
Abstract
Cbl is an adaptor protein and E3 ligase that plays both positive and negative roles in several signaling pathways that affect various cellular functions. Tyrosine 737 is unique to Cbl and phosphorylated by Src family kinases. Phosphorylated CblY737 creates a binding site for the p85 regulatory subunit of phosphatidylinositol 3 kinase (PI3K) that also plays an important role in the regulation of bone homeostasis. To investigate the role of Cbl–PI3K interaction in bone homeostasis, we examined knock-in mice in which the PI3K binding site on Cbl was ablated due to the substitution of tyrosine 737 to phenylalanine (CblYF/YF, YF mice). We previously reported that bone volume in these mice is increased due to decreased osteoclast function (Adapala et al., J Biol Chem 285:36745–36758, 19). Here, we report that YF mice also have increased bone formation and osteoblast numbers. In ex vivo cultures bone marrow-derived YF osteoblasts showed increased Col1A expression and their proliferation was also significantly augmented. Moreover, proliferation of MC3T3-E1 cells was increased after treatment with conditioned medium generated by culturing YF bone marrow stromal cells. Expression of stromal derived factor-1 (SDF-1) was increased in YF bone marrow stromal cells compared to wild type. Increased immunostaining of SDF-1 and CXCR4 was observed in YF bone marrow stromal cells compared to wild type. Treatment of YF condition medium with neutralizing anti-SDF-1 and anti-CXCR4 antibodies attenuated MC3T3-E1 cell proliferation. Cumulatively, these results show that abrogation of Cbl–PI3K interaction perturbs bone homeostasis, affecting both osteoclast function and osteoblast proliferation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Bone is a dynamic organ that provides support and protection to vital organs. Bone also plays a role in the regulation of calcium homeostasis. The effects of two cell types, osteoblasts and osteoclasts, are critical for maintaining proper bone mass and shape. Osteoclasts are a member of the monocyte/macrophage family, while osteoblasts are of mesenchymal origin and responsible for mineralizing bone matrix [1].
Cbl and Cbl-b are highly homologous members of a family of adaptor proteins that promote assembly of signaling complexes downstream of several receptors [2]. Both function as E3 ubiquitin ligases that regulate ubiquitylation of proteins. Cbl proteins are known to participate in growth factor, cytokine, and integrin-mediated signaling pathways in various cell types. Tyrosine phosphorylation of Cbl upon receptor activation is one of the most common signaling phenomena shown to promote both its E3 ubiquitin ligase activity and adaptor function [2].
Despite the marked effect of mutating Cbl [3, 4] or reducing its expression in vitro to affect osteoclast activity [5], Cbl−/− mice have a mild bone phenotype manifested primarily during endochondral bone formation [6]. In contrast, absence of Cbl-b protein in mice results in osteopenia due to osteoclast hyperactivity [7]. In spite of significant homology between Cbl and Cbl-b proteins, single knockouts of Cbl and Cbl-b have distinct bone phenotypes, suggesting specific roles for each protein in bone homeostasis. However, Cbl/Cbl-b double knockout mice die in utero around E10.5 [8], suggesting that Cbl and Cbl-b also have overlapping functions. The major structural difference that could contribute to the unique function(s) of Cbl proteins includes a tyrosine present only in Cbl (CblY737), which when phosphorylated binds to the SH2 domain of the p85 subunit of phosphatidylinositol 3 kinase (PI3K) [9–11]. Other investigators have shown that Tyr737 is among several Tyr residues that are phosphorylated by Src family kinases [12] and that when phosphorylated binds to the SH2 domain of the regulatory p85 subunit of PI3K [10, 11]. The absence of this residue in Cbl-b highlights a potential divergence in the role and mode of action of these two highly similar regulatory proteins, although this has not been examined with respect to osteoclast or osteoblast biology.
PI3Ks are a class of enzymes that phosphorylate phosphatidylinositol and its derivatives and regulate cell growth, the cytoskeleton, and cell survival [13]. PI3K activation in various cell types, including osteoblasts, is often associated with increased tyrosine phosphorylation induced by growth and differentiation factors. The importance of PI3K in osteoblast function has been established [14, 15]. In several cell types, Cbl is known to be a major protein that interacts with PI3K and regulates its activity [11, 16]. Downregulation of Cbl induced by twist haploinsufficiency in Saethre-Chotzen syndrome results in increased PI3K/AKT signaling and osteoblast proliferation [17]. In humans, osteoblasts expressing mutated FGFR2 levels of PI3K are decreased. The decreased PI3K signaling in response to FGFR2 activation is due to increased Cbl–PI3K molecular interaction mediated by the Cbl Y731 residue, which results in increased PI3K ubiquitylation and proteasome degradation [18]. We recently reported that, on the one hand, the Cbl–PI3K complex negatively regulates osteoclast differentiation and survival while, on the other hand, it positively influences osteoclast function [19].
In this report, we continued to study the role of Cbl–PI3K interaction in the skeletal system. Our results indicated that abrogation of the Cbl–PI3K interaction resulted in increased bone mass and increased trabecular and cortical bone volumes. The altered geometry of bone due to increased volume also resulted in increased mechanical properties. While osteoblast numbers were increased in mice, ex vivo differentiation and survival of the YF osteoblast was not significantly affected. Analysis of the conditioned medium derived from culturing YF bone marrow stromal cells, as well as the expression analysis of cells, showed increased expression of stromal cell derived factor-1 (SDF-1) and its receptor, CXCR4. Antibody-mediated ablation of SDF-1 activity attenuated cell proliferation. Taken together, these data and our previously published work suggest that abrogation of Cbl–PI3K interaction perturbs bone homeostasis, affecting both osteoclast function and osteoblast proliferation.
Materials and Methods
Mice Colony
The generation of CblY737F mice has been previously described [20]. Mice were maintained in a mixed C57BL/6 × 129SvJ background, and experiments were performed in compliance with the Institutional Animal Care and Use Committee at Temple University.
Histological Analysis
To label the skeleton, calcein (i.p., 10 mg/kg body weight) was injected 9 and 2 days before death. After death, tibiae and femora of 12-week-old male mice were excised and cleaned of soft tissue. Bones were processed as previously described [21]. Longitudinal sections (5 μM) were used to measure the fluorochrome label and then deplasticized and stained with Masson trichrome, as described previously [21]. Histomorphometry was performed on three tissue sections per limb using a Nikon 800 fluorescence microscope linked to a Bioquant Osteo II system (R&M Biometrics, Nashville, TN). Dynamic parameters of bone formation were collected using unstained sections from the trabecular surface in a defined region of interest (100 μM below the growth plate and 50 μM in from the cortical bone) using a ×20 objective. Bone formation rate (BFR) was assessed by measuring single-labeled surface for single perimeter (sL.Pm), double-labeled surface (dL.Pm), and the interlabel distance in the dL.Pms. The same sections were then evaluated under polarized white light after Masson’s trichrome staining to determine static parameters of bone formation: bone surface (BS), osteoid volume (OV), and osteoblast number (Ob.N). The following were then calculated: mineralizing surface (MS/BS, %), mineral apposition rate (MAR, mM/day), BFR (BFR/BS, MAR × [MS/BS]), OV (OV/BS), and (Ob.N/BS), as described previously [22].
