
Abstract
Extracellular vesicles (EVs) play crucial roles in physiological and pathophysiological processes. Although studies have described muscle–bone interactions via humoral factors, we reported that EVs from C2C12 muscle cells (Myo-EVs) suppress osteoclast formation. Current clinical evidence suggests that inflammation induces both sarcopenia and osteoporosis. Although tumor necrosis factor-α (TNF-α) is a critical proinflammatory factor, the influences of TNF-α on muscle–bone interactions and Myo-EVs are still unclear. In the present study, we investigated the effects of TNF-α stimulation of C2C12 cells on osteoclast formation and osteoblastic differentiation modulated by Myo-EVs in mouse cells. TNF-α significantly decreased the protein amount in Myo-EVs, but did not affect the Myo-EV size distribution. TNF-α treatment of C2C12 myoblasts significantly decreased the suppression of osteoclast formation induced by Myo-EVs from C2C12 myoblasts in mouse bone marrow cells. Moreover, TNF-α treatment of C2C12 myoblasts in mouse preosteoclastic Raw 264.7 cells significantly limited the Myo-EV-induced suppression of osteoclast formation and decreased the Myo-EV-induced increase in mRNA levels of osteoclast formation-related genes. On the other hand, TNF-α treatment of C2C12 muscle cells significantly decreased the degree of Myo-EV-promoted mRNA levels of Osterix and osteocalcin, as well as ALP activity in mouse mesenchymal ST-2 cells. TNF-α also significantly decreased miR196-5p level in Myo-EVs from C2C12 myoblasts in quantitative real-time PCR. In conclusion, TNF-α stimulation of C2C12 muscle cells blunts both the osteoclast formation suppression and the osteoblastic differentiation promotion that occurs due to Myo-EVs in mouse cells. Thus, TNF-α may disrupt the muscle–bone interactions by direct Myo-EV modulation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Increases in muscle mass and strength positively correlate with decreased fracture risk and increased bone mineral density [1]. This raises questions about the role that muscle–bone interactions play in the physiological and pathophysiological environment. Various myokines regulate bone metabolism by affecting distant bones in muscle–bone interactions under different physiological and pathophysiological states [2]. Increased fracture risk and decreased bone mineral density are reported in sepsis and chronic obstructive pulmonary disease (COPD) patients with systemic inflammation [3, 4]. Moreover, sarcopenia correlates with systemic inflammation in COPD and chronic liver diseases [5, 6]. These findings suggest that inflammation induces both sarcopenia and osteoporosis. Tumor necrosis factor-α (TNF-α), a typical proinflammatory factor, is secreted from various cells under inflammatory environments, including macrophages, lymphocytes, and fibroblasts, triggers NF-κB and AP-1 pathway activation, both critical for proinflammatory cytokine expression [7]. Higher TNF-α serum levels are reported in patients with sarcopenia [8]. Numerous studies indicate the catabolic effects of TNF-α on bone [9, 10]. TNF-α induces muscle protein degradation in mouse C2C12 myotubes through enhanced muscle-specific ubiquitin ligase expression [11]. However, the detailed effects of inflammation on muscle–bone interactions remain unknown. Moreover, the effects of TNF-α on muscle–bone interactions are not yet reported.
Extracellular vesicles (EVs) are secreted by cells, circulate in body fluids, and play important roles in physiological and pathophysiological processes by transporting their contents to distant tissues. These EVs, secreted from osteoblasts, osteoclasts, and osteocytes, are also reported to regulate bone metabolism [12,13,14,15,16,17]. We recently reported the suppression of osteoclast formation in mouse bone marrow and Raw 264.7 cells due to EVs secreted from C2C12 muscle cells [18]. In addition, we revealed that fluid shear stress to mouse muscle cells enhances the suppressive effects of muscle cell-derived EVs on osteoclast formation [19]. These findings suggest that EVs secreted from skeletal muscle may mediate muscle–bone interactions.
Plasma EVs from septic mice induce peritoneal neutrophil migration [20]. Cheng et al. reported that myocardium released EVs, after acute myocardial infarction, induced circulating myocardial progenitor cells from bone marrow in mice [21]. These findings suggest that inflammatory stimulation of cells affects EV bioactivity. Kang et al. recently reported that TNF-α-stimulated mesenchymal stem cell (MSC)-derived EVs suppress pro-M1 and elevate M2 markers in primary mouse macrophages, although TNF-α does not affect the size of MSC-derived EVs [22]. However, whether muscle cell inflammatory stimulation, such as TNF-α, influences the effects of muscle cell-derived EVs (Myo-EVs) on bone cells remains unknown. Therefore, in the present study, we investigated the effects of TNF-α stimulation of mouse muscle C2C12 cells on the osteoclast formation and osteoblastic differentiation modulated by Myo-EVs.
Materials and Methods
Materials
Fetal bovine serum (FBS; Sigma–Aldrich, St. Louis, MO) was heat-inactivated at 56 °C for 30 min in advance. EVs contained in the FBS were depleted by ultracentrifugation at 130,000 g for 16 h at 4 °C by Himac CP80NX system (Hitachi, Tokyo, Japan). Mouse muscle C2C12 cells were obtained from American Type Culture Collection (ATCC: Manassas, VA). Recombinant human TNF-α was purchased from FUJIFILM Wako Pure Chemical Corporation (FUJIFILM Wako, Osaka, Japan). Anti-CD9 (ab92726), anti-CD81 (ab109201), and anti-KDM1/LSD1 (ab129195) antibodies were from Abcam (Cambridge, MA, USA), and anti-β-actin (#4970) antibody was from Cell Signaling Technology Inc. (Danvers, MA, USA).
