
Abstract
Connective tissue diseases, including osteogenesis imperfecta (OI) and Ehlers-Danlos syndrome (EDS), exhibit a high degree of clinical and genetic heterogeneity. We report two sisters with blue sclerae, joint hypermobility and hearing loss. Whole-exome sequencing identified two compound heterozygous ZNF469 loss-of-function mutations due to a frameshift. Since these findings indicate the presence of brittle cornea syndrome (BCS), we performed ocular optical coherence tomography (OCT) and pachymetry, which revealed a moderate decrease in corneal thickness. While only one traumatic fracture was observed in each of the patients, a detailed skeletal assessment indicated no specific patterns of bone mass and microstructure reduction as well as normal bone turnover markers. Taken together, our findings point to a mild form of brittle cornea syndrome with a phenotype compatible with the extraskeletal features of OI but also with EDS.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Connective tissue diseases, including osteogenesis imperfecta (OI) and Ehlers-Danlos syndrome (EDS), exhibit not only a high degree of clinical but also genetic heterogeneity. OI is a well-described genetic skeletal disorder defined by increased bone fragility, which is most commonly caused by heterozygous mutations in two genes encoding type 1 collagen (COL1A1/COL1A2), however, many other forms and gene mutations have been identified [1]. Besides bone fragility, a number of extraskeletal symptoms including blue sclerae, joint hypermobility and hearing loss have been described [1]. Joint hypermobility is primarily a typical sign of EDS, a genetic connective tissue disorder caused by specific collagen mutations (type I, III or V) or other mutations in genes involved in collagen production or processing [2, 3]. While many atypical forms (both clinically and genetically) of OI and EDS have been reported, molecular analyses may also result in the detection of other connective tissue diseases. Zinc-finger proteins (ZNFs) are involved in a variety of cellular processes that occur via different molecular mechanisms [4]. Mutations in ZNF469 cause brittle cornea syndrome (BCS) [5, 6], a multisystem connective tissue disorder primarily associated with corneal thinning but also blue sclerae and joint hypermobility [7]. BCS may also be caused by mutations in the gene encoding the transcription factor PRDM5. Although the exact molecular mechanisms for BCS pathogenesis remain unknown, ZNF469 and PRDM5 have been found to result in reduced expression of the same extracellular matrix (ECM) genes (e.g., COL11A1) [8]. In this report, we present the clinical and molecular features of two sisters with suspect of an inherited connective tissue diseases, who were later correctly diagnosed with a mild form BCS by extended diagnostics including whole exome sequencing.
Patients and Methods
Here, we report two sisters (II.1, 47 years and II.2, 44 years) with blue sclerae, joint hypermobility and hearing impairment since childhood. Due to these specific clinical manifestations, which occurred at an early onset, the suspicion of a hereditary disorder was raised. Initially, OI had been suspected due to the presence of extraskeletal OI features, but had not been genetically confirmed. However, the sisters had suffered from only one traumatic fracture each (II.1 forearm, age 13; II.2 tibia, age 42). The further clinical history was uneventful. Both sisters were premenopausal, had a balanced diet, received no medication (especially no medication that could affect bone turnover or hearing) and were both normally physically active.
After exclusion of known disease genes for osteogenesis imperfecta and other skeletal and connective tissue diseases in the index case using a custom designed gene panel (Sure Select, Agilent) including 386 genes, whole-exome sequencing was carried out in the index patient and the unaffected parents (Human all exon V6, Agilent). Sequencing was performed on a HiSeq 2500 sequencing machine (Illumina, San Diego, CA, USA). Data were analyzed by the software tools GeneTalk [9] and MutationDistiller [10]. The variants were prioritized using the phenotype terms blue sclerae (HP:0000592), hypermobility of joints (HP:0001382), and hearing impairment (HP:0000365). ZNF469 variants were ranked among the top 10 variants. The pathogenicity of the prioritized variants was judged using MutationTaster [11].