Immunohistochemistry
Alternate sections of the above bones were deplasticized, rehydrated, and then stained for SDF-1 and CXCR4 using previously described methods for detecting tissue cytokines [23]. Specific antibodies for SDF-1 (rabbit SDF-1α, 1:250 dilution in PBS; Abcam, Cambridge, MA) and CXCR4 (rabbit CXCR4, 1:250 dilution in PBS; Abcam) were used. Primary antibodies were detected using anti-rabbit secondary antibodies tagged with Cy2 (green), Cy3 (red), or HRB (brown after enzymatic reaction with DAB). Sections were counterstained with DAPI or hematoxylin.
Micro-CT Analysis
High-resolution images of long bones from age-matched male mice were acquired using a Skyscan 1172, 12 MPix model (Microphotonics, Allentown, PA). Proximal tibiae and distal femora were scanned with the source voltage 59 kV, a source current of 167, and an isotropic voxel size of 6 μm. Imaging started at the distal end of the femur or the proximal end of the tibiae and included approximately 7 mm (1,335 slices) of the total bone length. After scanning, 3D image data were reconstructed using Sykscan (Aartselaar, Belgium) NRecon software; structural indices were calculated using the Skyscan CT Analyzer (CTAn) software. Using the CTAn software, trabecular bone was separated from cortical bone with a region of interest tool. Trabecular morphometric traits were computed from binarized images using direct 3D techniques that do not rely on prior assumptions from the underlying structures. The volume of interest for trabecular microarchitectural variables was bounded to the endocortical margin, starting 1.9 mm from the proximal tibial condyles, 0.2 mm distal to the growth plate, in the direction of the metaphysis, and then extending from this position for 100 slices (0.6 mm). Upper and lower thresholds of 255 and 80 were used to delineate each pixel as “bone” or “nonbone,” and trabecular bone volume per total volume (BV/TV), BS (BS/TV), mean trabecular thickness (Tb.Th), mean trabecular number (Tb.N), and mean trabecular separation (Tb.Sp) indices were computed. For the analysis of cortical parameters, the mid-shaft of the femur was determined as 40% of the entire length of the bone. The cortical region of interest began at the mid-shaft and extended 100 slices (0.6 mm) toward the femoral head. Using upper and lower thresholds of 255 and 80, total area (T.Ar), periosteal perimeter (Ps.Pm), endosteal area (En.Ar), endosteal perimeter (En.Pm), cortical bone area (Ct.Ar), cortical thickness (Ct.Th), and relative cortical area (RCA) were computed.
Mechanical Testing
Breaking strength of the right femur was measured under three-point bending using a material testing machine (ElectroForce Systems Group, Bose, Eden Prairie, MN) fitted with a 1,000 N load cell as previously described [24, 25]; to minimize the effect of shear loading, the distance between the lower support points was maximized. Femora were placed on the loading fixture anterior side down and loaded in the anterior–posterior plane at a span length of 19.26 mm. Prior to testing, bones were thawed in saline at room temperature to ensure hydration. Femora were loaded to failure at a rate of 0.05 mm/s, during which displacement and force were collected (100 Hz). Force and displacement values were normalized using terms derived from engineering analysis of three-point bending. Bending moments were calculated from the force (F) data (M = FL/4) (Nmm). Displacement data were divided by L 2/12 (mm/mm2), where L is the distance between the lower supports (19.26 mm). Whole-bone mechanical properties were then determined from the moment versus normalized displacement curves, including peak moment (Nmm, ultimate load the specimen sustained), yield moment (Nmm), stiffness (Nmm2, the slope of the initial linear portion of the moment–displacement curve), yield displacement (mm/mm2, displacement at the yield point), postyield displacement (mm/mm2), work to failure (Nmm-mm/mm2, the area under the moment–displacement curve before failure), and work to failure postyield (Nmm-mm/mm2). The yield point was calculated as the point where a 10% change in the slope of the moment versus normalized displacement curve occurred.
Cell Culture
Calvarial osteoblasts were obtained from pups 3–5 days old as previously described [26]. Typically, cells from three calvaria were plated in T-75 flasks in medium supplemented with 10% FBS. Culture medium was changed every third day until cells reached confluence. Cells were harvested and frozen until further use. For osteoblast differentiation in a typical experiment, 5 × 105 cells were plated in a 6 cm dish and cultured in the presence of 10% fetal bovine serum (FBS; Thermo Fisher Scientific, Pittsburg, PA). “Complete medium” is 10% FBS containing α-MEM. Differentiation of cells in the above medium was initiated by adding 50 mM ascorbic acid and 5 mM glycerophosphate (Sigma, St. Louis, MO). Osteogenic medium was changed every third day. On day 14 osteoblasts were stained for alkaline phosphatase (ALP) and assessed for ALP activity according to the manufacturer’s protocol (Sigma). ALP activity was normalized to the total protein content of cell lysates. Protein concentrations were quantified by a BCA protein assay (Pierce, Rockford, IL). On day 21 osteoblasts were stained with alizarin red, and staining was quantified using the Bioquant Osteo II (Nashville, TN) program.
Bone marrow stromal cells were isolated from the hind limbs of mice aged 6–8 weeks. The proximal and distal ends of the hind limbs were cut to allow the marrow to be flushed with complete medium. Bone marrow cells were filtered through a 70 μ nylon mesh filter (BD, Falcon, Oxnard, CA). Following lysis of RBCs using ammonium chloride lysis buffer, cells were collected by centrifugation and resuspended in complete medium. Cells (25 to 30 × 107) were plated into T-75 tissue culture flasks. Nonadherent cells were removed after 16–18 h. Adherent cells were cultured until they became 70–80% confluent. Cell culture was treated with 0.025% trypsin containing 0.02% EDTA for 2 minutes at room temperature. To generate conditioned medium, cells (1 × 107) were seeded in a 6 cm dish and cultured in the presence of complete medium for 5 days at 37°C, 5% CO2. On the fifth day, medium was removed, centrifuged at 1,500 rpm for 5 min, filtered through 22 μ filters, and either used immediately or stored at −80°C. Cells were harvested for analysis in protein markers by real-time PCR as described below. For colony-forming unit (CFU) assay, bone marrow-derived cells were used as previously described [27]. Briefly, bone marrow cells (1.5 × 105 cells cm−2) were plated in complete medium and cultured for 5 days. Then, one-third of the medium was replaced with complete medium every third day. After 20 days, cells were fixed with 3.7% formaldehyde and stained with KaryoMax Giemsa stain or ALP (Sigma) according to the manufacturer’s protocol. Cells were visualized using an E300 microscope at 10× magnification to count the number of colonies. Cells in clusters of 20 or more were counted as one colony.
