
Abstract
In recent years study of rare human bone disorders has led to the identification of important signaling pathways that regulate bone formation. Such diseases include the bone sclerosing dysplasias sclerosteosis and van Buchem disease, which are due to deficiency of sclerostin, a protein secreted by osteocytes that inhibits bone formation by osteoblasts. The restricted expression pattern of sclerostin in the skeleton and the exclusive bone phenotype of good quality of patients with sclerosteosis and van Buchem disease provide the basis for the design of therapeutics that stimulate bone formation. We review here current knowledge of the regulation of the expression and formation of sclerostin, its mechanism of action, and its potential as a bone-building treatment for patients with osteoporosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Osteoporosis is characterized by low bone mass and microarchitectural deterioration of bone tissue with a consequent increase in bone fragility and susceptibility to fractures [1]. The balance between bone resorption and bone formation determines the mass and structural integrity of the skeleton and is disturbed in osteoporosis. Current therapies of osteoporosis, with the exception of parathyroid hormone (PTH), decrease the risk of osteoporotic fractures by reducing bone resorption and preserving its architecture but cannot stimulate bone formation. Elucidating the mechanisms regulating bone formation may lead to the development of therapeutics able to rebuild bone mass and architecture.
In recent years, study of rare human bone disorders and of genetically manipulated animal models has led to the identification of signaling pathways that regulate bone formation, which provide potential targets for the development of novel therapeutics. Fundamental for this progress have been studies of two rare bone sclerosing dysplasias, sclerosteosis and van Buchem disease, that led to the identification of sclerostin, an important negative regulator of bone formation.
Sclerosteosis and van Buchem Disease
Sclerosteosis (OMIM 269500) and van Buchem disease (OMIM 239100) are two rare sclerosing bone disorders, first described in the 1950s as distinct clinical entities, with closely related phenotypes [2]. Sclerosteosis has been mainly diagnosed among Afrikaners of Dutch descent in South Africa, while most patients diagnosed with van Buchem disease come from a small fishing village in The Netherlands. A few individuals and families with sclerosteosis or van Buchem disease have been reported in other parts of the world, including Spain, Brazil, the United States, Germany, Japan, Switzerland, and Senegal [3].
The skeletal manifestations of sclerosteosis and van Buchem disease are the result of endosteal hyperostosis and are characterized by progressive generalized osteosclerosis [3–8]. The manifestations are most pronounced in the mandible and skull, with characteristic enlargement of the jaw and facial bones leading to facial distortion, increased intracranial pressure, and entrapment of cranial nerves, often associated with facial palsy, hearing loss, and/or loss of smell (Fig. 1). Patients with sclerosteosis have a more severe phenotype compared to patients with van Buchem disease and usually have syndactyly. In a limited number of bone biopsies of affected individuals, there is evidence of increased bone formation including predominance of cuboidal active osteoblasts, increased double tetracycline label spacing, and increased osteoid that mineralizes normally, while no consistent pattern of osteoclast numbers or activity has been reported [9–12]. Information about markers of bone turnover in such patients is also limited. Beighton’s group reported elevated serum alkaline phosphatase (AP) activity in the majority of patients with sclerosteosis [13, 14], while Wergedal et al. [15] found significantly higher levels of bone formation (AP, procollagen type 1 amino-terminal propeptide [P1NP], osteocalcin) and resorption (urinary amino-terminal type I collagen telopeptide [NTX]) markers in six patients with van Buchem disease compared to carriers of the disease.

