based on the literature review and earlier experience (Głowińska and Datta 2015b, 2016; Kasprzyk et al. 2020), it was stated that cellulose (in micro- and nanocellulose form) has a positive influence on some properties of polyurethane composites provided their amount is not higher than 5 wt.%. That is why, in this current study, the powdered cellulose (MC) was incorporated into the bio-based polyurethane matrix (obtained by using novel bio-based diisocyanate mixtures) in an amount of 5 wt.%. The influence of cellulose addition and the effect of a different [NCO]/[OH] molar ratio on the chemical structure, thermo-mechanical, thermal and mechanical properties and morphology of obtained PU composites were studied.
Experimental
Preparation of materials and bio-based polyurethane composites
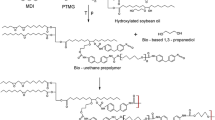
The bio-based polyurethane (bio-PU) composites were obtained via the prepolymer method. Prior to the usage, polyester polyol as α,ω-oligo(ethylene-butylene adipate)diol with the trade name Polios 55/20, Mn ~ 2000 (Purinova, Poland) was dried and degassed at 90 °C for 1 h. Dried cellulose filler (coded as MC) in the form of powdered cellulose (Alpha-Cel BH65, average particles size approx. 50 microns, International Fiber Corporation, Belgium) was added in the amount of 5 wt% to the polyol prior to the pre-polymerization process. The present experience shows that this sequence is important because it provided a better distribution of cellulose in the final materials, avoiding the creation of agglomerates. Otherwise, the chosen amount of MC did not have a significant effect on the processing of bio-PU system, making it easier to mix all ingredients. The prepolymer was synthesized from an isocyanate mixture containing 75 wt% of hexamethylene diisocyanate HDI, equivalent to a weight of approx. 84.1 g/mol (Vencorex Chemicals, France), and 25 wt.% of aliphatic bio-based diisocyanate Tolonate™ X Flo 100, equivalent to a weight of approx. 341 g/mol and functionality ~ 2 (Vencorex Chemicals, France), and the above-mentioned polyester polyol. The pre-polymerization was conducted at 80 °C for 4 h. Next, the free NCO content was determined by a back titration method (ASTM D 2572–97) and equaled 8.06%. Next, the prepolymer chain was extended with bio-based 1,4-butanediol (BASF, Germany) containing 0.2 wt.% of dibutyltin dilaurate (DBTDL, Sigma-Aldrich, Poland) as the catalyst. The polyurethane composites were synthesized within the narrow range of [NCO]prepolymer/[OH]chain extender molar ratio of 0.95, 1.0, 1.05 and 1.1, respectively. The bio-based polyurethane composites were molded and cured at 100 °C for 24 h.
The reference sample (coded as Ref. 1.0) was synthesized according to a similar procedure but without the addition of cellulose powder. In Scheme 1, the synthesis of bio-based polyurethane composites is presented, where substituent R can occur as residue of oleic acid or palmitic acid.

Preparation of bio-based polyurethane composites
Measurements
Fourier transform infrared spectroscopy (ATR-FTIR)
Spectroscopic analysis was conducted by using attenuated total reflectance Fourier transform infrared spectroscopy technique (ATR-FTIR) with a FTIR Nicolet 8700 spectrophotometer (Thermo Electron Corporation). During the measurement, a Specac Heated Golden Gate single reflection diamond ATR accessory was used. Each spectrum was registered for wavenumbers ranging from 500 to 4500 cm−1 at 4 cm−1 nominal resolution. The mean number of scans was 64. The measurements were taken at room temperature. Based on FTIR spectra, the degree of phase separation (DPS) and the degree of phase mixing (DPM) were studied and calculated using Eqs. (1), (2), and (3):
where R is the carbonyl hydrogen bonding index; Ab—absorption intensity of hydrogen-bonded carbonyl; and Af—absorption intensity of free carbonyl.