For the proliferation assay, 10,000 cells were plated/well in a 96-well plate in quadruplets. After 18–20 h in culture in complete medium, cells were washed with serum-free medium and then incubated with appropriate medium or growth factors. After 72 h, cells were washed three times with cold PBS. A proliferation assay was performed using the Cyquant Cell Proliferation Kit (Molecular Probes, Eugene, OR) per the manufacturer’s protocol. Fluorescence was measured using a Wallac 1420 fluorometer (PerkinElmer and Analytical Sciences, Turku, Finland), and cell proliferation was expressed as a percent of proliferation in 10% FBS-containing medium. In some experiments, prior to addition to cells, conditioned medium was treated with appropriate antibodies (10 μg/ml) and this solution was mixed end on end for 1 h at 4°C. In other experiments, cells were incubated with antibodies (10 μg/ml) for 1 h at 37°C prior to the addition of condition medium. Cell lysates were prepared in mRIPA lysis buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 0.5% NP40, 0.25% sodium deoxycholate, protease inhibitors, 1 mM PMSF, 1 mM Na2VO3, 50 mM NaF) and lysates were processed for electrophoresis and Western blotting analysis as previously described [19].
Real-Time PCR Analysis
The expression levels of osteoblast markers were analyzed by quantitative real-time PCR. RNA isolation was performed using TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. RNA (1 μg) was transcribed using Super Script II reverse transcriptase (Invitrogen), following the manufacturer’s protocol. Oligo(dT) and dNTPs were purchased from Promega (Madison, WI). Quantitative real-time PCR was performed using 1 μl of cDNA and SYBR Green PCR master mix (Applied Biosystems, Foster City, CA) on an Applied Biosystems 7500 real-time PCR system. Gene expression levels were normalized to GAPDH and calculated using the ΔΔCt method. Primers were designed using mRNA sequences obtained from Mouse Genomics Informatics and the Primer3 program (http://sourceforge.net/projects/primer3/). The following primers were used: SDF-1, AGTAGTGGCTCCCCAGGTTT (sense) and GAGACAGTCTTGCGGACACA (antisense); CXCR4, TCAGTGGCTGACCTCCTCTT (sense) and TTTCAGCCAGCAGTTTCCTT (antisense); Col1A, TGAACGTGACCAAAAACCAA (sense) and GCAGAAAAGGCAGCATTAGG (antisense); RUNX2, CCCAGCCACCTTTACCTACA (sense) and TATGGAGTGCTGCTGGTCTG (antisense); BSP, AAAGTGAAGGAAAGCGACGA (sense) and GTTCCTTCTGCACCTGCTTC (antisense); GAPDH, AACTTTGGCATTGTGGAAGG (sense) and ACACATTGGGGGTAGGAACA (antisense).
Analysis of Cytokines
Conditioned medium from bone marrow stromal cells was generated as described above and then sent for analysis to Searchlight Aushon Bio Systems (Billerica, MA) to determine the presence of cytokines and chemokines. Briefly, a multiplex ELISA was performed using the conditioned medium and the antibody to the protein of interest. Samples were diluted 1:2, 1:50, and 1:1,000 and tested in duplicate. A standard curve was established from the dilutions of the samples. Then, a mean value for the cytokine of interest was determined from the standard curve and reported as picograms per milligram total protein. Protein estimation of the conditioned medium was performed using the BCA kit. The results of the cytokine assays were normalized to the protein concentration for each sample.
Statistics
All experiments were performed at least three times, and differences between wild type (WT) and YF were determined using Student’s t test. All statistics were generated from Prism 4.0 program (GraphPad, San Diego, CA). Differences were considered significant at P < 0.05, and all data are represented as means ± SEM. For mechanical testing, body weights of mice in each group were normalized with a linear regression–based correction as previously described [28].
Results
Abrogation of Cbl–PI3K Interaction Results in Increased Cortical and Trabecular Bone
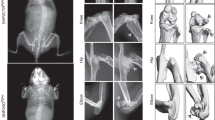
To understand the role of the Cbl–PI3K interaction in skeletal remodeling, we employed knock-in mice (CblY737F/Y737F, henceforth YF mice) in which the PI3K binding site on Cbl was ablated. Gross analysis revealed that YF mice were smaller in size and had decreased body weight compared to control pups (Fig. 1a, b). The delayed growth was first apparent 3 days after birth (data not shown) and continued for 9–18 days. The delay in growth occurred simultaneously with a lag in the growth of long bones (Fig. 1c). Histological analysis of bones from 9-day-old YF pups showed a significant delay in the formation of secondary spongiosa (data not shown). Histological analysis of tibial sections from 12-week-old mice stained with von Kossa showed that cancellous bone was increased in YF relative to age-matched control mice (Fig. 1d). However, weight and total tibial length were comparable between 12-week-old WT and YF mice (Fig. 1e, f). YF mice had a slight but significant decrease in body length from tail to snout (Fig. 1g). This matches prior reports of short stature on postnatal day 9, decreased bone length, and delayed formation of secondary ossification centers in Cbl−/− mice [6], although adult Cbl−/− mice do not exhibit any overt skeletal phenotype. We also examined bone architecture of 12-week-old mice by micro-CT analysis (Fig. 2a, b) and, in agreement with our histological analysis, found increased trabecular bone volume in YF mice (Table 1). Similarly, Ct.Ar was significantly increased due to the increased Ps.Pm and Ct.Th in YF mice (Table 1). Taken together, these results suggest that lack of interaction between Cbl and PI3K results in increased bone volume.

Disruption of Cbl–PI3K interaction results in a growth delay in young mice. a At 9 days old, YF mice are smaller than their counterpart WT mice. Delayed growth in 9-day-old YF mice resulted in decreased total body weight (b) and decreased tibial length (c). d Representative histological sections of proximal tibiae of 12-week-old WT and YF mice stained with von Kossa and counterstained with toluidine blue show increased mineralized bone in YF mice. Images were taken using a 10× objective. Twelve-week-old male WT and YF mice have similar weights (e) and tibial lengths (f). g Twelve-week-old YF mice have shorter body lengths. n = 8 mice per genotype. Mean ± SEM. *P < 0.05 compared to WT

Disruption of Cbl–PI3K interaction in mice perturbs trabecular and cortical bone architecture. a The trabecular region of proximal tibiae was scanned starting at the proximal end and included approximately 7 mm (1,335 slices) of the total bone length. Top and bottom panels represent a 3D reconstruction of trabecular bone. b The cortical bone analysis began at 40% from the distal end of the femur and included 100 sections proximal from that point. Top panel represents a 3D reconstruction of the cortical bone at mid-shaft. Bottom panel represents a 2D section of the cortical bone, taken 0.3 mm (50 slices) from the mid-shaft toward the femur. Micro-CT was performed on six mice per genotype. Representative scanned images are shown
Static and Dynamic Parameters of Bone Formation are Increased in YF Mice
We recently reported that although YF osteoclasts are unable to resorb bone in vivo, their numbers are significantly increased in YF mice [19]. Given that bone volume can also be increased due to increased osteoblast numbers, we examined osteoblasts in YF and WT mice. Histological analysis of tibiae stained with Masson trichome (Fig. 3a) revealed a significant increase in osteoblast numbers and a 63% increase in osteoid (unmineralized bone) in YF sections (Fig. 3b, c). Histomorphometric analysis of calcein-labeled sections indicated that the normalized MS/BS, MAR, and BFR/BS were significantly increased in YF samples (Fig. 3d–g). Taken together, the above results indicate that in the absence of Cbl–PI3K interaction increased bone volume in mice is due not only to the decreased osteoclast-mediated bone resorption reported previously by our group [19] but also to increased bone formation by osteoblasts.