EV Isolation and Analysis
We collected Myo-EVs from the conditioned medium (CM) of C2C12 cells as previously described [18, 23] (Fig. 1, 2). Briefly, C2C12 myoblasts were seeded in 10 cm culture dishes coated with collagen (cell matrix type I-C, manufactured by Nitta Gelatin Co., Ltd., Osaka, Japan) according to the manufacturer's instructions. The cells were cultured in high-glucose Dulbecco's Modified Eagle's Medium (DMEM: FUJIFILM Wako) supplemented with 10% heat-inactivated FBS and 1% penicillin/streptomycin (PS) at 37 °C until confluent. C2C12 myoblasts were cultured in high-glucose DMEM supplemented with 2% horse serum and 1% PS for an additional 5 days for differentiation into myotubes. C2C12 myoblasts and myotubes were washed with PBS twice to remove culture medium components, then cultured in fresh high-glucose DMEM supplemented with 10% EV-depleted FBS and 1% PS with and without 50 ng/mL TNF-α at 37 °C for 48 h. TNF-α stock solution (100 µg/mL) was diluted 2000-fold with the culture medium to a final concentration of 50 ng/mL. CM of C2C12 cells was centrifuged at 3,000 g for 5 min, filtered using a 0.22 μm PVDF filter, and ultracentrifuged at 130,000 g for 70 min at 4 °C to isolate EVs (pellets). The supernatant containing TNF-α was carefully removed after ultracentrifugation. The EV pellets were resuspended in an excess of fresh PBS and ultracentrifuged again. The supernatant after ultracentrifugation was carefully removed to remove proteins and other culture medium components. The washed EV pellets were resuspended in a small amount of fresh PBS. The suspension of Myo-EVs obtained by these procedures is considered TNF-α-free. The Myo-EV suspensions were stored at − 80 °C until use. Total Myo-EV protein levels were determined using the BCA protein assay kit (#23227 Pierce, Rockford, Illinois). Myo-EV size distribution was analyzed by the NanoSight LM10V-HS system (Malvern Instruments, Malvern, UK). The quantification was repeated 5 times within a single analysis, and standard error values were based on variations across all Myo-EVs within the analysis.

A flowchart for the preparation of Myo-EVs and the study design

Experimental workflow
Western Blotting
Myo-EVs or C2C12 cells were lysed with Cell Lysis Buffer (Cell Signaling Technology) with protease inhibitors (FUJIFILM Wako). Equal amounts of protein aliquots from Myo-EVs and C2C12 cells were separated by electrophoresis at 150 V for 1 h under reducing conditions, then transferred at 30 V for 1 h. The membranes were blocked with Tris-Buffered saline/0.05% Tween 20 (TBS-T) containing 3% skim milk for 2 h at room temperature. After washing with TBS-T, the membranes were incubated with primary antibodies against CD9 (1:1000), CD81 (1:1000), β-actin (1:2000), and KDM1/LSD1 (1:5000) overnight at 4 °C. After washing with TBS-T, the membranes were incubated with a secondary antibody (anti-rabbit IgG-HRP antibody, 1:10,000) at room temperature for 1 h. The immune complexes on the membranes were visualized with ECL Western Blotting Detection Reagent (#RPN2235 GE Healthcare Japan) and analyzed with an Amersham Imager 600 (GE Healthcare Japan).
Animal
Male mice with a mixed C57BL/6 J (81.25%) and 129/SvJ (18.75%) genetic background were bred and used for experiments. The mice were fed a normal diet, with freely available food and water. The mice were maintained in a controlled pathogen-free environment with a 12-h light–dark cycle at 23 ± 1 °C and a humidity of 55 ± 10% (3 or 4 per cage). All experiments were performed in accordance with the guidelines of the National Institutes of Health and the institutional rules for laboratory animal use and care at Kindai University. All animal experiments were approved by the animal ethics committee of Kindai University (approval number: KAME-31–051). All surgery was performed under anesthesia with excess isoflurane, and all efforts were made to minimize suffering.
Osteoclast Formation
Analysis of osteoclast formation was performed as previously reported [18] (Fig. 2). Briefly, bone marrow cells isolated from the tibiae of the male mice (8–10 weeks old) were seeded on a multi-well plate (1 × 105 cells/cm2) and cultured in α-MEM (FUJIFILM Wako) supplemented with 10% FBS, 1% PS, and macrophage-colony stimulating factor (M-CSF) (FUJIFILM Wako) at 37 °C for 3 days for bone marrow-derived macrophage (BMMs) formation. For osteoclast formation, BMMs were cultured in the presence of receptor activator of nuclear factor κB ligand (RANKL; FUJIFILM Wako) and M-CSF with and without Myo-EVs for an additional 4–5 days. Mouse monocytic Raw 264.7 cells (ATCC) were seeded in multi-well plates (1 × 104 cells/cm2) and cultured in α-MEM supplemented with 10% FBS, 1% PS, and 50 ng/mL RANKL with and without Myo-EVs at 37 °C for 5 days. During osteoclast formation, the medium was changed once with Myo-EVs again added. Each Myo-EV addition was diluted with culture medium based on the quantified BCA assay concentration and added to the cells at a final concentration of 5 µg/mL. For the control group, equal culture medium quantities as the added Myo-EVs were used. The cells were stained with a tartrate-resistant acid phosphatase (TRAP) staining kit (#294–67001 FUJIFILM Wako) according to the manufacturer's instructions. TRAP-positive multinucleated cells (MNCs) containing three or more nuclei in each well of 96-well plates were counted as osteoclast-like cells.