Dynamic mechanical analysis (DMA)
The dynamic mechanical analysis was performed using a DMA Q800 Analyzer (TA Instruments). The measurements were performed under nitrogen atmosphere in a temperature range from − 100 to + 150 °C at an operating frequency of 10 Hz, with a heating rate of 4 °C /min. Rectangular samples with dimensions of approximately 18 × 10 mm and a thickness of 3 mm were used. Based on the recorded DMA measurements, the storage modulus and glass transition temperature were determined.
Thermogravimetric analysis (TGA)
Thermogravimetric analysis (TGA) was performed using a TG 209 F1 Libra Analyzer (NETZSCH) under nitrogen atmosphere at a heating rate of 10 K/min and temperature range from 25 to 700 °C. Mass of each sample was about 10 mg. The results of the TGA analysis were presented as TG and DTG curves.
X-ray diffraction (XRD)
To study the structural properties of the cellulose additive (MC) and obtained bio-based PU materials, Phillips X’Pert Pro diffractometer (XRD) with CuK α radiation (1.540 Å) was used. The measurements were taken at 40 kV and 30 mA at room temperature. The diffraction angle of 2θ ranged from 10° to 60°.
Scanning electron microscopy (SEM)
The microstructure analysis of bio-based polyurethane composites containing powdered cellulose was conducted using a Quanta FEG scanning electron microscope equipped with the Everhart–Thornley detector (ETD). The measurements were taken under high vacuum (20.00 kV) at a working distance of 10 mm. Each sample was cut with a scalpel in one quick motion, and then the cross sections of samples were gold-coated prior to the analysis.
Mechanical properties
Tensile test
The tensile test was performed using a standard tensile-testing machine Zwick Z020, in accordance with EN ISO 527–1:1996 and EN ISO 527–2:1996 standards. The dumbbell-shaped samples of standard dimensions were tested at a rate of 50 mm/min. The measurements were performed at room temperature. Tensile strength (TSb) and elongation at break (εb) were determined.
Cyclic compression tests
The compression test was carried out using a Zwick Z020 tensile-testing machine, in accordance with the EN ISO 604:2006 standard. Cylindrical samples sized approx. 10 ± 0.5 mm in diameter and 25 ± 1 mm high were tested. The cyclic compression tests consisted of three load–unload loops applied continuously up to the strain of 40%. The bio-PU composites were tested at a compression rate of 5 mm/min, at room temperature.
Hysteresis is expressed as a loss of energy during a deformation cycle:
where WH is the lost energy; Wloading—energy of loading (deformation).
Results and discussion
ATR-FTIR spectroscopy analysis
The chemical structure of bio-based polyester–urethane–urea materials containing cellulose powder as well as the reference material (Ref. 1.0) was determined by ATR-FTIR and is presented in Fig. 1a.

a ATR-FTIR spectra of bio-based polyurethane with cellulose and reference material (Ref. 1.0); b deconvoluted carbonyl peaks of bio-based polyurethane reference materials (raw matrix); and c bio-based polyurethane composite coded as 1.0
Prima facie, the registered spectra of all obtained materials seem to be similar (Fig. 1a). Changes in the intensity of absorption peaks in FTIR spectra at 3320, 1727, 1681, 1540, 1257, 1133 and 1066 cm−1 were observed in relation to the [NCO]/[OH] molar ratio. The vibration characteristics of urethane and urea groups registered at about 3320 cm−1 are attributable to the stretching vibrations of the NH bond, whose intensity decreases with increasing [NCO]/[OH] molar ratio in the PU sample. At this region, the characteristic peak of N–H groups at 3270–3400 cm−1 is observed for both the hydrogen-bonded and free N–H as a non-separate peak. The absorption band at 1540 cm−1 corresponds to the out-of-plane bending of the NH bond and the CN stretching vibrations (Das et al. 2015). In the mentioned range of 3320 cm−1, the urea group derived from bio-based polyisocyanate can be visible as a broad peak shifted toward higher value of wavenumbers. The presence of a peak at 3320 cm−1 may indicate that the majority of N–H groups