Chronological portraits of a patient with sclerosteosis from the age of 3 years onward. She was born with syndactyly at both hands and developed facial palsy, deafness, facial distortion, and maxillary overgrowth during childhood. By the age of 30, she had developed proptosis and elevated intracranial pressure due to overgrowth of the calvaria. Craniectomy was performed, but she died nevertheless because of elevated intracranial pressure at the age of 54 years (description of this case was previously published by Epstein et al. [13])
The genetic defect that leads to sclerosteosis was identified in a newly cloned gene called SOST, which is located on chromosome 17q12-21 and encodes for the protein sclerostin. Five mutations have so far been identified in patients with sclerosteosis, of which three introduce a premature termination codon and the others interfere with splicing of the gene [16–19]. No mutations within this gene could be found in patients with van Buchem disease, but instead a 52-kb deletion 35 kb downstream of the SOST gene was identified [20, 21]. The deleted region was later found to contain regulatory elements for SOST transcription, explaining its ability to induce a phenotype closely resembling that of patients with sclerosteosis [22]. The different defects of the SOST gene cannot readily explain the differences in clinical phenotypes between the two diseases. It may be that the genomic region deleted in van Buchem disease does not contain regulatory elements required for sclerostin expression during digit formation, which can be the reason for the absence of syndactyly (or other digit malformations) in these patients as opposed to patients with sclerosteosis.
SOST/Sclerostin Expression
SOST mRNA is, especially during embryogenesis, expressed in many tissues, whereas sclerostin protein expression has been reported only postnatally in terminally differentiated cells embedded within a mineralized matrix, i.e., osteocytes, mineralized hypertrophic chondrocytes, and cementocytes [11, 12, 23, 24]. SOST mRNA expression in unmineralized tissues has been detected during mouse embryogenesis in the otic vesicle and peridigital or interdigital regions of the limb buds, of which the latter may be implicated in the pathogenesis of syndactyly in patients with sclerosteosis [25]. In humans, SOST mRNA is expressed in the heart, aorta, and liver, with high levels in the kidney [16, 17, 26, 27]; but no sclerostin protein has been detected in these organs. Accordingly, patients with sclerosteosis or van Buchem disease do not have renal or cardiovascular abnormalities [3].
In adult murine and human bone, sclerostin expression is restricted to osteocytes, with diffuse staining representing dendrites in osteocytic canaliculi [11, 23, 24, 28]. Osteoclasts, osteoblasts, and bone lining cells do not express sclerostin. Due to the current inability to isolate osteocytes from mammalian bone, in vitro studies of SOST/sclerostin expression are technically difficult. Osteogenic cell cultures that form mineralized bone nodules are one of the few available methods for generating osteocyte-like cells in vitro [29]. In mouse primary osteogenic bone marrow and mouse mesenchymal KS483 cell cultures, SOST mRNA expression is induced at low levels after onset of bone nodule mineralization [11, 30]. Similar to the induction of SOST mRNA in vitro, newly embedded osteocytes within unmineralized osteoid in humans in vivo do not express sclerostin but become positive for the protein at, or shortly after, primary mineralization [24]. When mineralization of osteoid is inhibited by administration of the bisphosphonate etidronate in rats, osteocytes within the unmineralized matrix remain immature and do not express sclerostin [31]. However, SOST mRNA is expressed by some osteoblast-like osteosarcoma cell lines [32].
As expected, sclerostin is not expressed by osteocytes in bone biopsies of patients with sclerosteosis [11]. In addition, no sclerostin expression was found in bone biopsies from patients with van Buchem disease, supporting the function of the genomic region deleted in these patients in the regulation of sclerostin expression in bone [12].
Sclerostin Mechanism of Action
In patients with sclerosteosis, the combination of high bone mass due to increased bone formation with premature termination codons in the SOST gene suggested an inhibitory effect of the gene product sclerostin on bone formation. This is supported by the observation that addition of exogenous sclerostin to osteogenic cultures inhibited proliferation and differentiation of mouse and human osteoblastic cells [11, 23, 33]. In addition, sclerostin was shown to decrease the life span of osteoblasts by stimulating their apoptosis [33]. In vivo, overexpression of sclerostin using either the osteocalcin promoter or BAC recombination induced osteopenia in mice [22, 23]. Bone formation in these animals was decreased, while bone resorption was unaffected. Furthermore, analysis of SOST knockout mice showed significant increases in radiodensity, bone mineral density (BMD), cortical and trabecular bone volume, bone formation rate, and bone strength [34]. Together these data support a negative effect of sclerostin on bone formation.