ST-2 Cell Culture
Mouse mesenchymal ST-2 cells (ATCC) were seeded in multi-well plates (2 × 104 cells/cm2) and cultured in low-glucose DMEM (FUJIFILM Wako) supplemented with 10% FBS and 1% PS at 37 °C in the presence and absence of BMP-2 (FUJIFILM Wako), with and without Myo-EVs for 72 h. Each Myo-EV was diluted with culture medium based on the quantified BCA assay concentration and added at a final concentration of 10 µg/mL. For the control group, equal amounts of culture medium as the added Myo-EVs were used.
Alkaline Phosphatase (ALP) Activity Assay and Mineralization
ALP activity was measured using a Lab assay ALP kit (#633–51021 FUJIFILM Wako) according to the manufacturer's instructions and normalized to total protein measured using the BCA protein assay kit. For mineralization, ST-2 cells were cultured in 96-well plates in the presence and absence of 200 ng/ml BMP-2, with and without 10 µg/mL Myo-EVs for 1 week, then the cells were cultured in mineralization media (low-glucose DMEM/10% FBS and 1% PS supplemented with 10 mM β-glycerophosphate and 50 µg/mL ascorbic acid) with and without 10 µg/mL Myo-EVs for more than 10 days. To evaluate mineralization levels, the cells were fixed with 10% formaldehyde and stained with Alizarin red (Kishida Chemical, Osaka, Japan). The cells were destained with cetylpyridinium chloride (Wako Pure Chemical) and the absorbance at 570 nm was measured by the Multiskan GO microplate spectrophotometer (Thermo Fisher Scientific).
Quantitative Real-Time PCR
Total RNA was extracted from cells and Myo-EVs using Nucleo Spin® RNA Plus (#U0984C Takara Bio, Shiga, Japan) and miRNeasy Mini Kit (#217004 Qiagen, Hilden, Germany) to detect mRNA and miRNA, respectively. For mRNA quantification, 500 ng of total RNA was reverse transcribed into cDNA using the Prime Script RT Reagent Kit equipped with a gDNA Elaser (#RR047B Takara Bio). The incorporation of SYBR Green into double-stranded DNA, performed using an Applied Biosystems Step One Plus™ Real-Time PCR System (Applied Biosystems, Carlsbad, CA), was assessed by quantitative real-time PCR. The PCR primer sequence is shown in Table 1. The mRNA levels of target genes were normalized to the mRNA levels of the GAPDH housekeeping gene.
For miRNA quantification, 100 ng of total RNA was reverse transcribed into cDNA using the TaqMan MicroRNA Reverse Transcription Kit (#4366596 Thermo Fisher Scientific). The reverse transcription primer used for cDNA synthesis was provided in the TaqMan miRNA Assay Kit (#4427975 Thermo Fisher Scientific). Quantitative real-time PCR for miR-155-5p, miR196-5p, and small nucleolar RNA-202 (snoRNA202) was performed using the TaqMan MicroRNA Assay (Thermo Fisher Scientific, Inc). The levels of target miRNAs (miR155-5p (Assay ID: 002571) and miR196-5p (Assay ID: 241070)) were normalized against those of snoRNA202 (Assay ID: 001232) as an endogenous control.
Statistical Analysis
All data are expressed as mean standard error (SEM) ± mean. Statistical significance was assessed using a one-way ANOVA followed by Tukey–Kramer post-hook test for multiple comparisons. The data for the quantification of TRAP-positive MNCs are representative of 6 experiments in each group. All data for PCR experiments and measurements of ALP activity are representative of 4 experiments in each group. The significance level was set to p < 0.05. All statistical analysis is done by GraphPad PRISM 6 Software (GraphPad Software Inc., San Diego, CA, USA).
Results
Effects of TNF-α on the Protein Amount and Particle Size of Myo-EVs Secreted from Muscle Cells
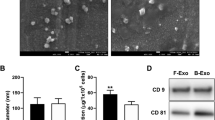
Myo-EVs were isolated from the CM of C2C12 myoblasts and myotubes treated with and without TNF-α. The Myo-EV protein quantity from C2C12 myoblasts was significantly greater than that of the Myo-EVs obtained from C2C12 myotubes (Fig. 3A). TNF-α significantly decreased the protein quantity in Myo-EVs secreted from both C2C12 myoblasts and myotubes (Fig. 3A). The particle sizes of Myo-EVs secreted from C2C12 myoblasts, with and without TNF-α treatment, were less than 200 nm (Fig. 3B), with a similar Myo-EV size distribution, independent of TNF-α treatment (Fig. 3B). Myo-EVs from C2C12 myoblasts treated with and without TNF-α were both positive for CD9 and CD81, well-defined EV markers, as shown by Western blot analysis, compared to the lysates of C2C12 myoblasts treated with and without TNF-α (Fig. 3C).