The Beighton Score was used to assess joint hypermobility [12]. To further investigate the ear and cornea phenotype, audiometry as well as ocular optical coherence tomography (OCT) and Pentacam corneal pachymetry were performed. In order to further analyze bone status, areal bone mineral density (aBMD) was evaluated in the lumbar spine and the left hip using dual energy X-ray absorptiometry (DXA, Lunar iDXA, GE Healthcare; Madison, WI, USA). Bone microstructure was analyzed in the non-dominant (left) distal radius and the contralateral distal tibia using high-resolution peripheral quantitative computed tomography (HR-pQCT; XtremeCT, Scanco Medical, Switzerland). Serum and urinary biochemical bone turnover markers including calcium, 25-hydroxyvitamin D, parathyroid hormone (PTH), osteocalcin, bone-specific alkaline phosphatase (BAP) and deoxypyridinoline (DPD) were assessed.
Results
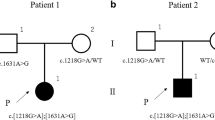
After all known OI genes were excluded by gene panel analysis, whole-exome sequencing of the index patient (II.1) and both parents revealed two heterozygous variants c.10664delC p.(Pro3556Glnfs*136) and c.10240delA p.(Arg3414Glyfs*59) in the ZNF469 gene (Fig. 1a, b). Each of the variants was found heterozygous in one parent, and both variants were also detected in the sister (II.2) (Fig. 1a, b). The variants, which were confirmed by Sanger sequencing in all four family members, have not been reported before, but the frameshift effect of the single nucleotide deletions make a loss-of-function very likely. Both parents did not show any phenotypic abnormalities supporting the recessive inheritance.

Compound heterozygous ZNF469 mutations of two patients with mild brittle cornea syndrome. a Integrated Genome Viewer presentation demonstrating the compound heterozygosity of the ZNF 469 mutations c.10664delC p.(Pro3556Glnfs*136) and c.10240delA p.(Arg3414Glyfs*59). b Pedigree
The variants were ranked class V (pathogenic) according to ACMG criteria (PVS1, PM2, PM3, PP1, PP3) [13].
The two affected sisters presented with a normal height (II.1: 157 cm, 10. to 25. percentile; II.2: 165 cm, 50. to 75. percentile), blue sclerae and notable joint hypermobility. In fact, the Beighton score was 9/9 (II.1) and 8/9 (II.2) indicating severe hypermobility including the ability to bend the knees and elbows backwards (hyperextension in both sisters, knee: 20°, elbow: 30°). Furthermore, both sisters were affected by mild hip dysplasia (Crowe type I), which was currently asymptomatic. Patient II.2 also suffered from recurrent patella luxation, which was treated surgically by reconstruction of the medial patellofemoral ligament.
After detection of the ZNF469 variants, the patients were closely reexamined for other typical features of brittle cornea syndrome (BCS). The blue sclerae were confirmed in both patients by several expert clinicians and by using a split lamp (Fig. 2a). Radiographs of the hands indicated rather long fingers without deformities (Fig. 2b). A combined sensorineural and conductive hearing loss was confirmed by audiometry (Fig. 2c). Ocular optical coherence tomography (OCT) and Pentacam pachymetry/tomography revealed a reduction in corneal thickness of 453 µm (II.1) and 456 µm (II.2) without the suspicion of keratoconus or keratoglobus (Fig. 2d).

Clinical phenotype. a Blue sclera exemplary shown for patient II.1 (Slit lamp photograph). b Hand radiograph demonstrating long fingers. c Audiometry showing a mixed sensorineural and conductive hearing loss. d Corneal pachymetry showing moderately reduced corneal thickness
DXA and HR-pQCT indicated no clear reductions of bone mass and microstructure compared to previously reported references [14]. In fact, only the BMD Z-score in the hip of patient II.2 was reduced (− 2.1) (Fig. 3a). Next to a slightly reduced trabecular number (Tb.N) in patient II.2 and normal Tb.N in patient II.1 in the distal radius and tibia, we detected a moderately reduced cortical thickness (Ct.Th) in both sisters in the distal radius and tibia (around 60–80% of the age-matched mean [14]) (Fig. 3a, b). Laboratory analyses excluded renal deficiency, hyperparathyroidism or thyroid disorders (Supplementary Table 1). The detected vitamin D deficiency in both sisters was balanced by 20,000 I.E. vitamin D weekly. Markers of bone formation (osteocalcin and BAP) and bone resorption (DPD) were within the normal range for both patients (Supplementary Table 1, Fig. 3c). A summary of the clinical features for both sisters is presented in Fig. 3d.