Bone formation is increased in YF mice. a Tibial sections from 12-week-old WT and YF were stained with Masson’s trichrome (400× magnification). Quantification of osteoblasts/bone surface (N.Ob/BS) (b) and osteoid/BS (c). d Dynamic parameters of bone formation were assessed using tibiae harvested from 12-week-old WT and YF that were injected with calcein as described in Materials and Methods. Representative photomicrographs of calcein double labeling (40×) in WT and YF in cortical bone (upper panels) and in trabeculae (lower panels) are shown. e Mineralized surface/bone surface (MS/BS, %). f Mineral apposition rate (MAR, μm2/day). g Bone formation rate/bone surface (BFR/BS, μm2/μm/day). n = 6 mice per genotype. Mean ± SEM. *P < 0.05, compared to WT
Lack of Cbl–PI3K Interaction Confers Increased Bone
While structural changes in bone are most likely to be detected in trabecular bone, we also observed structural changes in cortical bone in YF mice. A significant increase in total bone area (P < 0.05 vs. WT) as well as increased Ps.Pm, En.Pm, and En.Ar (P < 0.05 vs. WT, each) led to an increase in overall size of the cortical bone (Table 1). In addition, polar moment of inertia was significantly greater in YF mice. The RCA did not change between groups, although there was an overall increase in cortical thickness (Table 1). Imbalances between bone resorption and bone formation can affect bone geometry, affecting its ability to resist damage (fracture). Given that YF mice have increased cancellous bone (Fig. 1d; Table 1) and increased cortical bone size (Table 1), we examined the mechanical properties of the long bones using a three-point bending assay, as previously described [24, 25]. There was a significant increase in peak moment and yield displacement in YF mice compared to WT, but no differences were observed in the other parameters listed in Table 2. These data suggest that the structural and geometric changes in YF bones could potentially be a compensatory mechanism to maintain bone strength since both size and the quality of the bone contribute to whole-bone strength.
In the Absence of Cbl–PI3K Interaction Bone Marrow-Derived Osteoblasts Show Increased Col1A Expression
We previously reported that YF mice have increased osteoclast numbers and enhanced differentiation [19]. Given that the numbers of osteoblasts are also significantly increased in YF mice (Fig. 3b), we next examined if similar increases in numbers were seen in ex vivo culture conditions. To assess the ability of osteoblasts to differentiate in vitro, calvarial osteoblasts or bone marrow stromal cells from WT and YF mice were treated with osteogenic media for 14 or 21 days. No appreciable differences were observed in the expression levels of Col-1A, BSP, and Runx2 between WT- and YF-derived calvarial osteoblasts and no significant differences were observed in ALP and von Kossa staining (Fig. 4a–c and data not shown). In contrast, in YF bone marrow stromal cell cultures, expression levels of Col1A were significantly increased during differentiation compared to WT samples (Fig. 4d), while expression levels of BSP and Runx2 were not appreciably altered (Fig. 4e, f). Histochemical analysis of in vitro cultures demonstrated comparable levels of ALP and alizarin red activity between WT and YF samples (Fig. 4g–j). Taken together, these data suggest that, unlike YF osteoclasts, which exhibit enhanced differentiation due to the upregulation of transcription factors NFATc1 and NFκB [19], for the most part ex vivo osteoblast differentiation and the expression of transcription factor that regulates osteoblastogenesis are not significantly affected in the absence of Cbl–PI3K interaction.

Differentiation of calvarial and bone marrow stromal cells in ex vivo culture. a–f WT and YF primary calvarial or bone marrow stromal cells were differentiated in osteogenic media for 0, 14, or 21 days, as indicated. Osteogenic medium was changed every third day. Expression levels of Col-1A (a, d), BSP (b, e), and Runx-2 (c, f) were analyzed by real-time PCR in calvarial osteoblasts (COB) and bone marrow stromal cells (BM). g Bone marrow stromal cells stained for ALP activity. h ALP activity. i Bone marrow stromal cells stained with alizarin red S to assess calcium deposition. j Alizarin red S stain was released from the cell matrix and quantified. All experiments were performed three times. A representative sample of three experiments is shown. Mean ± SEM. *P < 0.05 compared to WT
Osteoblast Proliferation is Upregulated in the Absence of Cbl–PI3K Interaction
Trabecular and endocortical bone formation are directly correlated with the number of marrow-derived osteoprogenitors. Histological analysis of long bones indicated that in the absence of Cbl–PI3K interaction there is a twofold increase in osteoblast numbers (Fig. 3b) and increased MAR (Fig. 3f). Also, in contrast to calvarial cells, there was a significant increase in the expression of Col-1A when YF bone marrow-derived stromal cells were cultured in osteogenic medium (Fig. 4d). These data suggest that in the absence of Cbl–PI3K interaction, there might be increased potential for the bone marrow precursors to differentiate into osteoblasts and/or that osteoblasts have augmented proliferative capacity. To determine if the numbers of osteoblast progenitors were affected in YF mice, the number of CFUs was determined from bone marrow-derived cells. Although, the total numbers of marrow cells were comparable between the WT and YF mice (data not shown), there was a significant increase in CFUs for YF samples (Fig. 5a) and the potential of these cells to proliferate was increased compared to WT cells (Fig. 5b). We also observed a slight, but not statistically significant, increase in the proliferation rate of YF calvarial osteoblasts (data not shown).

Proliferation of YF osteoblasts is increased. a Bone marrow stromal cells were cultured for 20 days in 10% FBS-containing medium. Cells were fixed and stained with Giemsa; clusters of 20 or more cells were counted as a colony. b Bone marrow stromal cells from WT and YF were seeded in a 96-well plate in complete medium for 72 h, and proliferation was assessed by the Cyquant method, as described in Materials and Methods. c MC3T3-E1 cells were cultured in the presence of serum-free medium, 10% FBS, or osteoclast-conditioned medium derived from WT or YF osteoclasts. Cell proliferation was assessed as described above. d MC3T3-E1 were treated with serum-free medium, 10% FBS, or bone marrow conditioned medium derived from WT or YF bone marrow cultures. Cell proliferation was assessed as described above. e–h Calvarial and bone marrow stromal cells were cultured in complete medium for 5 days. Expression levels of SDF-1 (e, g) and CXCR4 (f, h) were measured by quantative real-time PCR. Data are presented as fold difference of expression between WT and YF culture. All experiments were performed three times, and a representative is shown. Mean ± SEM. *P < 0.05 compared to WT
PI3K is known to regulate cell survival, and we previously reported that abrogation of Cbl–PI3K resulted in enhanced osteoclast survival [19]; however, no differences were observed between the survival of WT and YF bone marrow-derived stromal cells (Supplementary Fig. 1). We also examined whether increased proliferation in YF osteoblasts was influenced by extracellular growth factors known to influence osteoblast proliferation. Treatment with several growth factors resulted in a similar level of proliferation in both WT and YF cells (Supplementary Fig. 2a). In response to addition of EGF, FGF, IGF, and PDGF to the cultures, phosphorylation of p85 and AKT was comparable in WT and YF samples (Supplementary Fig. 2b).