are hydrogen bonds. The difference between intensity of peaks at this region confirmed the structural changes caused by the molar ratio of [NCO]/[OH]. For all bio-PU materials, typically the stretching vibrations of CH2 group appeared at wavenumbers of 2867 and 2960 cm−1, while the deformation vibrations (symmetric and asymmetric) of CH groups at 1480 cm−1 and 1300 cm−1, respectively (Głowińska and Datta 2016). The structural changes were revealed as differences in band intensity in the region of C = O vibration were observed. The valent vibration of the carbonyl group derived from free and hydrogen-bonded urethane groups appeared as a multiplet peak in a wavenumber range from 1681 to 1727 cm−1, and the stretching vibration of the C–O group was registered in the range of approx. 1100–1260 cm−1 (1064, 1133 and 1257 cm−1). The low value of wavenumber of a multiplet peak corresponding to the hydrogen-bonded carbonyl groups indicates that the hydrogen-bonded C = O has derived from ordered crystalline domains (Głowińska and Datta 2016). The hydrogen bonds present between the carbonyl groups and NH in the urethane groups cause rigid domain formation and favor the process of microphase separation, especially in the case of samples containing cellulose. When the hydrogen bonds begin to form between the NH groups and the C(O)O or C–O–C groups, the latter being a structural element of elastic segments, the phase dispersion will occur. It is well established that the positions and intensities of these vibrations are extremely sensitive to the strength and specificity of the hydrogen bonds, which are formed.
Infrared spectroscopy was also used for determination of bio-PU phase separation (by DPS and DPM determination) by measuring the intensities and positions of the hydrogen-bonded and free hydrogen-bonded N–H stretching vibrations, of the hydrogen-bonded carbonyl groups (H-bonded C = O) and of the free hydrogen-bonded carbonyl (free bonded C = O) of the urethane groups. The ratio of the hydrogen-bonded carbonyl and free hydrogen-bonded carbonyl is known as ‘hydrogen bonding index’ and used to estimate the extent of hydrogen bonding (Chen et al. 1999, 2005). In the case of obtained bio-PU composites, in the urethane carbonyl region, the presence of urea carbonyl groups (residue derived from bio-based polyisocyanate) was also detected. Therefore, the carbonyl peak has been deconvoluted into four peaks: urea-bonded, urea-free, urethane-bonded and urethane-free. In Fig. 1b, deconvoluted multiplet peaks for the reference sample (Ref.1.0) and PU synthesized at [NCO]/[OH] molar ratio of 1.0 (Fig. 1c)) are presented. The hydrogen bonding index (R), the degree of phase separation (DPS) and the degree of phase mixing (DPM) were calculated using Eqs. 1, 2 and 3 and are listed in Table 1. Additionally, the location, fraction and absorbance of the –C = O peaks are also presented.
It was noticed that the addition of cellulose caused a decrease in DPS and the number of hydrogen bonds in the range of urethane groups because of the interaction with hydroxyl groups derived from cellulose. In general, the tendency toward phase separation increases with increasing [NCO]/[OH] molar ratio. Such an increase in the intramolecular attraction between the hard segments (urethane) with an increasing [NCO]/[OH] molar ratio tends to an increase in stress. These findings were confirmed by the results of the tensile test, which are described later.
Dynamic mechanical analysis
The results of a dynamic thermo-mechanical analysis of bio-based polyurethane composites containing cellulose obtained with different molar ratios of [NCO]/[OH], and also the chosen reference sample Ref. 1.0, are presented in Fig. 2. The main parameters, such as storage modulus (E’), recorded at glass transition temperature, loss modulus (E’’) and glass transition temperature (Tg), are presented in Table 2. Glass transition temperature was determined based on the curves of tanδ as a function of temperature. Additionally, the temperature range of efficient damping behavior and integral area were calculated and are presented in Table 2.