Skeletal analysis. a, b Bone density and microstructure assessed by DXA at the spine/hip and HR-pQCT at the distal radius/tibia, respectively. HR-pQCT values were compared to sex- and age-matched reference values from the literature [14]. c Bone turnover. Oc, osteocalcin, DPD, deoxypyridinoline. Bone-specific alkaline phosphatase values are not shown. Grey boxes indicate reference ranges. Black dot: patient II.1, red dot: patient II.2. d Summary of the clinical features in both patients affected by ZNF469 mutation
Discussion
In this case report, we present two novel compound heterozygous ZNF469 mutations causing a mild form of brittle cornea syndrome (BCS) that shows clinical resemblance with the extraskeletal manifestations of osteogenesis imperfecta (OI) type I. The phenotype was not only compatible with features of OI but primarily with EDS. In fact, since several other patients with BCS were previously clinically suspected to have a form of EDS [15], BCS was also included in the EDS classification in 2017 [16].
The patients reported here presented with a mild but characteristic phenotype of BCS including blue sclerae, hypermobility, corneal thinning and hearing loss [15]. While previously described as extraskeletal manifestations of OI, blue sclerae or joint hypermobility may also be present in other bone and connective tissue disorders (primarily EDS) [17, 18]. The differential diagnosis of blue sclerae includes particularly connective tissue disorders such as OI, Marfan's syndrome, EDS, and pseudoxanthoma elasticum, among others. The reduction of corneal thickness to around 450 µm in both sisters was moderate but relevant compared to previously published reference values (mean 537.4 µm; 95% confidence interval, 533.8 to 540.9 µm [19]). Nonetheless, patients with severe BCS usually present with a corneal thickness < 400 µm [15]. The auditory phenotype was also mild, however, combined sensorineural and conductive hearing loss was detected in both sisters. Deafness has been reported in two of three unrelated patients with ZNF469 mutations [20], but there is a wide phenotypic variability with both sensorineural and conductive components [15]. Given the fact that BCS represents a connective tissue disorder, musculoskeletal features have been found in most patients. Especially, hip dysplasia and scoliosis seem to be frequent features of BCS [6, 15]. Hip dysplasia was indeed present in both of our patients, but scoliosis was not diagnosed.
While low bone mineral density has been reported in one family with ZNF469 mutation [6], we here present the first comprehensive skeletal analysis in patients with ZNF469 mutations including DXA, HR-pQCT and bone turnover status. Bone mineral density and microstructure were overall not severely reduced, which is reflected by the low fracture rate in the two sisters reported herein. In our patients, the main musculoskeletal feature was hypermobility. In combination with the detected blue sclerae and hearing loss, these characteristics are compatible with EDS as well as the extraskeletal features of OI.
The detected ZNF469 variants have not been reported before, but similar deletions that lie very close within the ZNF469 gene have been reported [5, 20]. The majority of known ZNF469 mutations found in patients with brittle cornea syndrome have a loss-of-function effect [5, 20]. The molecular analysis was of particular importance to make the correct diagnosis and to rule out OI or EDS, especially with regard to the different inheritance pattern (i.e., autosomal dominant in most OI cases). The recurrence risk for BCS in the children of the two probands is negligible due to the recessive inheritance and the extreme rarity of the disorder. Although the variants were ranked as pathogenic, the limitation of this case study includes the fact that no further functional studies were carried out. It is interesting to note that mutations in another member of the ZNF genes, specifically ZNF687, have been reported in a peculiar subgroup of severe Paget’s disease of bone associated with giant cell tumor [21, 22].
In conclusion, we demonstrate novel compound heterozygous ZNF469 mutations in two middle-aged sisters. Our results indicate the existence of a mild phenotype of brittle cornea syndrome with blue sclerae, joint hypermobility and hearing loss, which expands the clinical spectrum of genetic connective tissue disorders that resemble OI and EDS.