In addition to their resorptive activity, osteoclasts are known to secrete factors that promote anabolic activity [29, 30]. Since YF mice had increased numbers of osteoblasts and osteoclasts (Fig. 3b and [19]), we next examined the possibility that osteoclast-dependent secretory factors may promote increased YF osteoblast proliferation. WT and YF osteoclasts were generated in ex vivo cultures as previously described [19] and the culture medium on day 5 of differentiation was collected. To analyze if the factors secreted from osteoclasts promote proliferation, MC3T3-E1 cells were grown in the osteoclast-derived culture medium. No significant differences were observed in the proliferation rate when cells were cultured in WT osteoclast-conditioned medium versus YF osteoclast-conditioned medium (Fig. 5c). This result suggests that, under these experimental conditions, factors secreted by YF osteoclasts may not contribute to the observed enhanced YF osteoblast proliferation.
Increased Levels of SDF-1 in YF Bone Marrow
The bone marrow niche contains a variety of cell types in various stages of maturation/differentiation. Many of these cell types secrete factors that can affect osteoblast proliferation. Therefore, we next examined if other factor(s) secreted by resident bone marrow cells could influence osteoblast proliferation. For this experiment, bone marrow stromal cells from WT and YF mice were cultured in the presence of 10% FBS-containing medium for 5 days. The resulting supernatant was used as the bone marrow conditioned medium. Proliferation of MC3T3-E1 cells was examined in response to WT and YF conditioned media. Compared to WT, treatment with YF conditioned medium induced increased proliferation of MC3T3-E1 cells (Fig. 5d), suggesting that a factor(s) present in the YF bone marrow-derived medium might contribute to the increased cell proliferation.
Analysis of the conditioned media using multiplex ELISAs indicated that the levels of IL-1β, IL-18, MCP-1, RANTES, and TGF-1β were comparable in WT and YF samples (Table 3). Only levels of SDF-1, a chemokine known to be present in the bone marrow milieu, were significantly increased in the YF bone marrow conditioned medium (Table 3). We next determined mRNA expression levels of SDF-1 and its receptor, CXCR4, in calvarial osteoblasts and bone marrow stromal cells. For these experiments, cells were cultured in the presence of 10% FBS for 5 days before harvesting. RT-PCR analysis showed that SDF-1 and CXCR4 levels were comparable in WT and YF calvarial cells (Fig. 5e, f). In contrast, compared to expression levels in WT bone marrow stromal cells, YF bone marrow stromal cells showed ~2.5- and 3.5-fold increases in the expression levels of SDF-1 and CXCR4, respectively (Fig. 5g, h). This expression was then verified by immunohistochemistry. Staining of YF tibial sections with anti-SDF-1 antibodies showed enhanced staining in many bone stromal cells but no staining in mature osteoblasts adjacent to the trabeculae (Fig. 6b, c, i). Both stromal cells and osteocytes in sections from YF samples demonstrated increased CXCR4 staining compared to WT samples (Fig. 6f, j, h, l). For validation, bone marrow proliferation assays were performed using neutralizing antibodies against SDF-1 or CXCR4. Enhanced proliferation of MC3T3-E1 cells in response to YF conditioned medium was significantly decreased in the presence of neutralizing antibodies (Fig. 7). Taken together, these data suggest that in YF mice cell proliferation is increased, at least in part, due to increased SDF-1 present in the bone marrow milieu. However, our results do not eliminate the possibility that other factors may also contribute to the increased osteoblast proliferation.

Bone marrow stromal cells from YF mice show enhanced SDF-1 and CXCR4 expression. Representative sections of proximal tibiae from 12-week-old WT and YF immunostained with either anti-SDF-1α (HRP-DAB or secondary antibody tag), anti-CXCR4, or DAPI. In all images, area indicated with asterisk is enlarged in inset. a WT tibial section showing only low-level SDF-1 staining in bone marrow stromal cells. Inset shows an absence of SDF-1 staining in osteocytes or quiescent bone lining cells on trabecular bone. b YF tibial section showing SDF-1 staining in many bone stromal cells. Inset shows SDF-1 staining in many stromal cells, including cells surrounding the trabecular bone, but no staining in osteoblasts (ob). Sections in a and b are lightly counterstained with hematoxylin and eosin (H&E). c YF tibial section with SDF and DAPI labeling showing SDF-1-stained cells in the bone marrow (bm) but not in cells lining the trabecular bone (tb). d YF tibial cortical bone showing increased numbers of osteocytes (os) expressing CXCR4. e WT tibial section showing SDF-1 staining in only a few of bone marrow cells but no staining in trabecular bone. Inset shows SDF-1 staining in only a few stromal cells. f Same section as shown in e showing an absence of CXCR4 staining. g Same section showing DAPI staining. h Merged e–g panels. Inset shows a low number of SDF + DAPI-stained stromal cells. i YF tibial section showing high levels of SDF-1 staining in many bone marrow stromal cells but none in trabecular bone. Inset shows SDF-1 in the cytoplasm of these cells. j Same section as in i showing increased CXCR4 staining in several stromal cells. Some, but not all, of the CXCR4-stained cells are double-labeled with SDF-1. Inset shows CXCR4 staining in the same cells as shown in inset in i. k Same section showing DAPI staining. l Merged i–k panels. Inset shows SDF + DAPI-stained stromal cells. All images were taken using a 40× objective. Scale bar in l = 20 μm. bv, blood vessel

Increased proliferation of cells in YF conditioned medium is mediated by SDF-1. MC3T3-E1 cells were cultured in the presence of serum-free medium, 10% FBS, or condition medium derived from WT or YF bone marrow cells. To assess the effect of SDF-1, 10 μg/ml of neutralizing anti-SDF-1 antibodies or isotype control antibodies was added to the conditioned medium. To determine the role of CXCR4, MC3TC-E1 cells were pretreated with 10 μg/ml neutralizing anti-CXCR4 antibodies. The cell proliferation assay was performed as described above. Cell proliferation is expressed as a percent of control (10% FBS-treated cells). Mean ± SEM. Symbols indicate P < 0.05 for the following comparisons: a FBS versus YF, b WT versus YF, c YF versus IgG, d YF versus YF + α-SDF-1, e IgG versus YF + α-SDF-1, f YF versus YF + α-CXCR4, g IgG versus YF + α-CXCR4
Discussion
Cbl proteins are known to play a role in osteoclast migration and function as well as in osteoblast differentiation downstream of the FGF pathway by ubiquitylating and degrading the Src family kinases Lyn and Fyn [31, 32]. However, loss of Cbl protein in adult mice did not result in an overt skeletal phenotype [6]. On the other hand, absence of Cbl-b, another member of the Cbl family, resulted in decreased bone volume [7]. These findings suggest that although Cbl and Cbl-b proteins share significant homologies and identical domain organization [2], they have different effects on cells that regulate bone mass. One major difference between the two proteins is the absence of Y737 in Cbl-b. Cbl protein phosphorylation of Y737 has been shown to recruit PI3K and regulate PI3K activity [16]. Studies on osteoblast cell lines have established that PI3K plays an important role in osteoblast function [15, 33, 34]. In an immortalized human osteoblast cell line, FGFR2–Cbl interaction in lipid rafts resulted in attenuation of PI3K signaling and osteoblast survival [18]. We recently reported that the lack of Cbl–PI3K interaction promotes decreased osteoclast function while increasing their survival [19]. In this report, we examined if the lack of this molecular interaction also had an effect on osteoblast differentiation and function.
