
Abstract
Melorheostosis is an exceptionally rare sclerosing hyperostosis that typically affects the appendicular skeleton in a limited segmental fashion. It occasionally occurs on a background of another benign generalised sclerosing bone condition, known as osteopoikilosis caused by germline mutations in LEMD3, encoding the inner nuclear membrane protein MAN1, which modulates TGFβ/bone morphogenetic protein signalling. Recent studies of melorheostosis lesional tissue indicate that most cases arise from somatic MAP2K1 mutations although a small number may arise from other genes in related pathways, such as KRAS. Those cases associated with MAP2K1 mutations are more likely to have the classic “dripping candle wax” appearance on radiographs. The relationship between these somatic mutations and those found in a variety of malignant conditions is discussed. There are also similar germline mutations involved in a group of genetic disorders known as the RASopathies (including Noonan syndrome, Costello syndrome and various cardiofaciocutaneous syndromes), successful treatments for which could be applied to melorheostosis. The diagnosis and management of melorheostosis are discussed; there are 4 distinct radiographic patterns of melorheostosis and substantial overlap with mixed sclerosing bone dysplasia. Medical treatments include bisphosphonates, but definitive guidance on their use is lacking given the small number of patients that have been studied. Surgical intervention may be required for those with large bone growths, nerve entrapments, joint impingement syndromes or major limb deformities. Bone regrowth is uncommon after surgery, but recurrent contractures represent a major issue in those with extensive associated soft tissue involvement.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Most radiologists, orthopaedic surgeons and rheumatologists have a special place in their case records for a group of rare musculoskeletal oddities to test their trainees. Melorheostosis must surely be somewhere near the top of any such list when it comes to rare, bizarre and poorly understood conditions with which to challenge all but the most dedicated students of medical curiosities. First described in 1922 as Leri’s disease (On-Line Mendelian Inheritance in Man catalogue-OMIM 155950), there have been fewer than 500 formal reports relating to this sclerosing bone disorder [1]. Even the ancient Greek origins of its name serve to deepen its mystery: melos-limb, rhein-to flow (as in river currents), osteon-bone. By any standards, melorheostosis is a rare condition, estimated to affect fewer than 1 in a million persons. It is not restricted to particular ethnic groups and has no recognised environmental contribution. The classic “dripping candle wax” appearance of the affected bone(s) and its segmental distribution is unmistakable but is a far from constant finding (Fig. 1). Indeed, the precise boundary between melorheostosis and other sclerosing bone conditions (osteopoikilosis, osteopathia striata and various other causes of hyperostosis and osteosclerosis) is somewhat blurred [2,3,4,5,6,7]. Many aspects of the condition are still unexplained or contentious. These include:

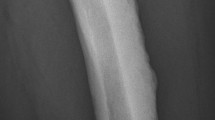
Typical features of melorheostosis showing (a) the “melting candle wax” appearance in the affected ulna of a middle-aged woman and (b) unilateral segmental involvement of the left upper limb in a young woman, including the second and third rays of the hand, the ulna and patchy involvement of the carpus (corresponding approximately to the 7th cervical “sclerotome” but bleeding into the 8th sclerotome)
-
(1)
The clinical and radiographic spectrum of melorheostosis and how to define the condition
-
(2)
How is it best treated and what are the risks of recurrent disease following surgery?
-
(3)
What is the full range of molecular mechanisms underlying melorheostosis and related diseases?
-
(4)
What useful therapeutic insights have arisen from understanding the molecular pathology?
This review is largely based on work published over many decades by others but also in part on our personal (and largely unreported) experience of 30 cases in the UK seen personally at our centre. We describe the clinical and radiographic features of melorheostosis and attempt to provide useful (but by no means definitive) advice about its management. We discuss some of the new genetic insights into this and related disorders that may help to explain the sporadic and segmental nature of the condition and its occasional association with other conditions. Finally, we explore briefly some of the potential clinical applications of these new findings.
Clinical Features
The presenting features of melorheostosis are variable, depending on the site and extent of the bone disease and whether there is any associated soft tissue involvement. Some cases are identified from incidental radiographic findings, but more typically there will be pain, bony swelling, localised growth abnormalities or joint deformities (often due to associated soft tissue involvement). Whether pain is an inherent characteristic of melorheostosis is a moot point; where it is present it can frequently be attributed to mechanical factors, including direct pressure on bony excrescences, joint impingement syndromes or abnormal joint dynamics resulting from deformities (Fig. 2). Various nerve entrapment syndromes have also been described, including spinal nerve root compressions and carpal tunnel syndrome (see below). Sometimes the first indications of underlying bone pathology may come from localised abnormalities in the overlying skin, which may have an appearance like scleroderma, or other soft tissues (tendons, ligaments and joint capsules) that may lead to severe joint contractures.

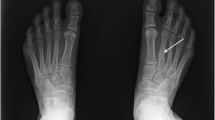
CT reconstruction of the foot of a 65-year-old woman with melorheostosis, showing a large mass of abnormal bone arising from the inferior border of the 2nd metatarsal
The typical features of melorheostosis, characterised by sclerotic cortical bone in a segmental distribution, should present few diagnostic difficulties [8,9,10], but a range of radiographic patterns have been reported and perhaps only one-third of cases exhibit the classic “dripping candle wax” appearance of the cortex (Fig. 1). There is dense, irregular and eccentric hyperostosis of both periosteal and endosteal bone surfaces, affecting either single or multiple adjacent bones. Typically, either the medial or lateral side of the bone is involved and there is usually quite clear demarcation between the affected and unaffected bone. For diagnostic purposes, four distinct patterns [9] are described (Table 1).