References
Forlino A, Marini JC (2016) Osteogenesis imperfecta. Lancet 387(10028):1657–1671
Cabral WA, Makareeva E, Colige A, Letocha AD, Ty JM, Yeowell HN, Pals G, Leikin S, Marini JC (2005) Mutations near amino end of alpha1(I) collagen cause combined osteogenesis imperfecta/Ehlers-Danlos syndrome by interference with N-propeptide processing. J Biol Chem 280(19):19259–19269
Malfait F, Symoens S, Goemans N, Gyftodimou Y, Holmberg E, Lopez-Gonzalez V, Mortier G, Nampoothiri S, Petersen MB, De Paepe A (2013) Helical mutations in type I collagen that affect the processing of the amino-propeptide result in an Osteogenesis Imperfecta/Ehlers-Danlos Syndrome overlap syndrome. Orphanet J Rare Dis 8(1):78
Cassandri M, Smirnov A, Novelli F, Pitolli C, Agostini M, Malewicz M, Melino G, Raschella G (2017) Zinc-finger proteins in health and disease. Cell Death Discov 3:17071
Abu A, Frydman M, Marek D, Pras E, Nir U, Reznik-Wolf H, Pras E (2008) Deleterious mutations in the Zinc-Finger 469 gene cause brittle cornea syndrome. Am J Hum Genet 82(5):1217–1222
Christensen AE, Knappskog PM, Midtbo M, Gjesdal CG, Mengel-From J, Morling N, Rodahl E, Boman H (2010) Brittle cornea syndrome associated with a missense mutation in the zinc-finger 469 gene. Investig Ophthalmol Vis Sci 51(1):47–52
Walkden A, Burkitt-Wright E, Au L (2019) Brittle cornea syndrome: current perspectives. Clin Ophthalmol 13:1511–1516
Burkitt Wright EMM, Spencer HL, Daly SB, Manson FDC, Zeef LAH, Urquhart J, Zoppi N, Bonshek R, Tosounidis I, Mohan M, Madden C, Dodds A, Chandler KE, Banka S, Au L, Clayton-Smith J, Khan N, Biesecker LG, Wilson M, Rohrbach M, Colombi M, Giunta C, Black GCM (2011) Mutations in PRDM5 in brittle cornea syndrome identify a pathway regulating extracellular matrix development and maintenance. Am J Hum Genet 88(6):767–777
Kamphans T, Krawitz PM (2012) GeneTalk: an expert exchange platform for assessing rare sequence variants in personal genomes. Bioinformatics 28(19):2515–2516
Hombach D, Schuelke M, Knierim E, Ehmke N, Schwarz JM, Fischer-Zirnsak B, Seelow D (2019) MutationDistiller: user-driven identification of pathogenic DNA variants. Nucleic Acids Res 47(W1):W114–W120
Schwarz JM, Rodelsperger C, Schuelke M, Seelow D (2010) MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 7(8):575–576
Beighton P, Solomon L, Soskolne CL (1973) Articular mobility in an African population. Ann Rheum Dis 32(5):413–418
Rivera-Munoz EA, Milko LV, Harrison SM, Azzariti DR, Kurtz CL, Lee K, Mester JL, Weaver MA, Currey E, Craigen W, Eng C, Funke B, Hegde M, Hershberger RE, Mao R, Steiner RD, Vincent LM, Martin CL, Plon SE, Ramos E, Rehm HL, Watson M, Berg JS (2018) ClinGen Variant Curation Expert Panel experiences and standardized processes for disease and gene-level specification of the ACMG/AMP guidelines for sequence variant interpretation. Hum Mutat 39(11):1614–1622
Burt LA, Liang Z, Sajobi TT, Hanley DA, Boyd SK (2016) Sex- and site-specific normative data curves for HR-pQCT. J Bone Miner Res 31(11):2041–2047
Burkitt Wright EM, Porter LF, Spencer HL, Clayton-Smith J, Au L, Munier FL, Smithson S, Suri M, Rohrbach M, Manson FD, Black GC (2013) Brittle cornea syndrome: recognition, molecular diagnosis and management. Orphanet J Rare Dis 8:68
Malfait F, Francomano C, Byers P, Belmont J, Berglund B, Black J, Bloom L, Bowen JM, Brady AF, Burrows NP, Castori M, Cohen H, Colombi M, Demirdas S, De Backer J, De Paepe A, Fournel-Gigleux S, Frank M, Ghali N, Giunta C, Grahame R, Hakim A, Jeunemaitre X, Johnson D, Juul-Kristensen B, Kapferer-Seebacher I, Kazkaz H, Kosho T, Lavallee ME, Levy H, Mendoza-Londono R, Pepin M, Pope FM, Reinstein E, Robert L, Rohrbach M, Sanders L, Sobey GJ, Van Damme T, Vandersteen A, van Mourik C, Voermans N, Wheeldon N, Zschocke J, Tinkle B (2017) The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C 175(1):8–26