Two processes are responsible for the construction and reconstruction of the skeleton throughout life, bone remodeling and modeling. Bone remodeling enables constant renewal of the skeleton. In this process, bone resorption by osteoclasts and formation by osteoblasts are tightly coupled within a basic multicellular unit (BMU) and bone resorption always precedes bone formation. Sclerostin expression by newly embedded osteocytes at the onset of mineralization of osteoid may serve as a negative feedback signal on osteoblasts to prevent overfilling of the BMU (Fig. 2a) [11, 24]. Data on the effect of sclerostin on osteoclastic bone resorption in humans are scarce and inconsistent, with unaffected, low, or increased bone resorption reported in patients with sclerosteosis and van Buchem disease [9, 10, 15]. In addition, during bone modeling sclerostin may keep bone lining cells in a quiescent state [24] and may thereby prevent activation of osteoblasts and bone formation without previous bone resorption (Fig. 2b) [8]. Sclerostin expression by osteocytes embedded in newly formed bone by modeling may serve a similar negative feedback mechanism on bone formation as in a BMU.

Schematic model of the mechanism of action of sclerostin in bone remodeling and modeling. In remodeling a, sclerostin produced and secreted by newly embedded osteocytes may be transported to the bone surface, where it inhibits osteoblastic bone formation and prevents overfilling of the BMU. In modeling b, sclerostin may serve two actions. First, it may keep bone lining cells in a state of quiescence and prevent, thereby, initiation of de novo bone formation. In addition, sclerostin produced and secreted by newly embedded osteocytes may inhibit osteoblastic bone formation, as in a BMU (reproduced from van Bezooijen et al. [8])
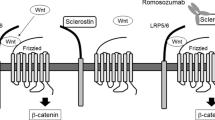
On the basis of its amino acid sequence, which indicates a cystine knot structure, sclerostin was classified as a member of the DAN (differential screening–selected gene aberrant in neuroblastoma) family of glycoproteins [16, 17, 35, 36]. This family consists of a group of secreted proteins that share the ability to antagonize bone morphogenetic protein (BMP) activity. The available data, however, suggest that sclerostin is not a classical BMP antagonist [11]. Some DAN family members have also been reported to antagonize canonical Wnt signaling, among which Wise is the most closely related to sclerostin [37]. Wnts are secreted glycoproteins that bind to seven transmembrane-spanning receptors of the Frizzled family. Stimulation of these receptors causes the intracellular signaling molecule β-catenin to accumulate and translocate into the nucleus, where it initiates transcription of target genes via complex formation with TCF/Lef1 transcription factors. Conversely, in the absence of Wnt, β-catenin forms a complex with the tumor-suppressor proteins APC and Axin and the kinases glycogen synthase kinase 3 (GSK3) and casein kinase I (CK1), which facilitates phosphorylation and proteosomal degradation of β-catenin [38].
The identification of gain-of-function mutations in the first β-propeller of the low-density lipoprotein receptor-related protein LRP5, an essential membrane-bound cofactor in canonical Wnt signaling, in patients with high bone mass (HBM) phenotype [39, 40] and loss-of-function mutations in LRP5 in patients with the osteoporosis pseudoglioma syndrome [41] demonstrated the importance of LRP5-mediated canonical Wnt signaling in regulating bone formation. Sclerostin has been shown to bind LRP5 and its closely related coreceptor LRP6 and, thereby, inhibit the canonical Wnt signaling (Fig. 3) [42–44]. However, although sclerostin binds LRP5/6 to antagonize Wnt signaling, sclerostin and Wnts do not appear to compete for binding of this coreceptor [42]. It may be that sclerostin exerts its effect through binding to a coreceptor and inducing internalization of LRP5/6 as has been shown for Dkk1, another Wnt antagonist. Characterization of the structure of sclerostin showed that it indeed consists of a cystine knot and three loops [45, 46]. One of these loops is high in positively charged residues, showing a possible site of interaction with the predicted binding site on the first β-propeller of LRP5, which is negatively charged. In addition, the binding site of a neutralizing antibody to sclerostin was mapped to this loop, suggesting a functional role of this region in the inhibition of Wnt signaling. A potential binding site for heparin was also found within sclerostin, which may mediate localization of sclerostin at the cell surface of target cells and possibly facilitate inhibition of Wnt signaling.