Osteoma-like lesions in a teenager with melorheostosis. a Radiograph showing enlargement of the right clavicle with extensive cortical thickening. There are also patchy sclerotic lesions in the right humeral head (b) Both hands showed extensive involvement

Variants of melorheostosis. a There is widespread osteopathia striata-like disease in the right tibia and fibula of this 40-year-old woman. She had extensive disease elsewhere in her body, including the skull, but the left lower leg was unaffected. b Myositis ossificans-like lesions of melorheostosis around the hip of a 65-year-old woman with extensive involvement of the left lower limb. There is also patchy sclerosis and thickening of the left ischial cortex. c Extensive soft tissue ossification is present around the left knee which exhibits a 45° fixed flexion deformity. There is also obvious thickening of the tibial cortex
Where the neighbouring soft tissues are also affected there may be linear scleroderma-like appearance of the involved skin with subcutaneous fibrosis, oedema, hypertrichosis, fibromas, fibrolipomas, capillary haemangiomas, lymphangiectasia and/or aneurysms. Tendon and joint capsule involvement may cause severe joint contractures and deformities, such as talipes equinovarus or flexion contractures of the hip and knee [11]. In some cases, the skin overlying the affected bones has a mottled appearance somewhat reminiscent of livedo reticularis.
In the largest published clinical review (n = 24) from the Mayo Clinic [12], pain was the commonest presenting symptom (83%), followed by limb deformity (54%), limitation of movement (46%), numbness (38%) and weakness (25%). Somewhat surprisingly, this case series included four times as many females as males although the evidence from other reports overall does not suggest a gender bias. Obviously such hospital-based studies could potentially overestimate the level of symptomatic disease, but since there are no population-based studies it is not known how many cases may be asymptomatic. In the Mayo Clinic series, the legs were involved in 16/24 cases, feet/ankles 9/24, arms 8/24, hands/wrists 7/24, spine 4/24 and head only 2/24. Our Oxford experience is very similar (head involvement in only 2/30) although we saw evidence of spinal involvement (often very mild) in 7 of our 30 cases. However, although the appendicular skeleton appears to be much more commonly affected than the spine or skull there may also be an element of ascertainment bias in this because asymptomatic involvement of the axial skeleton could quite easily be missed. Expression of the disease in the vertebrae is also less obvious, perhaps because they contain a preponderance of trabecular rather than cortical bone. The condition usually presents in childhood or adolescence (50% are diagnosed by 20 years of age) and the bony lesions typically progress relatively rapidly in childhood but with active and quiescent periods. Intrinsic bone pain seems to be particularly associated with active periosteal bone formation. In contrast, the bone lesions appear to progress relatively slowly, if at all, after skeletal maturity. Some cases may be complicated by early fusion of the affected epiphyses causing significant limb length discrepancy, which is a potentially severe complication that may require major reconstructive surgery (Fig. 5). This represents a particular management challenge because such surgery is by no means always successful and may need repeating over time to ensure sustained benefit. Very rarely, melorheostosis may be complicated by malignant transformation to osteosarcoma [13].

Progressive limb length inequality in a child with melorheostosis. a Leg length radiographs aged 5 years, showing melorheostosis clearly in both distal femurs and apparent shortening partly due to flexion deformity of the right knee. b Surgical correction of flexion deformity of right knee aged 7.5 years. c Epiphysiodesis of distal left femur aged 10 years to equalise leg length. d Equalisation of leg length by aged 12.10 years (note the surgical fixation of a supracondylar fracture of the left femur sustained in a fall). e Extensive melorheostosis in the right foot
Differential Diagnosis
The differential diagnosis of melorheostosis with a brief list of some of the most obvious distinguishing features is given in Table 2. Typical melorheostosis should be relatively easy to identify but by no means all cases conform to its classic description so that even distinction from Paget’s disease might occasionally be problematic [14]. Conditions from which melorheostosis should be distinguished include: (1) those that cause increased bone density (osteopetrosis, osteosclerosis and hyperostosis) and (2) others that cause ectopic ossification or bony tumours (benign or otherwise) [3, 8]. Some of the conditions that mimic melorheostosis radiographically are illustrated in Fig. 6. In practice, the asymmetry of melorheostosis and the fact that it tends to be restricted to the medial or lateral side of the affected bones contrasts with the often generalised nature of the osteopetroses/hyperostoses/osteoscleroses. There should be relatively little difficulty in distinguishing these disorders once a careful history, clinical examination and appropriate radiology have been completed. However, there is significant overlap with certain sclerosing conditions of bone, such as osteopathia striata (Voorhoeve disease), osteopoikilosis [3, 6, 7, 9], progressive diaphyseal dysplasia (Camurati–Englemann disease) [15, 16], and its transforming growth factor β (TGFβ) allelic lookalike multiple diaphyseal sclerosis (Ribbing disease) [17, 18]. Ghosal haematodiaphyseal dysplasia is a rare autosomal dominant condition causing a symmetric metadiaphyseal dysplasia of the long bones that can be distinguished also by the associated defective haematopoiesis due to marrow fibrosis [19, 20]. In the syndrome of mixed sclerosing bone, dystrophy/dysplasia matters are more complicated because melorheostosis coexists with other forms of bone sclerosis [4, 5, 7, 21,22,23,24]. This condition is itself heterogeneous since at least 4 different variants have been described depending on the relative preponderance of melorheostosis, osteopoikilosis, osteopathia striata and osteosclerosis [7]. Genetic disorders associated with bony outgrowths, such as multiple hereditary exostoses, multiple enchondromatosis (Ollier’s disease) or ectopic ossification, such as fibrodysplasia ossificans progressive or progressive osseous heteroplasia sometimes bear superficial resemblance to melorheostosis but should be quite straightforward to distinguish. Non-hereditary conditions to be considered include intramedullary osteosclerosis that typically affects the mid-diaphyseal region of one or both tibias (although the fibula and femur may also be affected) in the absence of alternative causes, such as infection, trauma or systemic illness. Some other conditions that are associated with a significant degree of periosteal reaction may be occasionally confused with melorheostosis. These include malignancy (osteogenic sarcoma and Ewing’s sarcoma), Caffey disease, infection, trauma and vitamin C deficiency.