Analyses of isolated extracellular vesicles (EVs) from C2C12 cells. A The protein levels of Myo-EVs secreted from C2C12 myoblasts (MB) or myotubes (MT) treated with or without TNF-α (50 ng/mL) cultured in 10 cm dishes for 48 h were measured. C2C12 cells were cultured in three 10 cm dishes in a single experiment. Data represent mean ± SEM of 3 technical replicates within one experiment in each group. B The distribution of particle size of Myo-EVs from MBs treated with or without TNF-α (50 ng/mL) for 48 h were analyzed using a NanoSight LM10V-HS system. Data represent mean ± SEM of 5 determinations within one experiment in each group. C Total proteins were extracted from Myo-EVs secreted from MBs treated with or without TNF-α (50 ng/mL) and MBs (Cell) treated with or without TNF-α (50 ng/mL) for 48 h, and Western blot analyses for CD9, CD81, β-actin, and KDM1/LSD1 (nuclear marker) were performed
Effects of Myo-EVs Secreted from C2C12 Cells Treated with TNF-α on Osteoclast Formation
We examined the effects of Myo-EVs from C2C12 cells treated with and without TNF-α on osteoclast formation from mouse bone marrow cells, in the presence of RANKL and M-CSF, to clarify whether TNF-α influences osteoclast formation through Myo-EVs by affecting muscle cells. As shown in Fig. 4, both Myo-EVs from C2C12 myoblasts and myotubes significantly suppressed the number of TRAP-positive MNCs from bone marrow cells enhanced by RANKL and M-CSF, which agreed with our previous study [18]. However, TNF-α treatment of C2C12 myoblasts significantly decreased the suppressive effect of Myo-EVs on osteoclast formation (Fig. 4). In addition, TNF-α treatment of C2C12 myotubes slightly decreased the normal suppression effect of Myo-EVs on osteoclast formation, however, this was not significant (Fig. 4).

Effects of Myo-EVs on osteoclast formation from mouse bone marrow cells. Osteoclast precursors were induced from mouse bone marrow cells by culturing with M-CSF (50 ng/mL) for 3 days and then cultured with and without Myo-EVs (5 μg/mL) secreted from MBs or MTs treated with and without TNF-α (50 ng/mL) in the presence and absence of M-CSF (50 ng/mL) and RANKL (50 ng/mL) for an additional 5 days. Numbers of TRAP-positive MNCs in a 96-well plate were quantified. Data represent mean ± SEM of 6 technical replicates within one experiment in each group. **p < 0.01 versus the RANKL ( +)/Myo-EV (-) group. Scale bar = 200 μm. N.S.: not significant
As bone marrow cells include cells other than osteoclast precursors, we next examined the effects of Myo-EVs from C2C12 cells treated with and without TNF-α on osteoclast formation in the presence of RANKL in mouse monocytic Raw 264.7 cells. As shown in Fig. 5A, Myo-EVs from both C2C12 myoblasts and C2C12 myotubes significantly suppressed the number of TRAP-positive MNCs induced by RANKL. Also, the TNF-α treatment of C2C12 myoblasts, but not C2C12 myotubes, significantly decreased the suppressive effect of Myo-EVs on RANKL-induced osteoclast formation (Fig. 5A). Moreover, the TNF-α treatment of C2C12 myoblasts, but not C2C12 myotubes, significantly decreased the suppressive effect of Myo-EVs on the mRNA levels of TRAP, nuclear factor of activated T-cells cytoplasmic 1 (NFATc1), cathepsin K, and peroxisome proliferator-activated receptor gamma coactivator 1β (PGC1β) (Fig. 5B).

Effects of Myo-EVs on osteoclast formation in Raw 264.7 cells. Raw 264.7 cells were cultured with and without Myo-EVs (5 μg/mL) secreted from MBs and MTs treated with and without TNF-α (50 ng/mL) in the presence and absence of RANKL (50 ng/mL) for 5 days. A Numbers of TRAP-positive MNCs in a 96-well plate were quantified. Data represent mean ± SEM of 6 technical replicates within one experiment in each group. B Total RNA of Raw 264.7 cells was extracted for gene expression analysis of TRAP, NFATc1, cathepsin K, PGC1β, or Gapdh by quantitative real-time PCR. Data represent mean ± SEM of 4 technical replicates within one experiment in each group and are expressed relative to Gapdh mRNA values. *p < 0.05 and **p < 0.01 versus the RANKL ( +)/Myo-EV (-) group. Scale bar = 200 μm. N.S.: not significant
Effects of Myo-EVs Secreted from C2C12 Cells Treated with TNF-α on Osteoblastic Differentiation
We next examined the effects of Myo-EVs from C2C12 cells treated with TNF-α on the differentiation of mouse mesenchymal ST-2 cells into osteoblastic cells induced by BMP-2 to clarify whether TNF-α influences osteoblastic differentiation through Myo-EVs by affecting muscle cells. Myo-EVs from C2C12 myoblasts significantly augmented the mRNA levels of Osterix and osteocalcin, but not Runx2 and type 1 collagen, enhanced by BMP-2, although the augmentations of the mRNA levels of Osterix and osteocalcin promoted by Myo-EVs from C2C12 myotubes enhanced by BMP-2 were not significant (Fig. 6A). TNF-α treatment of C2C12 myoblasts and myotubes significantly decreased the mRNA levels of Osterix and osteocalcin promoted by Myo-EVs in ST-2 cells, respectively (Fig. 6A). Moreover, TNF-α treatment of C2C12 myoblasts significantly decreased ALP activity promoted by Myo-EVs in ST-2 cells (Fig. 6B). Myo-EVs from C2C12 myoblasts significantly decreased mineralization enhanced by BMP-2, and TNF-α treatment of C2C12 myoblasts and myotubes significantly decreased mineralization in the presence of Myo-EVs in ST-2 cells (Fig. 6C).