Cbl is a major PI3K binding protein in several cell types. With a Y > F mutation on the Cbl–PI3K binding site, the expectation is that loss of this interaction would perturb PI3K-mediated signaling and cell survival. In thymocytes loss of Cbl–PI3K results in decreased AKT phosphorylation [35], whereas in osteoclasts abrogation of this interaction results in increased AKT phosphorylation, leading to enhance survival [19]. Others have shown that in human osteoblast cells that express mutated forms of FGFR2 FGFR2–Cbl interaction in lipid rafts triggers attenuation of PI3K/AKT signaling and osteoblast survival [18]. On the other hand, stimulation of growth factor receptors in YF osteoblasts did not modulate proliferation or PI3K-mediated signaling (Supplementary Fig. 2a, b). Thus, it appears that Cbl–PI3K interaction plays distinct roles in different cell types. In YF osteoblasts, the lack of impact on PI3K-mediated signaling suggests that Cbl-b and/or other proteins (e.g., Src) that are capable of interacting with PI3K might be able to compensate for the lack of Cbl–PI3K interaction and preserve the signaling. It is also likely that another SH2 domain-containing protein might interact with phosphorylated Y737 Cbl and that the observed effects in the YF mice are a consequence of loss of that interaction.
Histological and micro-CT analysis of bones from YF mice indicated that there was an increase in trabecular bone volume (Fig. 2; Table 1), osteoblast numbers (Fig. 3b), BFR, mineralizing surface, and MAR compared to WT mice (Fig. 3e–g). Also, culturing YF bone marrow stromal cells resulted in increased numbers of CFUs and increased proliferation (Fig. 5a, b). The increase in Col1A expression during differentiation of YF bone marrow cells (Fig. 4d) suggests that the increase in MAR is driven at least in part by increased matrix protein production. However, other markers of differentiation were unaffected in YF bone marrow cells (Fig. 4). These combined data suggest that the increased numbers of YF osteoblasts in bone marrow may be in part also due to a secondary effect.
The increased bone volume and increased numbers of nonresorbing osteoclasts and osteoblasts are indicative of an osteopetrotic phenotype in YF mice (Fig. 2 and [19, 36]). In several osteopetrotic mouse models where bone resorption is compromised due to the absence of osteoclast (c-Fos−/−) [37] or due to impaired osteoclast function (e.g., Src−/− [38] and Pyk2−/− [39]), bone formation is also impaired due to a cell autonomous defect in osteoblast activity [37, 40, 41]. In v-ATPase knockout mice, in which osteoclasts are incapable of degrading the bone matrix, both bone formation and osteoblast numbers increase due to uncoupling [42]. Osteoclast activity has been regarded as restricted to bone resorption; however, recent studies have suggested that osteoclasts might release anabolic signals that are not linked to their resorption activity [29, 30]. Our results indicate that cell proliferation was not significantly affected when cells were grown in conditioned medium generated from YF osteoclasts (Fig. 5c). This could be due to several possibilities. First, in an ex vivo culture system, a secreted factor(s) may not be in sufficient quantity or concentration to influence cell proliferation. Second, cell–cell contact between osteoclasts and osteoblasts may be required for the desired effect on proliferation since direct contact of osteoblasts with the bone marrow precursors is a prerequisite for the generation of mature osteoclasts and to facilitate interaction of the membrane-bound RANKL on osteoblasts with RANK on osteoclasts [43]. Furthermore, recent studies have indicated that the interaction between the transmembrane ephrinB2 ligand that is present on osteoclasts mediates anabolic signals via the ephB4 receptor present on osteoblasts [44]. Given that YF osteoclasts are not functional in in vivo and in vitro cultures [19], we could not test the possibility that a bone matrix-derived factor(s) could influence increased proliferation of osteoblasts in vivo. Given this scenario, it is not likely that YF osteoclasts modulate enhanced proliferation of YF osteoblasts, although more studies are needed to explore this further.
Bone marrow contains several different types of hematopoietic and stromal cells that secrete a variety of growth factors and cytokines that can influence the properties of cells residing in the marrow space [45, 46]. Our result show that there was a significant increase in the rate of proliferation of MC3T3-E1 cells cultured in YF conditioned medium compared to WT conditioned medium (Fig. 5d). Comparative analysis of WT and YF condition media showed that the level of the chemokine SDF-1 was elevated in the YF conditioned medium (Table 3). SDF-1, also known as CXCL2, is produced in bone marrow stromal cells and directs the migration of hematopoietic cells to the bone marrow during development [47]. The SDF-1/CXCR4 signaling axis is essential for several normal physiological functions, including bone remodeling [48]. For example, SDF-1 has been implicated as a regulator of bone resorption [49]. SDF-1 also regulates BMP2-mediated osteogenic differentiation in mesenchymal cell lines [50]. More recently, it has been demonstrated that conditional inactivation of CXCR4 in osteoblast precursors reduces postnatal bone formation due to impaired osteoblast development [51]. Our results show that expression of both SDF-1 and CXCR4 was substantially increased in YF bone marrow stromal cells (Fig. 6). In contrast, SDF-1 was not increased in mature osteoblast and bone lining cells (Fig. 6c, d). Interestingly, the increased expression was restricted to the YF bone marrow milieu since expression levels of SDF-1 and CXCR4 were comparable in WT and YF calvarial cells (Fig. 5e, f). It is likely that increased SDF-1 in the YF bone marrow environment promotes osteoblast proliferation since both Cbl [52] and PI3K [53] are known to participate in SDF-1-mediated signaling. Further studies are needed to specifically analyze the role of Cbl–PI3K interaction in the effects of SDF-1-mediated signaling events that induce proliferation, attachment, and migration of bone marrow stromal cells.
In conclusion, our studies establish that abrogation of Cbl–PI3K interaction results in increased bone volume. While our past studies show that this is primarily due to defective osteoclast function, these new data also suggest that the increased numbers of osteoblasts in vivo and increased proliferation of osteoblasts are due to a secondary effect probably mediated by factors in the marrow cavity, such as SDF-1.