A number of sclerotic bone conditions mimic melorheostosis (Table 2). a progressive diaphyseal dysplasia (Camurati–Englemann disease), demonstrating symmetric cortical thickening of the long bones in this pelvic radiograph and (b) osteopoikilosis in which there is localised symmetric sclerosis in the epiphyses/metaphyses as indicated here in the femoral neck (arrow). c Pachydermoperiostosis showing thickening of the distal part of the tibia and fibula due to periosteal reaction
Aetiology and Molecular Pathogenesis
Many ideas about the cause of melorheostosis reflect its limited segmental distribution although more extensive disease is also well described [25]. Sometimes the disease appears to jump anatomical segments. For example, in one of our cases the disease was evident in the pelvis and throughout both lower limbs but elsewhere the only evidence of disease anywhere in the body was limited to the 5th metacarpal in the right hand. The segmental distribution of melorheostosis led to the “sclerotomal” theory advanced by Murray and Mcredie [26] that has influenced many ideas about its pathogenesis [27, 28], including the possibility that foetal or childhood infections affecting particular nerve roots (and the sclerotomes that they innervate) might be responsible. However, this sclerotomal distribution has been challenged by a recent study where PET/CT was used to define the anatomical distribution of melorheostosis in 30 affected individuals [29]. Only 5/30 had disease restricted to a single sclerotome; 12/30 had disease roughly conforming to contiguous sclerotomes, but there was significant “bleeding” of disease across sclerotomes. These authors favour an explanation implicating “clonal proliferation of a mutation-positive skeletal progenitor cell along the limb axis”. In an extension of these studies, the same group provide detailed histological analysis of the abnormal bone in melorheostosis; they suggest that the condition reflects a periosteal reaction to the gradual deterioration in bony microarchitecture somewhat akin to the situation in local infection or trauma [30].
The vast majority of cases are sporadic, but in a small minority the condition appears against a background of other hyperostotic bone disease, such as osteopoikilosis, Buschke–Ollendorf syndrome (BOS–OMIM 166700) or mixed sclerosing bone dysplasia (MSBD) [31, 32]. The latter is particularly intriguing because it has a very variable phenotype combining features of melorheostosis, osteopoikilosis, osteopathia striata and osteosclerosis, the relative amounts of which vary not only between affected individuals but also between affected areas of the skeleton [4]. Some cases are also associated with extensive soft tissue pathology, including unilateral MSBD lymphangiectasis and capillary haemangioma [33], and arteriovenous malformations [22]. Its aetiology and precise relationship to most sporadic cases of melorheostosis is unclear to the extent that some authors have recommended that where MSBD and melorheostosis coexist the condition should be classified as melorheostosis. There are occasional reports of MSBD variants segregating as an autosomal dominant trait in families, but these cases are atypical with additional features, such as generalised ichthyosis and premature ovarian failure in one 3-generation family [34] or facial dysmorphism and cranial sclerosis in another [35]. The gene loci responsible for these rare familial cases have not been identified. In contrast, most cases of both osteopoikilosis and BOS are caused by germ-line mutations in LEMD3 (LEM domain-containing protein 3), which encodes an integral inner nuclear membrane protein, MAN1 involved in the TGFβ signalling pathway (see below) [36,37,38]. Endochondral ossification during the seventh week of embryogenesis forms the primary spongiosa, resorptive failure of which (in conditions like osteopetrosis) leads to generalised accumulation of calcified cartilage matrix in the medullary cavity. Subsequently, during the ninth week of embryogenesis, osteoclasts normally remodel the spongiosa into trabeculae and medullary cavity; failure to do this results in the focal sclerotic lesions of osteopoikilosis or striations in the medullary cavity (osteopathia striata) [reviewed in reference 2]. Where melorheostosis occurs in the context of osteopoikilosis/BOS it is localised and segmental (as in the sporadic form) but there will also be radiographic evidence of osteopoikilosis elsewhere [31, 32]. Recurrent melorheostosis has not been described in these families, but other relatives may show the characteristic changes of osteopoikilosis since this is a dominantly inherited condition.
Abnormalities of the TGFβ signalling pathway lie at the pathogenic heart of osteopoikilosis and BOS. The LEMD3 loss-of-function mutations that are found in the vast majority of cases are entirely consistent with this [36,37,38]. The MAN1 inner nuclear membrane protein encoded by LEMD3 modulates TGFβ/bone morphogenetic protein (BMP) superfamily signalling through negative regulation of SMAD transcription factors downstream of the receptor activation that is crucial for activating genes involved in bone formation (Fig. 7). The TGFβ superfamily of multifunctional cytokines plays wide-ranging roles in embryonic development and adult tissue homeostasis. Members of the family include BMPs and Activin as well as TGFβ itself. Signals from these proteins are transduced into cells through heteromeric serine/threonine kinase receptor complexes (including TGFβ Types 1 and 2 receptors) and their corresponding SMAD intermediaries, which function as transcription factors after translocation to the cell nucleus [39]. The activated TGFβ receptor phosphorylates the conserved C-terminal serine-X-serine motif of receptor-regulated SMAD (R-SMAD) proteins (SMAD2 & 3 in the case of TGFβ or Activin receptor signalling or SMAD1, 5 and 8 in the case of BMP signalling). Two molecules of the relevant R-SMADs form a heterotrimeric complex with one common mediator SMAD (SMAD4) before translocation to the cell nucleus where they regulate the expression of many genes. The R-SMAD N-terminal MH1 (MAD homology 1) domain is involved in DNA binding (in keeping with the transcription factor function of these proteins), while the C-terminal MH2 domain is involved in protein–protein interactions (of which between 62 and 347 have been described for each individual R-SMAD). Among the scores of proteins binding to R-SMADs at least some influence their biological activity. Thus, MAN1 (see above) binds to the MH2 domain of R-SMADs facilitating their dephosphorylation and thereby downregulating SMAD-mediated signalling (such as through the TGFβ receptor family). This explains the deleterious effects of loss-of-function LEMD3 mutations resulting in excessive signalling to genes involved in bone morphogenesis and soft tissue modelling/fibrosis. In the former, the abnormalities are restricted to bone (osteopoikilosis), whereas in the latter (BOS) there are additional connective tissue naevi related to abnormalities of elastin (juvenile elastomas) or collagen (dermatofibrosis lenticularis disseminata). The excessive amounts of collagen and elastin produced by affected skin fibroblasts are consistent with excessive TGFβ signalling in BOS [40]. However, in affected families the phenotype is variable with some individuals expressing only bone lesions and some only skin disease [41], while others have more extensive soft tissue involvement [42]. There also appears to be at least some molecular heterogeneity in the pathology of BOS since cases have been described without LEMD3 mutations [43]. It remains to be seen whether other R-SMAD - protein interactions might have similar functional effects on the bone or soft tissues. Germline LEMD3 mutations in the absence of osteopoikilosis or BOS do not occur in sporadic melorheostosis [38, 44, 45].