Schematic model of antagonized canonical Wnt signaling. Canonical Wnt signaling involves the formation of complexes of Wnts with Frizzled receptors and LRP5/6 coreceptors, resulting in the accumulation of β-catenin in the cytoplasm and translocation into the nucleus. The antagonist Dkk1 inhibits canonical Wnt signaling by the formation of complexes with LRP5/6 and Kremen, resulting in the removal of LRP5/6 from the membrane. Dkk1 binds to the first and third β-propellers of LRP5/6. The antagonist sclerostin inhibits canonical Wnt signaling by binding to probably the first β-propeller of LRP5/6. Whether sclerostin requires a cofactor like Kremen for Dkk1 to exert its antagonistic effect remains to be established (reproduced from van Bezooijen et al. [8])
The precise mechanism by which sclerostin secreted by osteocytes inhibits Wnt-mediated bone formation is still unclear. It may be transported to the bone surface via the canaliculi, or it may induce another signal in osteocytes that is transported to osteoblasts to inhibit bone formation. In support of the latter, Wnt signaling has been found in osteocytes [47, 48].
Recently, the role of bone-expressed LRP5 in the regulation of bone formation was questioned since targeted deletion of LRP5 in osteoblasts using the collagen type 1 promoter failed to induce osteopenia and targeted knock-in of LRP5 with an HBM mutation (G171V) using the same promoter did not increase bone mass in mice [49]. It was shown that LRP5-mediated signaling in the duodenum inhibited the expression of Tph1, the rate-limiting enzyme for serotonin production outside the brain, and thereby decreased serum levels of serotonin. Conversely, LRP5 knockout mice that have low bone mass had high serum serotonin levels. In addition, reduction of these elevated serotonin levels by administration of parachlorophenylalanine or a low-tryptophan diet normalized bone-formation parameters and bone mass. These findings suggest a new mechanism for the effect of LRP5-mediated signaling on bone mass, the importance of which in human physiology is currently unknown.
Regulation of SOST/Sclerostin Expression
Due to their location and morphology, osteocytes have been long implicated in mechanosensing and initiation of the bone anabolic response to mechanical load [50, 51]. In support of this, specific ablation of osteocytes in mice resulted in fragile bone, and these mice did not respond with bone loss to unloading [52]. Wnt signaling may play an important role in the anabolic response to deformation and loading since increased Wnt signaling has been found after loading of osteoblastic cells in vitro and of tibiae in vivo [47, 53, 54] and the Wnt coreceptor LRP5 was found to be essential for the increase in bone mass after loading [55]. Since sclerostin is produced by osteocytes in bone and inhibits bone formation by antagonizing canonical Wnt signaling, it may play a role in regulating Wnt signaling in response to mechanical loading. Consistent with this hypothesis, loading decreased SOST mRNA and sclerostin levels, while unloading increased SOST mRNA expression in vivo (Fig. 4) [54, 56, 57]. Interestingly, reduction of sclerostin staining intensity was most pronounced in areas with the highest strain, indicating a response to local loading conditions. Furthermore, SOST knockout mice do not exhibit bone loss after unloading nor is canonical Wnt signaling altered [54].