Storage modulus (E’) and tanδ as a function of temperature of bio-based polyurethane composites containing cellulose and reference materials (Ref. 1.0)
Analyzing Fig. 2 it can be concluded that an increase in the [NCO]/[OH] molar ratio had a positive effect on the thermo-mechanical properties of the obtained bio-PU composites containing cellulose over a broad range of temperature. The maximum value of E’ was observed for the bio-PU composites with the [NCO]/[OH] molar ratio of 1.1. An increase in the [NCO]/[OH] ratio from 0.95 to 1.1 resulted in changes in the properties of polyurethanes. The behavior of the material coded as 0.95 can be described as a rubber-toughened plastic. This is the reason why the E´ value of material with a molar ratio of 0.95 started to decrease at a lower temperature (ca. 50 °C) compared to the rest of the samples.
In general, the glass transition temperature (Tg) of hard segments in polyurethanes ranges between 50 and 100 °C. However, in the case of bio-based polyurethane composites synthesized at the [NCO]/[OH] molar ratio of 0.95, a lower ability to form physical and chemical cross-links was observed. These particular bio-PUs composites showed noticeable thermoelastic properties that typically occur in polyurethanes with the [NCO]/[OH] molar ratio below 1.0. From the comparison of the storage modulus of samples 1.0 and Ref. 1.0, it can be stated that cellulose acted as a reinforcing filler, as indicated by the higher value of E’ at room temperature registered for sample 1.0.
It is noteworthy that the tanδ values were similar for all samples of polyurethane composites. In Fig. 2 (tanδ as a function of temperature), well-formed peaks can be observed, which indicates a homogeneous structure of the matrix. The glass transition temperature was defined as a temperature at which the maximum value of tanδ occurred. It was found that both cellulose powder and [NCO]/[OH] molar ratio did not significantly influence glass transition temperature and tanδ. The Tg values for the obtained bio-PU composites containing cellulose powder ranged from − 26.7 to − 29.7 °C, which corresponds to the temperature range of phase transition of soft segments. With increasing [NCO]/[OH] molar ratio, glass transition temperature decreased. It was observed that cellulose caused a slight decrease in glass temperature in comparison to the material without cellulose, which was − 25.7 °C (Ref. 1.0). Generally, the rubbery plateau modulus of polyurethane composites increased with increasing [NCO]/[OH] molar ratio, which corresponds to the increased share of hard segments. These findings suggest that the formation of hard domains is characterized by better continuity increase. Moreover, based on the obtained DMA data, it was observed that an increasing [NCO]/[OH] molar ratio causes the value of storage modulus and the tanδ to increase. Similar findings were described in the literature (Xu et al. 2008).
It was observed that all obtained materials exhibit a loss factor higher than 0.3 indicating efficient damping properties. Based on the obtained results, the temperature range of efficient damping behavior of bio-PU composites was determined and is presented in Table 2 as △T. The integral area from − 50 to 35 °C in tanδ temperature curves was also calculated (coded as S − 50 − 35 °C). Analyzing these results, it was observed that efficient energy dissipation is higher in the case of bio-PU composites synthesized at a lower [NCO]/[OH] molar ratio. These findings are related to the increase in the polymer-based matrix chain mobility and were also confirmed by other measurements.
Thermogravimetric analysis
Thermal stability of bio-based polyurethane composites was determined by thermogravimetric analysis, and the results are presented as temperature-dependent on weight and derivative weight (Fig. 3). Based on the obtained data, the effect of [NCO]/[OH] molar ratio and powdered cellulose content on the thermal decomposition of the materials was described (Table 3).

Mass changes and DTG as a function of temperature of bio-based polyurethane composites containing cellulose and the reference material chosen (Ref. 1.0)
In general, the thermal decomposition of the obtained materials took place in two main steps, which is typical for polyurethanes materials. What is more, the peaks of these steps are well separated indicating good phase separation regardless of cellulose addition. Because of the small content of powdered cellulose, which was dispersed in the matrix, their degradation peaks were not visible on the thermograms. Based on the literature, cellulose degraded with the maximum speed at a temperature of approx. 360 °C (Głowińska et al. 2017).