Cartoon demonstrating some of the molecular pathways incriminated in melorheostosis, the Buschke–Ollendorff syndrome and the RASopathies (see text). Germline mutations in MAN1 (LEMD3) cause osteopoikilosis/BOS. Somatic mutations in MEK1 (MAP2K1) and KRAS have been implicated in melorheostosis. Germline mutations in PTPN11 & KRAS have been incriminated in Noonan syndrome, neurofibromin in neurofibromatosis type 2/CFC overlap syndrome, HRAS in Costello syndrome, BRAF in CFC1, KRAS in CFC2, MAP2K1 in CFC3, and MAP2K2 in CFC4. Abbreviations: BMPR (bone morphogenetic protein/TGFβ receptor), PTPN11 (Protein tyrosine phosphatase 11), for others see text
Much of the focus on the molecular pathogenesis of melorheostosis in recent years has been on its rare coexistence with osteopoikilosis/BOS. It has been widely postulated that, in those cases of osteopoikilosis with coexistent melorheostosis, a second hit in LEMD3, causing a second loss-of-function mutation in the other allele or deleting it altogether, might account for the more severe segmental disease [27, 28]. However, there is no direct evidence either for this or the existence of de novo LEMD3 mutations in lesional tissue from sporadic cases of isolated melorheostosis. Instead, the majority of cases of sporadic melorheostosis are caused by cell type-specific post-zygotic mutations in the MAP2K1 gene, encoding the protein kinase MEK1 [45]. Mutations in this gene have also been implicated in many types of cancer, including lung, melanoma and hairy cell leukaemia [46,47,48]. Whole exome sequencing of lesional tissue from 15 cases (none of whom had evidence of additional osteopoikilosis or BOS) compared with unaffected bone, revealed eight individuals with tightly clustered MAP2K1 mutations in a negative regulatory domain in MEK1 (Q56P, K57E, K57N). The extent and distribution of the condition reflect the degree of somatic chimerism for these mutations. Further, it has been suggested in a follow-up study that cases with MAP2K1 mutations are more likely to have the classic “dripping candle wax” phenotype or characteristic skin lesions overlying the affected bone than those with no evidence of such mutations [49]. MEK1 is a component of the RAS signalling pathway that plays a role in skeletal development. The effect of these loss-of-function MEK1 mutations is to increase pERK1/2 signalling, which results in increased osteoblast proliferation but reduces their differentiation; this also leads to reduced production of RUNX2 (the key osteoblast transcription factor), type 1 collagen, alkaline phosphatase and reduced BMP2-mediated mineralisation [45]. There is also a significant increase in the RANKL/OPG ratio, consistent with the observed overactive osteoclastogenesis. These functional effects are also consistent with the histological findings in melorheostosis where there is a marked increased unmineralised osteoid, an increase in osteoblast and osteoclast numbers and markedly increased bone remodelling. In passing, it is worth noting that mutations in RUNX2, which plays such a key role in osteoblast biology, usually have a rather less dramatic phenotype than might be expected and also one that, like LEMD3 is variably expressed within and between families [50,51,52,53].
Of interest, in one of the cases described by Kang et al. where no MAP2K1 mutation was evident there was a K61R mutation in the KRAS oncogene, incriminated in a wide variety of cancers [45]. This mutation was present in both abnormal and normal bone, and there were prominent skin lesions consistent with a “RASopathy”, leading the authors to suggest that this case may represent a complex early post-zygotic mosaicism, of which melorheostosis was part. Germline gain-of-function mutations in the RAS-MAPK-ERK pathway affect cell differentiation, proliferation and apoptosis and are the unifying biochemical feature of the RASopathies, an uncommon set of genetic disorders, in which the variable clinical features include congenital heart defects, facial dysmorphism, skin abnormalities and learning difficulties among others [54, 55]. As shown in Fig. 7, these disorders include different forms of Noonan syndrome (OMIM163950) variously associated with the protein tyrosine phosphatase PTPN11, KRAS or neurofibromin (in a Noonan variant associated with type 2 neurofibromatosis – OMIM601321); cardiofaciocutaneous (CFC) syndromes 1–4 (OMIM115150, 615278, 615280) associated with BRAF, KRAS, MAP2K1 or MAP2K2 mutations; and Costello syndrome (OMIM218040) associated with the HRAS oncogene. A similar KRAS mutation (Q61H) to the above case and also to those in the germline of some RASopathies has recently been described in lesional tissue (scleroderma-like) from the skin of one patient with features of melorheostosis, Buschke–Ollendorf syndrome and osteopoikilosis [56]. In this case, an LEMD3 mutation was also present in the proband and other affected family members; the KRAS mutation was restricted to the more severely affected skin and not seen in unaffected skin (the bone was not biopsied in this case). The authors suggest that this KRAS mutation might function synergistically with the underlying LEMD3 mutation to produce areas of more severe skin involvement and melorheostosis in a segmental fashion. Mosaicism for the same MAP2K1 mutations (Q56P and K57E) that have been described in melorheostosis osteoblasts [45] has also been described in cases of extracranial arteriovenous malformations where their distribution is restricted to endothelial cells [57].