Schematic model for the regulation of the control of bone formation by sclerostin. Sclerostin may exert its inhibitory effect on bone formation by preventing the activation of lining cells as well as the inactivation of active osteoblasts. Glucocorticoids stimulate sclerostin expression and, thereby, inhibit bone formation, whereas intermittent PTH and loading inhibit sclerostin expression in osteocytes and, thereby, stimulate bone formation
Several systemic and local factors have been suggested as possible regulators of SOST/sclerostin expression by osteocytes. Recombinant human PTH and active fragments of this protein are used in the treatment of osteoporosis [58]. In contrast to the bone resorption–stimulating effect of continuous elevation of endogenous PTH, as is seen in patients with hyperparathyroidism, intermittent increases of PTH provided by daily injections are associated with distinct anabolic effects. The mechanisms by which PTH mediates this bone anabolic effect are not completely understood. Part of it may be mediated via sclerostin as PTH has been shown to inhibit its expression both in vitro and in vivo (Fig. 4). In vitro, PTH decreased SOST transcription by osteoblastic and osteocytic cells within 4 h. This was not affected by the protein synthesis inhibitor cyclohexamide but was decreased by the cAMP inducer forskolin [28, 32]. These observations suggest a direct and cAMP-dependent regulatory effect of PTH on the expression of SOST. Within the 52-kb genomic region deleted in van Buchem disease, an MEF2 response element has been identified that is essential for the PTH-induced downregulation of SOST expression [59]. In vivo, PTH administration resulted in a decrease in SOST mRNA and sclerostin expression in mice and rats [28, 32, 59, 60]. In addition, a constitutively active PTH receptor 1 (caPTHR1) exclusively expressed in osteocytes resulted in increased remodeling with decreased osteoblast apoptosis and suppression of SOST expression [61]. This effect was blunted in mice lacking LRP5, suggesting that the effect of caPTHR1 was mediated by increased Wnt signaling due to suppression of SOST. The importance of SOST regulation by PTH is further supported by the observations that the anabolic effect of PTH is blunted in SOST-deficient mice as well as in mice overexpressing SOST using a constitutive active promoter [62].
Two other systemic factors have also been shown to affect SOST/sclerostin expression. 1,25-Dihydroxyvitamin D3 alone or in combination with retinoic acid increased SOST expression in human osteoblastic cells in vitro [30, 63]. The specific effect of glucocorticoids on SOST expression depends on the experimental conditions. In vitro, dexamethasone suppressed SOST expression in osteoblastic cells [30], while in vivo treatment of mice with prednisolone increased SOST expression in tibiae, suggesting that suppression of Wnt signaling by the upregulation of sclerostin may account for the glucocorticoid-induced suppression in bone formation (Fig. 4) [64].
BMP2, -4, and -6 are local growth factors shown to stimulate SOST expression in osteoblastic cells in vitro [25, 30]; and putative BMP responsive elements are present within the SOST promoter region [65]. Decreased BMP signaling due to osteoblast-specific knockout of BMPR1A decreased SOST mRNA and sclerostin protein expression in embryonic mice calvariae and was associated with increased bone mass [65]. In these mice, however, both bone formation and resorption were inhibited. The authors proposed that the decrease in bone formation was independent of sclerostin expression and a direct result of decreased BMP signaling. The decrease in bone resorption, however, may be an effect of increased Wnt signaling due to the decrease in sclerostin expression. This in turn may be due to upregulation of osteoprotegerin in mature osteoblasts by Wnts and, thereby, inhibition of RANKL-induced osteoclastogenesis [66].
Despite the rapid progress in our understanding of the regulation of the production and function of sclerostin, there are still important questions that need to be addressed in future research. These include the identification of factors that regulate sclerostin/SOST expression and determine its highly restricted expression pattern. Furthermore, the mechanism by which sclerostin binding to LRP5/6 interferes with canonical Wnt signaling as well as potential additional functions of sclerostin, besides antagonizing canonical Wnt signaling, need to be elucidated. More detailed and structured analysis of bone metabolism in patients with sclerosteosis and van Buchem disease, sclerostin expression in pathological conditions, and a genotype–phenotype characterization of SOST are required to better understand its function and regulation in humans.