Effects of Myo-EVs on osteoblastic differentiation of ST-2 cells. ST-2 cells were cultured with and without Myo-EVs (10 μg/mL) secreted from MBs and MTs treated with and without TNF-α (50 ng/mL) in the presence and absence of BMP-2 (200 ng/mL) for 72 h. A Total RNA of ST-2 cells was extracted for gene expression analysis of Runx2, Osterix, osteocalcin, type 1 collagen, or Gapdh by quantitative real-time PCR. Data are expressed relative to Gapdh mRNA values. B ALP activity of ST-2 cells was measured. C ST-2 cells were cultured with 200 ng/ml BMP-2, with and without 10 μg/mL Myo-EVs for 1 week, and then the cells were cultured with and without Myo-EVs (10 μg/mL) in the presence of 10 mM β-glycerophosphate and 50 µg/mL ascorbic acid for further 1 or 2 weeks. Alizarin red staining and quantification then performed. All data represent the mean ± SEM of 4 or 8 technical replicates within one experiment in each group. *p < 0.05 and **p < 0.01 versus BMP-2 ( +)/Myo-EV (-) group. Scale bar = 1 mm. N.S.: not significant
Effects of TNF-α Treatment of C2C12 Myoblasts on miRNA Level in Myo-EVs
EVs contain various miRNAs that vary by cell type and origin, which are transferred, via EVs to distant tissues [21, 24]. Our previous study suggested that fluid flow shear stress enhances the expression of miR155-5p and miR196-5p and suppresses osteoclast formation and low expression in mouse bone cells [19]. We, therefore, examined the effects of TNF-α treatment on the levels of these miRNAs in Myo-EVs from C2C12 myoblasts. As shown in Fig. 7, TNF-α significantly decreased miR196-5p level in Myo-EVs from C2C12 myoblasts in quantitative real-time PCR, although TNF-α did not affect miR155-5p level.

Effects of TNF-α on levels of miRNA 155-5p and miRNA 196-5p in Myo-EVs. Total RNA from Myo-EVs secreted from C2C12 MBs treated with and without TNF-α (50 ng/mL) for 48 h was extracted for the analysis of miRNA expression by quantitative real-time PCR. Data represent mean ± SEM of 4 technical replicates within one experiment in each group and are expressed relative to miRNA values of snoRNA202. N.S.: not significant
Discussion
In the present study, we revealed that TNF-α treatment of C2C12 muscle cells limits the suppressive effect of Myo-EV upon osteoclast formation in mouse bone marrow cell cultures and Raw 264.7 cells. Moreover, TNF-α treatment of C2C12 muscle cells increased the expression of osteoclast differentiation-related genes, such as TRAP, NFATc1, cathepsin K, and PGC1β suppressed by Myo-EVs in Raw 264.7 cells. These data indicate that TNF-α blunts the induced inhibition of osteoclast formation by Myo-EVs by affecting muscle cells. Moreover, we showed that TNF-α treatment of C2C12 muscle cells decreased the Myo-EV-induced promotion of the expression of Osterix and osteocalcin as well as the ALP activity induced by BMP-2 in ST-2 cells, suggesting that TNF-α blunts the Myo-EV augmentation of osteoblastic differentiation. Taken together, we speculated that TNF-α, in mice, blunts the bone anabolic effects of muscle-derived EVs in muscle–bone interactions. Inflammation may induce osteopenia partly by antagonizing the osteotropic effects of Myo-EVs through direct effects on muscle cells.
The EV protein amount is generally thought to reflect the EV particle number. In our study, TNF-α treatment of C2C12 cells significantly decreased the protein amount in Myo-EVs from both C2C12 myoblasts and myotubes, although it did not affect the particle-size distribution of Myo-EVs. This suggests that TNF-α treatment of C2C12 cells decreases Myo-EV particle number, but not particle size. It is reported that lipopolysaccharide stimulation of macrophages decreases the amount of EVs secreted from these cells [25]. In addition, TNF-α stimulation of human bronchial epithelial cells triggers the release of its receptor contained within secreted EVs through sphingomyelinase related to EV production in cells [26]. Taken together, TNF-α may directly modulate the EV secretion from muscle cells.