Management
It is not precisely known what proportion of individuals with melorheostosis actually require medical treatment since some cases may go undetected if they are asymptomatic. Those cases cited in the literature are, of course, biased toward those who are symptomatic. Published case series are few, and the rarity of the condition also means that evidence-based guidelines for the management of these patients are difficult to produce. Consequently, many of the recommendations for treatment are based on very limited data. About half of the Mayo Clinic series had undergone some form of surgery [12], which is similar to our experience. However, as previous reviewers have highlighted the wide variety of presentations of this condition makes it difficult to draw many definitive conclusions about how it should be managed surgically or medically [58]. A multidisciplinary approach to the assessment of patients is logical; in addition to potential surgical options, it should often include non-operative measures, such as specialist imaging, pharmacologic approaches, physical therapy and psychological assessments. It is important not to attribute all the patient’s symptoms hastily to melorheostosis; people with rare diseases also suffer from common coexistent conditions. The precise origin of any pain should be ascertained before proceeding with treatment; people with melorheostosis are just as likely to suffer from shoulder capsulitis, tennis elbow or cervical spondylosis! In surgical decision making, the potential for recurrent disease and also the degree of involvement of neighbouring soft tissues are important considerations [59].
Pharmacological treatments of the condition mainly focus on the relief of pain, which may be multifactorial in origin. A clear definition of the origin of the pain is essential if treatment success is to be achieved; is it related to pressure effects, nerve compression, joint dysfunction, limb deformity or hyperactive bone itself? As with many causes of chronic pain psychological factors may also become important and input from a specialist pain clinic may be valuable. Bisphosphonates have proved effective in the management of pain in some individuals with melorheostosis, but there are no controlled trials of their efficacy. Given the hyperactive state of the involved bone with markedly increased remodelling and increased numbers both of active osteoblasts and osteoclasts a trial of bisphosphonates may be appropriate in some cases. Their benefits are recorded in a series of case reports but, as such, are subject to the problems of positive ascertainment bias. Nevertheless, there is sufficient evidence to justify their use where other causes of pain have been excluded first. In one case, zoledronate not only improved pain but also reduced bone turnover as judged by scintigraphy; these authors also nicely review the evidence for positive results from etidronate and pamidronate [60]. In a more recent case, the efficacy of bisphosphonates is reviewed and the successful use of denosumab after failure of zoledronate is reported [61]. However, there are potential pitfalls to the excessive use of bisphosphonates in this condition. One case known to us (with pain from melorheostosis affecting the 5th metatarsal, cuboid and calcaneus) was given regular infusions of zoledronate over a period of more than 1 year resulting in severe atypical fractures of multiple ribs bilaterally and a flail chest. Unfortunately, the zoledronate appeared to have little impact on the local ankle pain, which was almost certainly mechanical in nature as a result of lateral ankle joint impingement from the enlarged affected bones.
In many cases, pain may arise from direct pressure on the abnormal bone or arise from joint impingement effects (Fig. 2). Concerns about possible recurrent bone growth after surgery, perhaps even more severe than the primary tumour, are natural. In our experience removal of such growths is justified where the symptoms are severe and there appears to be a relatively low risk of recurrence (Figs. 4, 8). Nerve entrapment syndromes are well described, including carpal tunnel syndrome and cervical radiculopathy [62, 63]. These should be investigated and treated as appropriate but do not appear to be common. The prevalence of spinal involvement appears to be relatively low, but we have seen asymptomatic disease in several of our patients, suggesting that it may be more common than reported. At the age of 60 years, one of our cases had symptoms and signs of cervical radiculopathy from encroachment of the enlarged lamina on the emerging right C6 nerve root. This was managed conservatively and did not appear to have had any major impact on his sporting prowess as a younger man when he had a successful amateur rugby football career as a prop forward. The patchy nature of the lesions in the spine may require CT for clearer definition (Fig. 9).

In this example (a) from a 45-year-old male there is segmental involvement approximately corresponding to C5/6 sclerotomes in the humerus, radius, scaphoid and trapezium. b There are two areas of ectopic ossification (arrowed) adjacent to the elbow which were associated with “tennis elbow”-like symptoms. Surgical excision of the lesions was successful

Spinal melorheostosis is often patchy and best demonstrated by computed tomography. In this example, there is limited involvement of the posterior elements of the lumbar and 1st sacral vertebrae
Where there is significant limb deformity major soft tissue dissection may be required in addition to correcting any bone lesions. Even with the most intensive and diligent postoperative care success cannot be guaranteed in the short-, medium- or long-term. In our experience, limb reconstruction techniques can partially correct even major degrees of joint contracture at the knee and help to restore limb length inequality and function (Fig. 5). However, where there is significant soft tissue involvement the restoration of a normal range of movement is relatively unlikely and the risk of recurrence is high due to recurrent soft tissue contractures. In a case report of the treatment of recurrent intra-articular melorheostosis affecting the knee, the authors include a most informative review of the surgical literature and also indications for medical treatment. They highlight the risk of recurrent disease following excision and the possibilities for joint replacement surgery in the event of repeat recurrences or significant joint contractures [59]. Nevertheless, even where there is severe widespread disease pain can be alleviated and function restored in some cases by multiple joint replacement [64]. In contrast, there will still be cases even today where the symptoms, severe loss of function, technical difficulty of surgery and relatively low chance of a successful outcome ultimately justify amputation [11]. For the more complex cases, referral to tertiary centres with experience of managing the condition is highly desirable.
Future Directions
Because it is such a rare disease the data relating to medical and surgical treatment of melorheostosis are inevitably limited and subject to reporting bias. The establishment of a formal register of the efficacy of medical and surgical treatments used for individuals with the condition would be useful in redressing this imbalance. For example, in the UK, the RUDY electronic patient platform could serve such a function [65] and has previously been used for generating similar data relating to osteogenesis imperfecta, X-linked hypophosphataemia and fibrous dysplasia [66]. This could be particularly helpful in collecting individual responses to surgical treatments that are often unique.