At the first stage of the thermal decomposition, the urethane groups undergo the dissociation. In higher temperature ranges, the decomposition of polyol residue was assigned. Based on the analysis of Fig. 3, it was noticed that the TG curve of Ref. 1 sample is shifted to the lower temperature in comparison to bio-PU composites. These findings suggest that cellulose caused the improvement of thermal stability.
The beginning of the decomposition, determined as 5% of mass loss for each sample, was in the temperature range from approx. 308 to 315 °C and increased in the case of bio-PU materials containing powdered cellulose (Table 3). Analyzing the rate of decomposition, it was observed that the addition of cellulose had a slight influence on this parameter. The degradation in the second step occurred with a slightly lower rate. The influence of the changes of [NCO]/[OH] molar ratio on the decomposition rate was not observed. The ash residue at 600 °C in the case of bio-PU composites is higher and increased with increasing [NCO]/[OH] molar ratio.
X-ray diffraction (XRD)
X-ray diffraction indicated a degree of ordering in the prepared materials. XRD patterns of micro cellulose fibers with the main diffraction signals at 2θ 15.2°, 16.0°, 22.1° and 34.4° that correspond to the crystalline form were registered (Fig. 4). According to the literature, this is a typical crystal lattice of cellulose type I (Liu et al. 2010; Lin et al. 2013). In the case of bio-PU composites coded as 1.0 and 1.1, peaks assigned to the cellulose additive (MC) were slightly visible because of their small amount. However, the XRD patterns obtained indicate that all the samples showed partially crystalline structures. The main diffraction signals observed at 20.2, 21.8, 23.8° correspond to the crystalline forms derived from hard and soft segments, which agrees with the literature (Saralegi et al. 2013). There is visible intensification of diffraction signals at 22.1°, which is assigned to both TPU and cellulose. The increase in the intensity of this peak indicates that the native crystalline structure of cellulose remained in the obtained composites (Lin et al., 2013).

X-ray diffractograms of MC and selected bio-based polyurethane composites
Scanning electron microscopy (SEM)
The SEM images of bio-based polyurethane composites containing cellulose are presented in Fig. 5. All pictures show an almost homogeneous distribution of cellulose powder particles, visible as similarities in the shapes of white objects. Based on the SEM analysis, the sample structure seemed to get more regular with increasing [NCO]/[OH] molar ratio, which can be related to an interaction that occurred between OH groups from the cellulose powder and NCO groups. A similar finding was described by Tay et al. (2003). The dark regions in the images consist of soft segments of bio-PU, whereas lighter regions mark the presence of hard segments. For all the samples, good interfacial adhesion between matrix and filler was observed.

SEM images of bio-based polyurethane composites containing cellulose with the [NCO]/[OH] molar ratio of 0.95, 1.0, 1.05 and 1.1
Mechanical properties
Mechanical properties were tested by means of tensile tests and compression tests, and the obtained results are given in Table 4.
Tensile properties
The results of tensile tests point to the strong influence of the [NCO]/[OH] molar ratio on the tensile strength (TSb) and elongation at break (εb) of the obtained bio-based polyurethane composites. According to the literature, with increasing molar ratio, TSb of bio-PU materials increased due to relatively high chemical cross-linking that can restrict the chain mobility and increase the intermolecular attraction between hard segments (Huang and Zhang, 2002). Some correlations can be found for the obtained tensile strength of the produced materials (see Fig. 6). Based on the elongation at break results, it can be concluded that the decreasing [NCO]/[OH] molar ratio favored the thermoelastic behavior of bio-PU because of the low value of εb (Table 4), which is characteristic for thermoplastics. The highest values of elongation at break and tensile strength were observed for the polyurethane composites obtained with the [NCO]/[OH] molar ratios of 1.05 and 1.1. These materials exhibited Eb values above 100%, which is characteristic for thermoplastic elastomeric materials. The polyurethane composites synthesized at molar ratios lower than 1.05 displayed the poorest mechanical properties. Lin et al. (2013) reported similar results for materials with a [NCO]/[OH] molar ratio of 1.0 and containing 5 wt.% cellulose. The authors confirmed that the addition of cellulose improves the mechanical properties of bio-PU composites. The observed improvement shows that there is good compatibility between matrix and cellulose, which is most likely due to the hydrophilic character of both components (Özgür Seydibeyoğlu and Oksman 2008). The tensile strength and elongation at break for reference material Ref. 1.0 equaled 25.7 ± 1.9 MPa and 883 ± 52%, respectively.