In the present study, the blunting effects of TNF-α treatment of C2C12 cells on the osteoclast formation suppressed by Myo-EVs and osteoblastic differentiation augmented by Myo-EVs were more pronounced in Myo-EVs from C2C12 myoblasts, compared with Myo-EVs from C2C12 myotubes. Moreover, the protein amount of Myo-EVs from C2C12 myoblasts was significantly higher than that of Myo-EVs from C2C12 myotubes. It was reported that human primary myoblasts can be easily infected by Zika virus, compared with myotubes [27]. In addition, it was reported that plasma membrane permeability enhancement and the suppression of cellular ATP content by simvastatin are stronger in C2C12 myoblasts, compared with C2C12 myotubes [28]. Although the mechanism for the differences between myoblasts and myotubes in the sensitivity to external stimulation is unknown, we speculated that the sensitivity to TNF-α treatment is different between myoblasts and myotubes, which may lead to the difference between myoblasts and myotubes in the physiological action of Myo-EVs on bone metabolism.
The osteogenic differentiation of MSCs and osteoclastogenesis inhibition of bone marrow macrophages are promoted by miR196 [29,30,31]. Moreover, miR155 inhibits osteoblastic differentiation of MC3T3-E1 cells by suppressing Smad5 and RANKL-induced osteoclast formation through TGF-β signaling [32, 33], suggesting that miR155 is related to a decrease in bone turnover. We recently reported that miR196-5p included in EVs secreted from C2C12 myoblasts suppresses osteoclast formation in mouse cells [29]. In addition, the application of shear stress to C2C12 cells significantly elevated miR196 and miR155 levels in EVs from C2C12 cells and these miRNAs suppress osteoclast formation in mouse cells [19]. In the present study, TNF-α stimulation of C2C12 myoblasts significantly reduced miR196 levels in Myo-EVs, although it did not affect miR155 levels in Myo-EVs. These findings suggest that miR196 may be involved in the attenuation of TNF-α treatment of C2C12 muscle cells on osteoclast formation, suppressed by Myo-EVs in mouse bone marrow cell cultures and Raw 264.7 cells, although further studies are necessary to clarify the critical factors responsible for TNF-α effects on Myo-EVs.
In the present study, Myo-EVs from C2C12 myoblasts significantly decreased mineralization in ST-2 cells, although Myo-EVs from C2C12 myoblasts did not affect the osteoblast phenotypes and mineralization in mouse osteoblasts in our previous study [18]. The reason of different effects of Myo-EVs on mineralization between ST-2 cells and mouse primary osteoblasts remains unknown. However, the effects of Myo-EVs on osteoblast differentiation are different due to osteoblastic differentiation stage, which might influence the Myo-EVs effects on mineralization. Alternatively, Myo-EVs might suppress mineralization process, but not osteoblast differentiation, in ST-2 cells. On the other hand, TNF-α treatment of C2C12 myoblasts and myotubes significantly decreased mineralization in the presence of Myo-EVs in ST-2 cells, supporting our findings that TNF-α negatively affects the muscle–bone interactions by directly modulating Myo-EVs. The potent suppression of TNF-α treatment of C2C12 cells on mineralization were compatible with its suppression on osteoblastic differentiation by Myo-EVs in ST-2 cells.
There are several study limitations. Although we provided significant evidence for the TNF-α stimulation of muscle cells blunting the effects of Myo-EVs on bone cells in vitro, further in vivo studies using muscle-specific EV-deleted inflammatory mice are necessary to show the pathophysiological significance of TNF-α effects on bone through skeletal muscle-derived EVs. Secondly, EVs contained in horse serum were not completely depleted by ultracentrifugation in the present study, although we cultured C2C12 myotubes with 10% EV-depleted FBS, but not horse serum, with and without TNF-α for 48 h for the preparation of conditioned medium to isolate Myo-EVs. Thus, we cannot completely rule out the possibility that EVs contained in the horse serum could modulate the effects of Myo-EVs from C2C12 myotubes on osteoclast formation and osteogenic differentiation.
In conclusion, our study showed that TNF-α stimulation of C2C12 muscle cells attenuates the osteoclast formation suppression by Myo-EVs and osteoblastic differentiation promoted by Myo-EVs in mouse cells. The level of miR196 in EVs may be involved in the attenuation of TNF-α treatment of C2C12 muscle cells on osteoclast formation that is suppressed by Myo-EVs. TNF-α may negatively affect the muscle–bone interactions by directly modulating Myo-EV secretion.