Now that it is known that most sporadic cases of melorheostosis are caused by MAP2K1 mutations interest has focused on whether the RAS/ MAPK/ERK pathway can be targeted for treatment of this disorder. There is considerable enthusiasm for this as a result of the success of inhibiting components of this pathway in various cancers [67] and potentially in a range of rare genetic RAS/MAPK developmental disorders [55]. The hydrophobic pocket adjacent to the ATP-binding site of MEK1 and 2 allows the binding of potent allosteric inhibitors. For example, the MEK1 inhibitor trametinib is used to treat BRAF mutant melanoma in combination with first-generation BRAF inhibitors and a range of MEK1 inhibitors are now in development. Pre-clinical studies of MEK1/2 inhibitors for the treatment of type 2 neurofibromatosis (associated with neurofibromin mutations—see Fig. 7) are promising [68]. However, the abnormally active mutation is cell type specific but any therapy targeting the RAS/MAPK/ERK pathway will not be restricted to the mutant cells alone and there is a substantial risk of off-target side-effects. Whether, such drugs can be developed for the treatment of melorheostosis in future is never the less an intriguing prospect.
Finally, there appears to be at least some genetic heterogeneity in melorheostosis since a significant minority of cases appear to lack MAP2K1 mutations in lesional tissue [45]. Further investigation of these cases may uncover alternative pathogenic mechanisms and further insights into the pathology of this and other bone diseases.
References
Leri A, Joanny J (1922) Une affection non décrite des os hyperostose “en coulée” sur toute la longeur d’un member ou “melorhéostose”. Bull Mem Soc Med Hosp Paris 46:1141–1145
Boulet C, Madani H, Lenchik L, Vanhoenacker F, Amalnath DS, de Mey J, De Maeseneer M (2016) Sclerosing bone dysplasias: genetic, clinical and radiology update of hereditary and non-hereditary disorders. Br J Radiol 89(1062):20150349
Abrahamson MN (1968) Disseminated asymptomatic osteosclerosis with features resembling melorheostosis, osteopoikilosis and osteopathia striata. J Bone Joint Surg (Am) 50:991–996
Whyte MP, Murphy WA, Fallon MD, Hahn TJ (1981) Mixed sclerosing bone dystrophy. Skeletal Radiol 6:95–102
Ghai S, Sharma R, Ghai S (2003) Mixed sclerosing bone dysplasia: a case report with literature review. Clin Imaging 27:203–205
Green AE, Ellswood WH, Collins JR (1962) Melorheostosis and osteopoikilosis with a review of the literature. Am J Roentgenol 87:1096–1111
Pacifici R, Murphy WA, Teitelbaum SL, Whyte MP (1986) Mixed sclerosing bone dystrophy; 42 year follow up of a case reported as osteopetrosis. Calc Tissue Int 38:175–185
Smith and Wordsworth (2016) Clinical and biochemical disorders of the skeleton. Oxford University Press, Oxford
Freyschmidt J (2001) Melorheostosis: a review of 23 cases. Eur Radiol 11(3):474–479
Lachman RS (1996) Skeletal dysplasias. In: Taybi H & Lachman RS (eds) Radiology of syndromes, metabolic disorders and skeletal dysplasias. Mosby, St Louis 745–751
Nevin NC, Thomas PS, Davis RI, Cowie GH. Melorheostosis in a family with autosomal dominant osteopoikilosis. Am J Med Genet 199(82): 409–14
Smith GC, Pingree MJ, Freeman LA, Matsumoto JM, Howe BM, Kannas SN, Pyfferoen MD, Struss LT, Wenger DE, Amrami KK, Matsumoto M, Jurisson ML (2017) Melorheostosis: a retrospective clinical analysis of 24 patients at the mayo clinic. PM&R 9: 283–288
Bostman OM, Holmstrom T, Riska EB (1987) Osteosarcoma arising in a melorheostosis femur: a case report. J Bone Joint Surg Am 69:232–237
Gaudio A, Zanoli L, Fiore CE (2017) Paget’s disease is not always the culprit. Eur J Int Med 43:e3. https://doi.org/10.1016/j.ejim.2017.02.001. Epub 2017 Feb 6
Kinoshita A, Saito T, Tomashita H, Makita Y, Yoshida K, Ghadami M, Yamada K et al (2000) Domain-specific mutations in TGFB1 result in Camurati–Engelmann disease. Nat Genet 26:19–20
Nishimura G, Nishimura M, Tanaka Y et al (2002) Camurati–Englemann disease type II: progressive diaphyseal dysplasia with striations of the bones. Am J Med Genet 107:5–11
Savoie A, Gouin F, Maugars Y, Isidor B, Larrose C, Berthelot JM (2013) Treatment responses in five patients with Ribbing disease including two with 466 C> T missense mutations in TGFβ. Joint Bone Spine 80:638–644
Beals RK, Pearson JM, Mansoor A (2002) Ribbing disease: a case report, a review of the literature and a description of novel treatment. J Bone Joint Surg 84A:2050–2055
Gumruck F, Besin A, Altay L (1993) Ghosal haemo-diaphyseal dysplasia: a new disorder. Eur J Paediatr 152:218–221
Arora R, Aggarwal S, Deme S (2015) Ghosal haematodiaphyseal dysplasia: a concise review including an illustrative patient. Skelet Radiol 44:447–450
Walker GF (1964) Mixed sclerosing bone dystrophies. J Bone Joint Surg (Br) 46:546–552
Kanis JA, Thomson JG (1974) Mixed sclerosing bone dystrophy with regression of melorheostosis. Br J Radiol 48:400–402
Cantatone FP, Carrozzo M, Loperfido MC (1991) Mixed sclerosing bone dystrophy with regression of melorheostosis. Clin Rheumatol 10:191–195