Tensile strength as a function of elongation for bio-based polyurethane composites containing cellulose and reference material
Compression properties
The hysteresis loops and values of 25 and 40% deformation moduli obtained from compression testing are given in Table 4. In the cyclic compression tests, the material absorbs energy when compressive loads are applied, and they release energy when the loads are removed (Datta and Włoch 2016). The values of E loading and E unloading were determined using a mathematical program. All composite materials exhibit permanent deformation after the compression processes, which might be caused by changes in their microstructure due to energy dissipation. It was noted that bio-PU composites containing cellulose, synthesized at higher [NCO]/[OH] molar ratios, required the highest amount of energy during the compression processes. The bio-PU composites synthesized with a molar ratio of 0.95 had the largest hysteresis due to the elevated values of energy dissipation regardless of the number of compression cycles. These findings were also confirmed by DMA results.
The hysteresis decreased with increasing [NCO]/[OH] molar ratio, which resulted from the increase in structural order, and the subsequent lower energy loss during the compression processes. After the first cycle, double energy loss was recorded. The differences between the hysteresis loops in the second and fourth cycles were not significant (Datta and Kopczyńska 2015).
All analyzed bio-PU materials with the addition of cellulose exhibited a large hysteresis effect in the first cycle for 40% deformation due to a chain reorganization process. (The polyurethane molecular chains undergo a reorganization process, involving molecular orientation.) In the first loading/unloading cycle, the polyester–urethane–urea materials with the [NCO]/[OH] molar ratios of 0.95, 1, 1.05 and 1.1 exhibited the hysteresis effect of 68.2, 63.9, 61.0 and 60.2, respectively. After five repeated 40% deformation cycles, the hysteresis values of polyester–urethane–urea materials with the [NCO]/[OH] molar ratios of 0.95, 1, 1.05 and 1.1 reached the values of 31.4, 28.7, 27.1 and 27.2, respectively. The occurrence of large hysteresis in the fabricated PU materials was also reported by others (Zhang et al. 2014; Alagi et al. 2016). The polyurethane composites exhibited a permanent deformation after each loading/unloading cycle. The hysteresis values were calculated according to Eq. (4).
Conclusion
In this work, it was confirmed that the [NCO]/[OH] molar ratio and the addition of cellulose influenced phase separation of novel bio-PU composites. Based on FTIR analysis, it was found that powdered cellulose caused a decrease in the DPS index. DMA results lead to conclude that bio-based polyurethane composites containing cellulose might be best described as rubber-toughened plastic materials. The rubbery plateau modulus of bio-based polyurethane composites increased with increasing [NCO]/[OH] molar ratio. TGA analysis showed that the amount of powdered cellulose did not affect degradation of bio-PU, which is an advantage. The XRD patterns confirmed that all the PU samples have partially crystalline structures. SEM images showed that materials have rather regular structure with increasing [NCO]/[OH] molar ratio, which can be related to the interaction that occurred between OH groups from cellulose powder and NCO groups. The [NCO]/[OH] molar ratio affects the mechanical properties of bio-based polyurethane composites.
It was also confirmed that powdered cellulose introduced into 5 wt.% did not influence polyurethane systems processing, which constitutes a positive prognosis for further investigations on a technical scale. What is important, the polyurethane matrix was synthesized with the use of partially bio-based polyisocyanate in the aliphatic diisocyanate mixture, which is a more environmentally friendly reagent compared to materials obtained using only petrochemical monomers. The authors believe that these materials, which exhibit thermoelastic behavior could be used as a bio-PU-cellulose-based filament for 3D printing.