References
Sims N, Baron R (2000) Bone cells and their function. In: Canalis E (ed) Skeletal growth factors. Lippincott Williams and Wilkins, Philadelphia, pp 1–16
Swaminathan G, Tsygankov AY (2006) The Cbl family proteins: ring leaders in regulation of cell signaling. J Cell Physiol 209:21–43
Miyazaki T, Sanjay A, Neff L, Tanaka S, Horne WC, Baron R (2004) SRC kinase activity is essential for osteoclast function. J Biol Chem 279:17660–17666
Sanjay A, Miyazaki T, Itzstein C, Purev E, Horne WC, Baron R (2006) Identification and functional characterization of an Src homology domain 3 domain-binding site on Cbl. FEBS J 273:5442–5456
Tanaka S, Amling M, Neff L, Peyman A, Uhlmann E, Levy JB, Baron R (1996) c-Cbl is downstream of c-Src in a signalling pathway necessary for bone resorption. Nature 383:528–531
Chiusaroli R, Sanjay A, Henriksen K, Engsig MT, Horne WC, Gu H, Baron R (2003) Deletion of the gene encoding c-Cbl alters the ability of osteoclasts to migrate, delaying resorption and ossification of cartilage during the development of long bones. Dev Biol 261:537–547
Nakajima A, Sanjay A, Chiusaroli R, Adapala NS, Neff L, Itzsteink C, Horne WC, Baron R (2009) Loss of Cbl-b increases osteoclast bone-resorbing activity and induces osteopenia. J Bone Miner Res 24:1162–1172
Naramura M, Jang I-K, Kole H, Huang F, Haines D, Gu H (2002) c-Cbl and Cbl-b regulate T cell responsiveness by promoting ligand-induced TCR down-modulation. Nat Immunol 3:1192–1199
Feshchenko EA, Shore SK, Tsygankov AY (1999) Tyrosine phosphorylation of C-Cbl facilitates adhesion and spreading while suppressing anchorage-independent growth of V-Abl-transformed NIH3T3 fibroblasts. Oncogene 18:3703–3715
Hunter S, Burton EA, Wu SC, Anderson SM (1999) Fyn associates with Cbl and phosphorylates tyrosine 731 in Cbl, a binding site for phosphatidylinositol 3-kinase. J Biol Chem 274:2097–2106
Ueno H, Sasaki K, Honda H, Nakamoto T, Yamagata T, Miyagawa K, Mitani K, Yazaki Y, Hirai H (1998) c-Cbl is tyrosine-phosphorylated by interleukin-4 and enhances mitogenic and survival signals of interleukin-4 receptor by linking with the phosphatidylinositol 3′-kinase pathway. Blood 91:46–53
Feshchenko EA, Langdon WY, Tsygankov AY (1998) Fyn, Yes, and Syk phosphorylation sites in c-Cbl map to the same tyrosine residues that become phosphorylated in activated T cells. J Biol Chem 273:8323–8331
Vanhaesebroeck B, Ali K, Bilancio A, Geering B, Foukas LC (2005) Signalling by PI3K isoforms: insights from gene-targeted mice. Trends Biochem Sci 30:194–204
Dufour C, Holy X, Marie PJ (2007) Skeletal unloading induces osteoblast apoptosis and targets alpha5beta1-PI3K-Bcl-2 signaling in rat bone. Exp Cell Res 313:394–403
Ghosh-Choudhury N, Abboud SL, Nishimura R, Celeste A, Mahimainathan L, Choudhury GG (2002) Requirement of BMP-2-induced phosphatidylinositol 3-kinase and Akt serine/threonine kinase in osteoblast differentiation and Smad-dependent BMP-2 gene transcription. J Biol Chem 277:33361–33368
Meng F, Lowell CA (1998) A β1 integrin signaling pathway involving Src-family kinases, Cbl and PI-3 kinase is required for macrophage spreading and migration. EMBO J 17:4391–4403
Guenou H, Kaabeche K, Dufour C, Miraoui H, Marie PJ (2006) Down-regulation of ubiquitin ligase Cbl induced by twist haploinsufficiency in Saethre-Chotzen syndrome results in increased PI3K/Akt signaling and osteoblast proliferation. Am J Pathol 169:1303–1311
Dufour C, Guenou H, Kaabeche K, Bouvard D, Sanjay A, Marie PJ (2008) FGFR2–Cbl interaction in lipid rafts triggers attenuation of PI3K/Akt signaling and osteoblast survival. Bone 42:1032–1039
Adapala NS, Barbe MF, Langdon WY, Nakamura MC, Tsygankov AY, Sanjay A (2010) The loss of Cbl–phosphatidylinositol 3-kinase interaction perturbs RANKL-mediated signaling, inhibiting bone resorption and promoting osteoclast survival. J Biol Chem 285:36745–36758
Molero JC, Turner N, Thien CB, Langdon WY, James DE, Cooney GJ (2006) Genetic ablation of the c-Cbl ubiquitin ligase domain results in increased energy expenditure and improved insulin action. Diabetes 55:3411–3417
Sims NA, Clement-Lacroix P, Da Ponte F, Bouali Y, Binart N, Moriggl R, Goffin V, Coschigano K, Gaillard-Kelly M, Kopchick J, Baron R, Kelly PA (2000) Bone homeostasis in growth hormone receptor-null mice is restored by IGF-I but independent of Stat5. J Clin Invest 106:1095–1103
Parfitt AM, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR (1987) Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res 2:595–610
Al-Shatti T, Barr AE, Safadi FF, Amin M, Barbe MF (2005) Increase in inflammatory cytokines in median nerves in a rat model of repetitive motion injury. J Neuroimmunol 167:13–22
Yingling V, Elle Saine M, Joshi R (2009) Hypothalamic suppression decreases bone strength before and after puberty in a rat model. Calcif Tissue Int 84:485–493
Yingling VR (2009) A delay in pubertal onset affects the covariation of body weight, estradiol, and bone size. Calcif Tissue Int 84:286–296
Sabatakos G, Davies GE, Grosse M, Cryer A, Ramji DP (1998) Expression of the genes encoding CCAAT-enhancer binding protein isoforms in the mouse mammary gland during lactation and involution. Biochem J 334:205–210
Hankenson KD, Bain SD, Kyriakides TR, Smith EA, Goldstein SA, Bornstein P (2000) Increased marrow-derived osteoprogenitor cells and endosteal bone formation in mice lacking thrombospondin 2. J Bone Miner Res 15:851–862
Di Masso RJ, Font MT, Capozza RF, Detarsio G, Sosa F, Ferretti JL (1997) Long-bone biomechanics in mice selected for body conformation. Bone 20:539–545
Karsdal MA, Martin TJ, Bollerslev J, Christiansen C, Henriksen K (2007) Are nonresorbing osteoclasts sources of bone anabolic activity? J Bone Miner Res 22:487–494
Karsdal MA, Neutzsky-Wulff AV, Dziegiel MH, Christiansen C, Henriksen K (2008) Osteoclasts secrete non-bone derived signals that induce bone formation. Biochem Biophys Res Commun 366:483–488
Kaabeche K, Guenou H, Bouvard D, Didelot N, Listrat A, Marie PJ (2005) Cbl-mediated ubiquitination of alpha5 integrin subunit mediates fibronectin-dependent osteoblast detachment and apoptosis induced by FGFR2 activation. J Cell Sci 118:1223–1232
Kaabeche K, Lemonnier J, Le Mee S, Caverzasio J, Marie PJ (2004) Cbl-mediated degradation of Lyn and Fyn induced by constitutive fibroblast growth factor receptor-2 activation supports osteoblast differentiation. J Biol Chem 279:36259–36267
Lai C-F, Cheng S-L (2002) Signal transductions induced by bone morphogenetic protein-2 and transforming growth factor-β in normal human osteoblastic cells. J Biol Chem 277:15514–15522
Stains JP, Civitelli R (2005) Gap junctions regulate extracellular signal-regulated kinase signaling to affect gene transcription. Mol Biol Cell 16:64–72
Thien CB, Dagger SA, Steer JH, Koentgen F, Jansen ES, Scott CL, Langdon WY (2010) c-Cbl promotes TCR-induced thymocyte apoptosis by activating the PI3K/Akt pathway. J Biol Chem 285:10969–10981