Ostrowski DM, Gilula LA (1992) Mixed sclerosing bone dystrophy presenting with upper extremity deformitie. A case report and review of the literature. J Hand Surg Br 17:108–112
Long H-T, Li K-H, Zhu Y (2009) Case report: severe melorheostosis involving the ipsilateral extremities. Clin Orthop Relat Res 467:2738–2743
Muray RO, McCredie J (1979) Melorheostosis and the sclerotomes: a radiological correlation. Skelet Radiol 4:57–71
Fryns JP (1995) Melorheostsosis and somatic mosaicism. Am J Med Genet 58:199
Happle R (2004) Melorheostosis may originate as a type 2 segmental manifestation of osteopoikilosis. Am J Med Genet A 125A:221–223
Jha S, Laucis N, Kim L, Malayeri A, Dasgupta A, Papadakis GZ, Karantaras A, Torres M, Bhattacharyya T (2018) CT analysis of anatomical distribution of melorheostosis challenges the sclerotome hypothesis. Bone 117:31–36
Fratzl-Zelman N, Roschger P, Kang H, Jha S, Roschger A, Blouin S, Deng Z et al. Melorheostotic bone lesions caused by somatic mutations in MAP2K1 have deteriorated microarchitecture and periosteal reaction. J Bone Miner Res 2019 Jan 22 https://doi.org/10.1002/jbmr.3656. (E.pub ahead of print)
Butkus CE, Michels VV, Lindor NM, Cooney WP (1998) Melorheostosis in a family with osteopoikilosis. Am J Med Genet 72:43–46
Debeer P, Pykels E, Lammens J, Devriendt K, Fryns JP (2003) Melorheostosis in a family with autosomal dominant osteopoikilosis: report of a third family. Am J Med Genet A 119A:188–193
Ewald FC (1972) Unilateral mixed sclerosing bone dystrophy associated with unilateral lymphangiectasis and capillary haemangioma. J Bone Joint Surg (Am) 54:878–880
Gunal I, Taymaz A, Karatosun V, Toylu A, Degirmenci B (2005) Mixed sclerosing dysplasia of the bone associated with ovarian and skin problems. Clin Orthop Relat Res 436:270–276
Murphy-Ryan M, Kirmani S, Thompson DM, Binkovitz LA, Thomas KB, Babrovic-Vuksanovic D (2012) A novel sclerosing skeletal dysplasia with mixed sclerosing bone dyplasia, characteristic syndromic features, and clinical radiographic evidence of male-male transmission. Am J Med Genet A 158A:2292–2296
Hellemans J, Preobraxhenska O, Willaert A, Debeer P, Verdonk PCM, Costa T, Janssens K, Merten B, Van Roy N, Vermeulen SJT, Savarirayan R, Van Hull W et al (2004) Loss of function mutations in LEMD3 result in osteopoikilosis, Buschke–Ollendorff syndrome and melorheostosis. Nat Genet 36:1213–1218
Couto AR, Bruges-Armas J, Peach CA, Chapman K, Brown MA, Wordsworth BP, Zhang Y (2007) A novel LEMD3 mutation common to patients with osteopoikilosis with and without melorheostosis. Calc Tissue Int 81:81–84
Zhang Y, Castori M, Ferranti G, Paradisi M, Wordsworth BP (2009) Novel and recurrent germline LEMD3 mutations causing Buschke–Ollendorff syndrome and osteopoikilosis but not isolated melorheostosis. Clin Genet 75:556–561
Miyazono K, Ohno Y, Wada H, Ito T, Fukatsu Y, Kuriski A, Asashima M, Tanokura M (2018) Structural basis for receptor-regulated SMAD recognition by MAN1. Nucleic Acids Res 46:12139–12153
Giro MG, Duvic M, Smith LT, Kennedy R, Rapini R, Arnett FC, Davidson JM (1992) Buschke–Ollendorff syndrome associated with elevated elastin production by affected skin fibrobasts in culture. J Invest derm 99:129–137
Woodrow SL, Pope FM, Handfield-Jones SE (2001) The Buschke–Ollendorff syndrome presenting as familial elastic tissue naevi. Br J Derm 144:890–893
Berlin R, Hedensio B, Lilja B, Linder L (1967) Osteopoikilosis: a clinical and genetic study. Acta Med Scand 181:305–314
Yadegari M, Whyte MP, Mumm S, Phelps RG, Shaneke A, Totty WG, Cohen SR (2010) Buschke–Ollendorf syndrome: absence of LEMD3 mutation in an affected family. Arch Dermatol 146:63–68
Hellemans J, Debeer P, Wright M, Janecke A, Kjaer KW, Savarirayan R, Basel L, Moss C, Roth J, David A, De Paepe A, Coucke P, Mortier GR (2006) Germline LEMD3 mutations are rare in sporadic patients with melorheostosis. Hum Mut 27:290
Kang H, Jha S, Deng Z, Fratzl-Zelman N, Cabral WA, Ivovic A, Meylan F, Hanson EP, Lange E, Katz J, Roschger P, Klaushofer K, Cowen EW, Siegel RM, Marini JC, Bhattacharyya T (2018) Somatic activating mutations in MAP2K1 cause melorheostsosis. Nat Commun 9:1390
Marks JL, Gong Y, Chitale D, Golas B, McLellan MD, Kasai Y, Ding L, Mardis ER, Wilson RK, Solit D, Levine R, Michel K, Thomas RK, Rusch VW, Ladanyi M, Pao W (2008) Novel MEK1 mutation identified by mutational analysis of epidermal growth factor receptor signaling pathway genes in lung adenocarcinoma. Cancer Res 15(14):5524–5528. https://doi.org/10.1158/0008-5472.CAN-08-0099
Murugan AK, Dong J, Xie J, Xing M (2009) MEK1 mutations, but not ERK2 mutations, occur in melanomas and colon carcinomas, but none in thyroid carcinomas. Cell Cycle 8:2122–2124. https://doi.org/10.4161/cc.8.13.8710
Waterfall JJ, Arons E, Walker RL, Pineda M, Roth L, Killian JK, Abaan OD, Davis SR, Kreitman RJ, Meltzer PS. (2014) High prevalence of MAP2K1 mutations in variant and IGHV4-34-expressing hairy-cell leukemias. Nat Genet 46(1):8–10. https://doi.org/10.1038/ng.2828. (Epub 2013 Nov 17)