References
Alagi P, Choi YJ, Hong SC (2016) Preparation of vegetable oil-based polyols with controlled hydroxyl functionalities for thermoplastic polyurethane. Eur Polymer J 78:46–60. https://doi.org/10.1016/j.eurpolymj.2016.03.003
Auad ML, Contos VS, Nutt S et al (2008) Characterization of nanocellulose- reinforced shape memory polyurethanes. Polym Int 57:651–659
Chen TK, Tien YI, Wei KH (1999) Synthesis and characterization of novel segmented polyurethane clay nanocomposite via poly(epsilon-caprolactone)/clay. J Polymer Sci Part a-Poly Chem 37:2225–2233. https://doi.org/10.1002/(sici)1099-0518(19990701)37:13%3c2225::aid-pola37%3e3.0.co;2-z
Chen CP, Dai SA, Chang HL et al (2005) Polyurethane elastomers through multi-hydrogen-bonded association of dendritic structures. Polymer 46:11849–11857. https://doi.org/10.1016/j.polymer.2005.06.127
Das S, Pandey P, Mohanty S, Nayak SK (2015) Influence of NCO/OH and transesterified castor oil on the structure and properties of polyurethane: Synthesis and characterization. Mater Express 5:377–389. https://doi.org/10.1166/mex.2015.1254
Datta J, Kopczyńska P (2015) Stiffness and energy dissipation of poly(etherurethane) resilient elements. Przemysł Chemiczny 94:1000–1003
Datta J, Włoch M (2016) Preparation, morphology and properties of natural rubber composites filled with untreated short jute fibres. Polym Bull 74:763–782. https://doi.org/10.1007/s00289-016-1744-x
Desai S, Thakore IM, Sarawade BD, Devi S (2000) Effect of polyols and diisocyanates on thermo-mechanical and morphological properties of polyurethanes. Eur Polymer J 36:711–725
Głowińska E, Datta J (2015a) Structure, morphology and mechanical behaviour of novel bio-based polyurethane composites with microcrystalline cellulose. Cellulose. https://doi.org/10.1007/s10570-015-0685-0
Głowińska E, Datta J (2015b) Structure, morphology and mechanical behaviour of novel bio-based polyurethane composites with microcrystalline cellulose. Cellulose 22:2471–2481. https://doi.org/10.1007/s10570-015-0685-0
Głowińska E, Datta J (2016) Bio polyetherurethane composites with high content of natural ingredients: hydroxylated soybean oil based polyol, bio glycol and microcrystalline cellulose. Cellulose 23:581–592. https://doi.org/10.1007/s10570-015-0825-6
Głowińska E, Datta J, Parcheta P (2017) Effect of sisal fiber filler on thermal properties of bio-based polyurethane composites. J Therm Anal Calorim 130:113–122. https://doi.org/10.1007/s10973-017-6293-5
Głowińska E, Wolak W, Datta J (2021) Eco-friendly route for thermoplastic polyurethane elastomers with bio-based hard segments composed of bio-glycol and mixtures of aromatic-aliphatic and aliphatic-aliphatic diisocyanate. J Polym Environ. https://doi.org/10.1007/s10924-020-01992-5
Huang J, Zhang L (2002) Effects of NCO / OH molar ratio on structure and properties of graft-interpenetrating polymer networks from polyurethane and nitrolignin. Polymer 43:2287–2294
Kabir MM, Wang H, Lau KT, Cardona F (2012) Composites : Part B Chemical treatments on plant-based natural fibre reinforced polymer composites : An overview. Compos B 43:2883–2892. https://doi.org/10.1016/j.compositesb.2012.04.053
Kasprzyk P, Błażek K, Datta J (2020) Microcrystalline cellulose management in the production of Poly ( ether-urethane ) s- structure, morphology, and thermal characteristic. Fibers and Polymers 21:690–700. https://doi.org/10.1007/s12221-020-9482-y