References
Kawao N, Kaji H (2015) Interactions between muscle tissues and bone metabolism. J Cell Biochem 116(5):687–695. https://doi.org/10.1002/jcb.25040
Kaji H (2016) Effects of myokines on bone. Bonekey Rep 5:826. https://doi.org/10.1038/bonekey.2016.48
Dam TT, Harrison S, Fink HA, Ramsdell J, Barrett-Connor E (2010) Bone mineral density and fractures in older men with chronic obstructive pulmonary disease or asthma. Osteoporos Int 21(8):1341–1349. https://doi.org/10.1007/s00198-009-1076-x
Lee YF, Tsou HK, Leong PY, Wang YH, Wei JC (2021) Association of sepsis with risk for osteoporosis: a population-based cohort study. Osteoporos Int 32(2):301–309. https://doi.org/10.1007/s00198-020-05599-3
Byun MK, Cho EN, Chang J, Ahn CM, Kim HJ (2017) Sarcopenia correlates with systemic inflammation in COPD. Int J Chron Obstruct Pulmon Dis 12:669–675. https://doi.org/10.2147/COPD.S130790
Kim Y (2021) Emerging treatment options for sarcopenia in chronic liver disease. Life 11(3):250. https://doi.org/10.3390/life11030250
Chu WM (2013) Tumor necrosis factor. Cancer Lett 328(2):222–225. https://doi.org/10.1016/j.canlet.2012.10.014
Li CW, Yu K, Shyh-Chang N, Li GX, Jiang LJ, Li DJ et al (2019) Circulating factors associated with sarcopenia during ageing and after intensive lifestyle intervention. J Cachexia Sarcopenia Muscle 10(3):586–600. https://doi.org/10.1002/jcsm.12417
Osta B, Benedetti G, Miossec P (2014) Classical and paradoxical effects of TNF-α on bone homeostasis. Front Immunol 5:48. https://doi.org/10.3389/fimmu.2014.00048
Zhao B (2020) Intrinsic restriction of TNF-mediated inflammatory osteoclastogenesis and bone resorption. Front Endocrinol 11:583561. https://doi.org/10.3389/fendo.2020.583561
Wang DT, Yin Y, Yang YJ, Lv PJ, Shi Y, Wei LB (2014) Resveratrol prevents TNF-α-induced muscle atrophy via regulation of Akt/mTOR/FoxO1 signaling in C2C12 myotubes. Int Immunopharmacol 19(2):206–213. https://doi.org/10.1016/j.intimp.2014.02.002
Cui Y, Luan J, Li H, Zhou X, Han J (2016) Exosomes derived from mineralizing osteoblasts promote ST2 cell osteogenic differentiation by alteration of microRNA expression. Febs Lett 590(1):185–192. https://doi.org/10.1002/1873-3468.12024
Li Q, Huang QP, Wang YL, Huang QS (2018) Extracellular vesicle-mediated bone metabolism in the bone microenvironment. J Bone Miner Metab 36(1):1–11. https://doi.org/10.1007/s00774-017-0860-5
Sun W, Zhao C, Li Y, Wang L, Nie G, Li Y et al (2016) Osteoclast-derived microRNA-containing exosomes selectively inhibit osteoblast activity. Cell Discov 2(1):1–23. https://doi.org/10.1038/celldisc.2016.15
Ikebuchi Y, Aoki S, Honma M, Hayashi M, Sugamori Y, Khan M, Suzuki H et al (2018) Coupling of bone resorption and formation by RANKL reverse signalling. Nature 561(7722):195–200. https://doi.org/10.1038/s41586-018-0482-7
Uenaka M, Yamashita E, Kikuta J, Morimoto A, Ochiya T, Ishii M et al (2022) Osteoblast-derived vesicles induce a switch from bone-formation to bone-resorption in vivo. Nat Commun 13(1):1–13. https://doi.org/10.1038/s41467-022-28673-2
Qin Y, Peng Y, Zhao W, Pan J, Ksiezak-Reding H, Cardozo C, Qin W et al (2017) Myostatin inhibits osteoblastic differentiation by suppressing osteocyte-derived exosomal microRNA-218: A novel mechanism in muscle-bone communication. J Biol Chem 292(26):11021–11033. https://doi.org/10.1074/jbc.M116.770941
Takafuji Y, Tatsumi K, Ishida M, Kawao N, Okada K, Kaji H (2020) Extracellular vesicles secreted from mouse muscle cells suppress osteoclast formation: roles of mitochondrial energy metabolism. Bone 134:115298. https://doi.org/10.1016/j.bone.2020.115298
Takafuji Y, Tatsumi K, Kawao N, Okada K, Muratani M, Kaji H (2021) Effects of fluid flow shear stress to mouse muscle cells on the bone actions of muscle cell-derived extracellular vesicles. PLoS One 16(5):e0250741. https://doi.org/10.1371/journal.pone.0250741
Xu J, Feng Y, Jeyaram A, Jay SM, Zou L, Chao W (2018) Circulating plasma extracellular vesicles from septic mice induce inflammation via microRNA-and TLR7-dependent mechanisms. J Immunol 201(11):3392–3400. https://doi.org/10.4049/jimmunol.1801008
Cheng M, Yang J, Zhao X, Zeng Q, Yu Y, Qin G et al (2019) Circulating myocardial microRNAs from infarcted hearts are carried in exosomes and mobilise bone marrow progenitor cells. Nat Commun 10(1):1–9. https://doi.org/10.1038/s41467-019-08895-7
Kang M, Huang CC, Lu Y, Shirazi S, Ravindran S, Cooper LF (2022) Extracellular vesicles from TNFα preconditioned MSCs: Effects on immunomodulation and bone regeneration. Front. Immunol 13:878194. https://doi.org/10.3389/fimmu.2022.878194
Takafuji Y, Hori M, Mizuno T, Harada-Shiba M (2019) Humoral factors secreted from adipose tissue-derived mesenchymal stem cells ameliorate atherosclerosis in Ldlr−/− mice. Cardiovasc Res 115(6):1041–1051. https://doi.org/10.1093/cvr/cvy271