Jha S, Fratzl-Zelman N, Roschger P, Papadakis GZ, Cowen EW, Kang H, Lehky TJ, Alter K, Deng Z, Ivovic A, Flynn L, Reynolds JC, Dasgupta A, Miettinen M, Lange E, Katz J, Klaushofer K, Marini JC, Siegel RM, Bhattacharyya T (2019) Distinct clinical and pathological features of melorheostosis associated with somatic MAP2K1 mutations. J Bone Miner Res 34:145–156
Otto F, Kaegane H, Mundlos S (2002) Mutations in the RUNX2 gene in patients with cleidocranial dysplasia. Hum Mutat 19:209–216
Mundlos S (1999) Cleidocranial dysplasia: clinical and molecular genetics. J Med Genet 36:177–182
Jaruga A, Hordyjewska E, Kandzierski G, Tylzanowski P (2016) Cleidocranial dysplasia and RUNX2-clinical phenotype-genotype correlations. Clin Genet 90:393–402
Chitayat D, Hodgkinson KA, Azouz EM (1992) Intrafamilial variability in cleidocranial dysplasia: a three generation family. Am J Med Genet 42:298–303
Roberts A, Allanson J, Jadico SK, Kavamura MI, Noonan J, Opitz JM, Young T, Neri G (2006) The cardiofaciocutaneous syndrome. J Med Genet 43:833–842
Stevenson DA, Schill L, Schoyer L, Andresen BS, Bakker A, Bayrak-Yoydemir P, Burkitt-Wright E et al. (2016) The fourth international symposium on genetic disorders of the Ras/MAPK pathway. Am J Med Genet A 170: 1959–1966
Whyte MP, Griffith M, Trani L, Mumm S, Gottesman GS, McAllister WH, Krysiak K et al (2017) Melorheostosis: exome sequencing of an associated dermatosis implicates postzygotic mosaicism of mutated KRAS. Bone 101:145–155
Couto JA, Huang AV, Konczyk DJ, Goss JA, Fishman SJ, Mulliken JB, Warman ML, Greene AK (2017) Somatic MAP2K1 mutations are associated with extracranial arteriovenous malformations. Am J Hum Genet 100:546–554
Kotwal A, Clarke B (2017) Melorheostsosis: a rare sclerosing bone dysplasia. Curr Osteoporos Rep 15:335–342
John B, Sharma A, Pandey RA (2017) Managing recurrence in intraarticular melorheostosis involving the knee joint: a case report. J Orthop Case Rep 7:29–33
Slimani S, Nezzar A, Makhloufi H (2013) Successful treatment of pain in melorheostosis with zoledronate with improvements in scintigraphy. BMJ Case Rep 2013pii:Bcr2013009820
Byberg S, Abrahamsen B, Kassem M, Ralston S, Schwartz P (2018) Clinical improvement in a patient with monostotic melorheostosis after treatment with denosumab: a case report. J Med Case Rep 12:278
Abdullah S, Mat Nor NF, Mohamed NH (2014) Melorheostosis of the hand affecting the C6 sclerotome presenting with carpal tunnel syndrome. Singap Med J 55:e54–e56
Auethavekiat P, Murali NS, Manek NJ (2004) Clinical images: melting candle wax, an uncommon presentation of an uncommon disease. Arthritis Rheum 50:4080
Shin SJ, Nam U, Kim SR, Kim HJ, Dimitriou D, Li G et al. (2015) Vascular malformations corresponding to sclerotomes in multifocal melorheostosis: painful hip and knee contractures treated with total joint arthroplasty. JBJS Case Connect 5: e40
Javaid MK, Forestier-Zhang L, Watts L, Turner A, Ponte C, Teare H, Gray D, Gray N, Popert R, Hogg J, Barrett J, Pinedo-Villanueva R, Cooper C, Eastell R, Bishop N, Luqmani R, Wordsworth P, Kaye J (2016) The RUDY study platform: a novel approach to patient driven research in rare musculoskeletal diseases. Orphanet J Rare Dis 11:150
Forestier-Zhang L, Watts L, Turner A, Teare H, Kaye J, Barrett J, Cooper C, Eastell R, Wordsworth P, Javaid MK, Pinedo-Villanueva R (2016) Health-related quality of life and a cost-utility simulation of adults in the UK with osteogenesis imperfecta, X-linked hypophosphatemia and fibrous dysplasia. Orphanet J Rare Dis 28(11): 160
Caunt CJ, Sale MJ, Smith PD, Cook SJ (2015) MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nat Rev Cancer 15(10):577–92. https://doi.org/10.1038/nrc4000
Fuse MA, Dinh CT, Vitte J, Kirkpatrick J, Mindos T, Plati SK, Young JI, Huang J, Carlstedt A, Franco MC, Brnjos K, Nagamoto J, Petrilli A, Copik AJ, Soulakova JN, Bracho O, Yan D, Mittal R, Shen R, Telischi FF, Morrison H, Giovannini M, Liu XZ, Chang LS, Fernandez-Valle C (2019) Preclinical assessment of MEK1/2 inhibitors for neurofibromatosis type 2-associated Schwannomas reveal differences in efficacy and drug resistance development. Neuro Oncol 21(4):486–497
Acknowledgements
The authors appreciate the support of the NIHR Oxford Comprehensive Biomedical Research Centre and NIHR Oxford Musculoskeletal Biomedical Research Unit.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Paul Wordsworth and Marian Chan declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
Patients were recruited following informed consent to the Rare UK Disease Study (RUDY - ratified by South Central Research Ethics Committee, LREC14/SC/0126).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wordsworth, P., Chan, M. Melorheostosis and Osteopoikilosis: A Review of Clinical Features and Pathogenesis. Calcif Tissue Int 104, 530–543 (2019). https://doi.org/10.1007/s00223-019-00543-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-019-00543-y