Adapala NS, Barbe MF, Langdon WY, Tsygankov AY, Sanjay A (2010) Cbl-phosphatidylinositol 3 kinase interaction differentially regulates macrophage colony-stimulating factor-mediated osteoclast survival and cytoskeletal reorganization. Ann NY Acad Sci 1192:376–384
Wang Z-Q, Ovitt C, Grigoriadis AE, Mohle-Steinlein U, Ruther U, Wagner EF (1992) Bone and haematopoietic defects in mice lacking c-fos. Nature 360:741–745
Soriano P, Montgomery C, Geske R, Bradley A (1991) Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell 64:693–702
Gil-Henn H, Destaing O, Sims NA, Aoki K, Alles N, Neff L, Sanjay A, Bruzzaniti A, De Camilli P, Baron R, Schlessinger J (2007) Defective microtubule-dependent podosome organization in osteoclasts leads to increased bone density in Pyk2−/− mice. J Cell Biol 178:1053–1064
Buckbinder L, Crawford DT, Qi H, Ke HZ, Olson LM, Long KR, Bonnette PC, Baumann AP, Hambor JE, Grasser WA 3rd, Pan LC, Owen TA, Luzzio MJ, Hulford CA, Gebhard DF, Paralkar VM, Simmons HA, Kath JC, Roberts WG, Smock SL, Guzman-Perez A, Brown TA, Li M (2007) Proline-rich tyrosine kinase 2 regulates osteoprogenitor cells and bone formation, and offers an anabolic treatment approach for osteoporosis. Proc Natl Acad Sci USA 104:10619–10624
Marzia M, Sims NA, Voit S, Migliaccio S, Taranta A, Bernardini S, Faraggiana T, Yoneda T, Mundy GR, Boyce BF, Baron R, Teti A (2000) Decreased c-Src expression enhances osteoblast differentiation and bone formation. J Cell Biol 151:311–320
Lee SH, Rho J, Jeong D, Sul JY, Kim T, Kim N, Kang JS, Miyamoto T, Suda T, Lee SK, Pignolo RJ, Koczon-Jaremko B, Lorenzo J, Choi Y (2006) v-ATPase V0 subunit d2-deficient mice exhibit impaired osteoclast fusion and increased bone formation. Nat Med 12:1403–1409
Teitelbaum SL, Ross FP (2003) Genetic regulation of osteoclast development and function. Nat Rev Genet 4:638–649
Zhao C, Irie N, Takada Y, Shimoda K, Miyamoto T, Nishiwaki T, Suda T, Matsuo K (2006) Bidirectional ephrinB2–EphB4 signaling controls bone homeostasis. Cell Metab 4:111–121
Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, Scadden DT, Ma’ayan A, Enikolopov GN, Frenette PS (2010) Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 466:829–834
Winkler IG, Sims NA, Pettit AR, Barbier V, Nowlan B, Helwani F, Poulton IJ, van Rooijen N, Alexander KA, Raggatt LJ, Levesque JP (2010) Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood 116:4815–4828
Nagasawa T, Hirota S, Tachibana K, Takakura N, Nishikawa S, Kitamura Y, Yoshida N, Kikutani H, Kishimoto T (1996) Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature 382:635–638
Yu X, Huang Y, Collin-Osdoby P, Osdoby P (2003) Stromal cell-derived factor-1 (SDF-1) recruits osteoclast precursors by inducing chemotaxis, matrix metalloproteinase-9 (MMP-9) activity, and collagen transmigration. J Bone Miner Res 18:1404–1418
Wright LM, Maloney W, Yu X, Kindle L, Collin-Osdoby P, Osdoby P (2005) Stromal cell-derived factor-1 binding to its chemokine receptor CXCR4 on precursor cells promotes the chemotactic recruitment, development and survival of human osteoclasts. Bone 36:840–853
Zhu W, Boachie-Adjei O, Rawlins BA, Frenkel B, Boskey AL, Ivashkiv LB, Blobel CP (2007) A novel regulatory role for stromal-derived factor-1 signaling in bone morphogenic protein-2 osteogenic differentiation of mesenchymal C2C12 cells. J Biol Chem 282:18676–18685
Zhu W, Liang G, Huang Z, Doty SB, Boskey AL (2011) Conditional inactivation of the CXCR4 receptor in osteoprecursors reduces postnatal bone formation due to impaired osteoblast development. J Biol Chem 286:26794–26805
Okabe S, Tauchi T, Ohyashiki K, Broxmeyer HE (2006) Stromal-cell-derived factor-1/CXCL12-induced chemotaxis of a T cell line involves intracellular signaling through Cbl and Cbl-b and their regulation by Src kinases and CD45. Blood Cells Mol Dis 36:308–314
Sotsios Y, Whittaker GC, Westwick J, Ward SG (1999) The CXC chemokine stromal cell-derived factor activates a Gi-coupled phosphoinositide 3-kinase in T lymphocytes. J Immunol 163:5954–5963
Acknowledgements
This study was supported by National Institutes of Health grant AR055601 (to A. S.). The authors also thank Ms. Mamta Amin for performing the immunohistochemical staining of the bone sections.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
A Sanjay has received funding from the NIH. All other authors have stated no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
223_2011_9531_MOESM1_ESM.tiff
Supplementary Figure 1. Osteoblast survival is not affected due to lack of Cbl-PI3K interaction. A. WT and YF osteoblasts were grown in complete culture medium for 24 h. Cells were then kept in serum-free media for 72 hrs to induce apoptosis. Cell viability was measured by MTT assay as previously described [19]. The graph represents % cell survival. B and C. Cells were treated with LY294002 (50 □M) for 72 hrs to induce cell death. TUNEL assay was performed to identify apoptotic cells. B. Blue is nuclear staining for DAPI and TUNEL positive cells are green. C. Cells that co stained with DAPI and TUNEL were quantified. The graph represents the % of TUNEL positive cells. Comparable numbers of apoptotic cells were seen for untreated and drug-treated WT and YF osteoblasts. The experiments were performed at least three times and a representative experiment is shown. (TIFF 550 kb)
223_2011_9531_MOESM2_ESM.jpg
{kind=link}
Supplementary Figure 2. CblY737F mutation does not affect growth factor induced proliferation or phosphorylation of AKT. A. WT and YF primary osteoblasts were grown for 18 hrs in complete media and then serum starved for 24 hrs. Cells were then treated with 100 ng/mL of indicated growth factor for 5 minutes. Black and white bars indicate WT and YF samples. B. Western blotting was used to determine the levels of Cbl protein, phosphorylated p85 subunit levels, and phosphorylated AKT levels in response to growth factor stimulation for both WT and YF osteoblasts. Levels of phosphorylated proteins were similar between the WT and YF osteoblasts in response to treatment with different growth factors. To quantify changes in the band density, densitometry of the phosphorylated and total protein bands was performed using the ImageJ (NIH, USA). Numbers at the bottom of the bands in each lane indicate the ratio between the phosphorylated and total protein. The experiments were performed at least three times and a representative experiment is shown. (JPG 297 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Brennan, T., Adapala, N.S., Barbe, M.F. et al. Abrogation of Cbl–PI3K Interaction Increases Bone Formation and Osteoblast Proliferation. Calcif Tissue Int 89, 396–410 (2011). https://doi.org/10.1007/s00223-011-9531-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-011-9531-z