Forterre A, Jalabert A, Chikh K, Pesenti S, Granjon A, Rome S et al (2014) Myotube-derived exosomal miRNAs downregulate Sirtuin1 in myoblasts during muscle cell differentiation. Cell Cycle 13(1):78–89. https://doi.org/10.4161/cc.26808
Chen L, Yao X, Yao H, Ji Q, Ding G, Liu X (2020) Exosomal miR-103-3p from LPS-activated THP-1 macrophage contributes to the activation of hepatic stellate cells. FASEB J 34(4):5178–5192. https://doi.org/10.1096/fj.201902307RRR
Sohda M, Misumi Y, Oda K (2015) TNFα triggers release of extracellular vesicles containing TNFR1 and TRADD, which can modulate TNFα responses of the parental cells. Arch Biochem Biophys 587:31–37. https://doi.org/10.1016/j.abb.2015.10.009
Legros V, Jeannin P, Chaze T, Gianetto QG, Butler-Browne G, Ceccaldi PE et al (2020) Differentiation-dependent susceptibility of human muscle cells to Zika virus infection. PLoS Negl Trop Dis 14(8):e0008282. https://doi.org/10.1371/journal.pntd.0008282
Sanvee GM, Bouitbir J, Krahenbuhl S (2021) C2C12 myoblasts are more sensitive to the toxic effects of simvastatin than myotubes and show impaired proliferation and myotube formation. Biochem Pharmacol 190:114649. https://doi.org/10.1016/j.bcp.2021.114649
Takafuji Y, Tatsumi K, Kawao N, Okada K, Muratani M, Kaji H (2021) MicroRNA-196a-5p in extracellular vesicles secreted from myoblasts suppresses osteoclast-like cell formation in mouse cells. Calcif Tissue Int 108(3):364–376. https://doi.org/10.1007/s00223-020-00772-6
Kim YJ, Bae SW, Yu SS, Bae YC, Jung JS (2009) miR-196a regulates proliferation and osteogenic differentiation in mesenchymal stem cells derived from human adipose tissue. J Bone Miner Res 24(5):816–825. https://doi.org/10.1359/jbmr.081230
Candini O, Spano C, Murgia A, Grisendi G, Piccinno MS, Dominici M et al (2015) Mesenchymal progenitors aging highlights a miR-196 switch targeting HOXB7 as master regulator of proliferation and osteogenesis. Stem Cells 33(3):939–950. https://doi.org/10.1002/stem.1897
Gu Y, Ma L, Song L, Li X, Chen D, Bai X (2017) miR-155 inhibits mouse osteoblast differentiation by suppressing SMAD5 expression. BioMed Res Int 2017:1893520. https://doi.org/10.1155/2017/1893520
Zhao H, Zhang J, Shao H, Liu J, Chen J, Huang Y et al (2017) Transforming growth factor β1/Smad4 signaling affects osteoclast differentiation via regulation of miR 155 expression. Mol Cells 40(3):211–221. https://doi.org/10.1434/molcells.2017.2303
Acknowledgements
The study was partially supported by All-Kindai University support project against COVID-19 to Y. Takafuji., the Cooperative Research Program (Joint usage/Research Center program) of the Institute for Frontier Life and Medical Sciences, Kyoto University to Y. Takafuji., Takeda Science Foundation to Y.Takafuji., Tokyo Biochemical Research Foundation to Y. Takafuji., a JSPS KAKENHI Grant-in-Aid for Early Career Scientists (19K18480) and Grants-in-Aid for Scientific Research (C: 21K09240) to Y. Takafuji., and Grants-in-Aid for Scientific Research (C:20K09514) to H.K. from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Funding
All-Kindai University support project against COVID-19, the Cooperative Research Program (Joint usage/Research Center program) of the Institute for Frontier Life and Medical Sciences, Kyoto University; Takeda Science Foundation; Tokyo Biochemical Research Foundation; JSPS KAKENHI Grant-in-Aid for Early Career Scientists, 19K18480, Yoshimasa Takafuji; JSPS KAKENHI Grants-in-Aid for Scientific Research, 21K09240, Yoshimasa Takafuji and 20K09514, Hiroshi Kaji
Author information
Authors and Affiliations
Contributions
YT, YT, NK, TO, and H K contributed to the conception and design of the research. YT, YT, YM, and K O performed the experiments. YT, YT, YM, and HK interpreted the results of the experiments. YT and YT analyzed data, prepared the figures, and drafted the manuscript. HK edited and revised the manuscript. All authors approved the final version of manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Yuto Takada, Yoshimasa Takafuji, Yuya Mizukami, Takashi Ohira, Naoyuki Kawao, Kiyotaka Okada, and Hiroshi Kaji declare that they have no conficts of interest.
Human and Animal Rights and Informed Consent
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted. This study does not contain any studies with human participants performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Takada, Y., Takafuji, Y., Mizukami, Y. et al. Tumor Necrosis Factor-α Blunts the Osteogenic Effects of Muscle Cell-Derived Extracellular Vesicles by Affecting Muscle Cells. Calcif Tissue Int 112, 377–388 (2023). https://doi.org/10.1007/s00223-022-01056-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-022-01056-x