Therapeutic Potential
The identification of sclerostin deficiency as the cause of sclerosteosis and van Buchem disease and the progress in our understanding of the action of sclerostin on bone formation have opened a new area in bone therapeutics. The restricted expression pattern of sclerostin and the exclusive bone phenotype of good quality of patients with sclerosteosis and van Buchem disease provide the basis for the design of therapeutics that specifically stimulate bone formation, an action relevant to the treatment of osteoporosis. As sclerostin is a secreted protein, one approach to achieve this is to develop antibodies capable of inhibiting the biological activity of sclerostin, mimicking, thus, its absence in sclerosteosis. Such antibodies have already been shown to increase BMD, bone volume, and bone strength in ovariectomized rats [67] and primates [68] and to reverse bone loss in a model of colitis [69]. Preliminary data from a blinded, placebo-controlled, dose-escalating single-dose study of 48 healthy postmenopausal women demonstrated that a single injection of a monoclonal antibody against sclerostin markedly increased bone-formation markers and BMD and was well tolerated [70]. Serum P1NP levels reached a peak 14–25 days after the antibody administration and progressively returned to baseline values after about 2 months. In contrast, the bone-resorption marker serum CTx decreased to a minimum about 14 days after the antibody injection and returned to baseline values after about 2 months. Clinical phase II studies are currently under way. Other approaches to inhibit sclerostin production or activity are also feasible. However, given the availability of efficacious treatments, any novel treatment for osteoporosis should not only be effective but also devoid of adverse effects. The absence of any extraskeletal complications of patients with sclerosteosis and van Buchem disease is reassuring. Furthermore, the finding of consistently higher BMD values in carriers of sclerosteosis with no skeletal complications [6] suggests that sclerostin inhibition can be titrated and can lead to the desired outcome without any side effects, but safety margins need to be determined. However, there have been concerns that stimulation of bone formation by increasing Wnt signaling may lead to unwanted skeletal effects [71, 72]. The Wnt inhibitor factor 1 (WIF1), for example, has been identified as a candidate tumor-suppressor gene in human osteosarcoma, suggesting that the susceptibility to osteosarcoma may be increased in patients receiving novel anabolic treatments targeting Wnt antagonists [73]. This is another issue that needs to be further investigated.
Study of the molecular defects of rare bone disorders such as sclerosteosis and van Buchem disease can, thus, lead to the development of new bone-forming agents, allowing us to tailor pharmacotherapy to the needs of the individual patient with osteoporosis. In addition, it may help in the management of the small group of patients with sclerosteosis or van Buchem disease, for whom the only available treatment is the technically difficult and often risky removal of excess bone tissue from the skull.