Janecke AR, Li B, Boehm M, Krabichler B, Rohrbach M, Müller T, Fuchs I, Golas G, Katagiri Y, Ziegler SG, Gahl WA (2016) The phenotype of the musculocontractural type of Ehlers-Danlos syndrome due to CHST14 mutations. Am J Med Genet A 170(1):103–115
Reversade B, Escande-Beillard N, Dimopoulou A, Fischer B, Chng SC, Li Y, Shboul M, Tham PY, Kayserili H, Al-Gazali L, Shahwan M, Brancati F, Lee H, O'Connor BD, Schmidt-von Kegler M, Merriman B, Nelson SF, Masri A, Alkazaleh F, Guerra D, Ferrari P, Nanda A, Rajab A, Markie D, Gray M, Nelson J, Grix A, Sommer A, Savarirayan R, Janecke AR, Steichen E, Sillence D, Hausser I, Budde B, Nurnberg G, Nurnberg P, Seemann P, Kunkel D, Zambruno G, Dallapiccola B, Schuelke M, Robertson S, Hamamy H, Wollnik B, Van Maldergem L, Mundlos S, Kornak U (2009) Mutations in PYCR1 cause cutis laxa with progeroid features. Nat Genet 41(9):1016–1021
Wolfs RC, Klaver CC, Vingerling JR, Grobbee DE, Hofman A, de Jong PT (1997) Distribution of central corneal thickness and its association with intraocular pressure: The Rotterdam Study. Am J Ophthalmol 123(6):767–772
Rohrbach M, Spencer HL, Porter LF, Burkitt-Wright EMM, Burer C, Janecke A, Bakshi M, Sillence D, Al-Hussain H, Baumgartner M, Steinmann B, Black GCM, Manson FDC, Giunta C (2013) ZNF469 frequently mutated in the brittle cornea syndrome (BCS) is a single exon gene possibly regulating the expression of several extracellular matrix components. Mol Genet Metab 109(3):289–295
Divisato G, Formicola D, Esposito T, Merlotti D, Pazzaglia L, Del Fattore A, Siris E, Orcel P, Brown JP, Nuti R, Strazzullo P, Benassi MS, Cancela ML, Michou L, Rendina D, Gennari L, Gianfrancesco F (2016) ZNF687 mutations in severe paget disease of bone associated with giant cell tumor. Am J Hum Genet 98(2):275–286
Qi X, Pang Q, Wang J, Zhao Z, Wang O, Xu L, Mao J, Jiang Y, Li M, Xing X, Yu W, Asan XW (2017) Familial early-onset paget's disease of bone associated with a novel hnRNPA2B1 mutation. Calcif Tissue Int 101(2):159–169
Acknowledgements
Open Access funding provided by Projekt DEAL. This project has received funding from the German Federal Ministry of Education and Research (BMBF) within the project “Detection and Individualized Management of Early Onset Osteoporosis (DIMEOs)”.
Author information
Authors and Affiliations
Contributions
TR collected the patient data collection and wrote the first draft. RO coordinated the diagnostic assessment and treatment of the patients. UK performed the genetic analysis. SL performed the ophthalmologic assessment. All authors critically reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Tim Rolvien, Uwe Kornak, Stephan Linke, Michael Amling and Ralf Oheim declare that they have no conflict of interest.
Research involving Human Participants & Informed Consent
This case report is in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards. Informed consent was obtained from all individual patients included in the case report.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rolvien, T., Kornak, U., Linke, S.J. et al. Whole-Exome Sequencing Identifies Novel Compound Heterozygous ZNF469 Mutations in Two Siblings with Mild Brittle Cornea Syndrome. Calcif Tissue Int 107, 294–299 (2020). https://doi.org/10.1007/s00223-020-00721-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-020-00721-3