Kasprzyk P, Głowińska E, Parcheta-Szwindowska P et al (2021) Green TPUs from prepolymer mixtures designed by controlling the chemical structure of flexible segments. Int J Mol Sci 22:7438. https://doi.org/10.3390/ijms22147438
Lee M, Haeng M, Lee H et al (2017) Tunable softening and toughening of individualized cellulose nanofibers-polyurethane urea elastomer composites. Carbohyd Polym 159:125–135. https://doi.org/10.1016/j.carbpol.2016.12.019
Lin S, Huang J, Chang PR et al (2013) Structure and mechanical properties of new biomass-based nanocomposite: Castor oil-based polyurethane reinforced with acetylated cellulose nanocrystal. Carbohyd Polym 95:91–99. https://doi.org/10.1016/j.carbpol.2013.02.023
Liu H, Liu D, Yao F, Wu Q (2010) Fabrication and properties of transparent polymethylmethacrylate/cellulose nanocrystals composites. Biores Technol 101:5685–5692. https://doi.org/10.1016/j.biortech.2010.02.045
Marcovich NE, Auad ML, Bellesi NE et al (2006) Cellulose micro/nanocrystals reinforced polyurethane. J Mater Res 21:870–881. https://doi.org/10.1557/jmr.2006.0105
Özgür Seydibeyoğlu M, Oksman K (2008) Novel nanocomposites based on polyurethane and micro fibrillated cellulose. Compos Sci Technol 68:908–914. https://doi.org/10.1016/j.compscitech.2007.08.008
Pattamaprom C, Wu CH, Chen PH et al (2020) Solvent-free one-shot synthesis of thermoplastic polyurethane based on bio-poly(1,3-propylene succinate) glycol with temperature-sensitive shape memory behavior. ACS Omega 5:4058–4066. https://doi.org/10.1021/acsomega.9b03663
Saralegi A, Rueda L, Fernández-D’Arlas B et al (2013) Thermoplastic polyurethanes from renewable resources: Effect of soft segment chemical structure and molecular weight on morphology and final properties. Polym Int 62:106–115. https://doi.org/10.1002/pi.4330
Tay GS, Rozman HD, Abusamah A (2003) The effect of glycol type, glycol mixture, and isocyanate/glycol ratio on flexural properties of oil palm empty fruit bunch-polyurethane composites. J Wood Chem Technol 23:249–260. https://doi.org/10.1081/wct-120026932
Trache D, Hussin MH, Hui Chuin CT et al (2016) Microcrystalline cellulose: Isolation, characterization and bio-composites application—A review. Int J Biol Macromol 93:789–804. https://doi.org/10.1016/j.ijbiomac.2016.09.056
Xu Y, Petrovic Z, Das S, Wilkes GL (2008) Morphology and properties of thermoplastic polyurethanes with dangling chains in ricinoleate-based soft segments. Polymer 49:4248–4258. https://doi.org/10.1016/j.polymer.2008.07.027
Zhang L, Huang M, Yu R et al (2014) Bio-based shape memory polyurethanes (Bio-SMPUs) with short side chains in the soft segment. J Mater Chem A 2:11490. https://doi.org/10.1039/c4ta01640h
Acknowledgements
Diisocyanates, bio-based—1,4-butanediol and powdered cellulose were delivered by Vencorex Chemicals (France), BASF (Germany) and International Fiber Corporation (Belgium). The authors wish to thank the aforementioned companies for kindly providing all components, NETZSCH company for making the TG 209 F1 Libra analyzer available and Dr J. Karczewski and Dr A. Mielewczyk-Gryń for conducting SEM and XRD measurements.
Funding
This work was supported partially by Ministry of Science and Higher Education in Poland (Decision number 4914/E-359/M?2016).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Głowińska, E., Kasprzyk, P. & Datta, J. Segmented bio-based polyurethane composites containing powdered cellulose obtained from novel bio-based diisocyanate mixtures. Wood Sci Technol 55, 1673–1691 (2021). https://doi.org/10.1007/s00226-021-01331-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00226-021-01331-4