References
National Osteoporosis Foundation (2008) Facts on osteoporosis. http://www.nof.org/osteoporosis/diseasefacts.htm
van Buchem FS, Hadders HN, Ubbens R (1955) An uncommon familial systemic disease of the skeleton: hyperostosis corticalis generalisata familiaris. Acta Radiol 44:109–120
Hamersma H, Gardner J, Beighton P (2003) The natural history of sclerosteosis. Clin Genet 63:192–197
Beighton P, Barnard A, Hamersma H, van der Wouden A (1984) The syndromic status of sclerosteosis and van Buchem disease. Clin Genet 25:175–181
Beighton P (1988) Sclerosteosis. J Med Genet 25:200–203
Gardner JC, van Bezooijen RL, Mervis B, Hamdy NAT, Löwik CWGM, Hamersma H, Beighton P, Papapoulos SE (2005) Bone mineral density in sclerosteosis; affected individuals and gene carriers. J Clin Endocrinol Metab 90:6392–6395
van Bezooijen RL, ten Dijke P, Papapoulos SE, Löwik CWGM (2005) SOST/sclerostin, an osteocyte-derived negative regulator of bone formation. Cytokine Growth Factor Rev 16:319–327
van Bezooijen RL, Papapoulos SE, Hamdy NAT, Lowik CWGM (2008) SOST/sclerostin; an osteocyte-derived inhibitor of bone formation that antagonizes canonical Wnt signaling. In: Raisz LG, Martin TJ, Bilezikian JP (eds) Principles of bone biology. Academic Press, New York, pp 139–152
Hill SC, Stein SA, Dwyer A, Altman J, Dorwart R, Doppman J (1986) Cranial CT findings in sclerosteosis. AJNR Am J Neuroradiol 7:505–511
Stein SA, Witkop C, Hill SC, Fallon MD, Viernstein L, Gucer G, McKeever P, Long D, Altman J, Miller NR, Teitelbaum SL, Schlesinger S (1983) Sclerosteosis: neurogenetic and pathophysiologic analysis of an American kinship. Neurology 33:267–277
van Bezooijen RL, Roelen BA, Visser A, van der Wee-Pals L, de Wilt E, Karperien M, Hamersma H, Papapoulos SE, ten Dijke P, Löwik CWGM (2004) Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med 199:805–814
van Bezooijen RL, Bronckers AL, Gortzak RA, Hogendoorn PC, van der Wee-Pals L, Balemans W, Oostenbroek HJ, van Hul W, Hamersma H, Dikkers FG, Hamdy NAT, Papapoulos SE, Löwik CWGM (2009) Sclerostin in mineralized matrices and van Buchem disease. J Dent Res 88:569–574
Epstein S, Hamersma H, Beighton P (1979) Endocrine function in sclerosteosis. S Afr Med J 55:1105–1110
Beighton P, Durr L, Hamersma H (1976) The clinical features of sclerosteosis. A review of the manifestations in twenty-five affected individuals. Ann Intern Med 84:393–397
Wergedal JE, Veskovic K, Hellan M, Nyght C, Balemans W, Libanati C, Vanhoenacker FM, Tan J, Baylink DJ, van Hul W (2003) Patients with van Buchem disease, an osteosclerotic genetic disease, have elevated bone formation markers, higher bone density, and greater derived polar moment of inertia than normal. J Clin Endocrinol Metab 88:5778–5783
Balemans W, Ebeling M, Patel N, van Hul E, Olson P, Dioszegi M, Lacza C, Wuyts W, van den Ende J, Willems P, Paes-Alves AF, Hill SC, Bueno M, Ramos FJ, Tacconi P, Dikkers FG, Stratakis C, Lindpaintner K, Vickery B, Foernzler D, van Hul W (2001) Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet 10:537–543
Brunkow ME, Gardner JC, van Ness J, Paeper BW, Kovacevich BR, Proll S, Skonier JE, Zhao L, Sabo PJ, Fu Y, Alisch RS, Gillett L, Colbert T, Tacconi P, Galas D, Hamersma H, Beighton P, Mulligan J (2001) Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet 68:577–589
Balemans W, Cleiren E, Siebers U, Horst J, van Hul W (2005) A generalized skeletal hyperostosis in two siblings caused by a novel mutation in the SOST gene. Bone 36:943–947
Kim CA, Honjo R, Bertola D, Albano L, Oliveira L, Jales S, Siqueira J, Castilho A, Balemans W, Piters E, Jennes K, van Hul W (2008) A known SOST gene mutation causes sclerosteosis in a familial and an isolated case from Brazilian origin. Genet Test 12:475–479
Balemans W, Patel N, Ebeling M, van Hul E, Wuyts W, Lacza C, Dioszegi M, Dikkers FG, Hildering P, Willems PJ, Verheij JB, Lindpaintner K, Vickery B, Foernzler D, van Hul W (2002) Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet 39:91–97
Staehling-Hampton K, Proll S, Paeper BW, Zhao L, Charmley P, Brown A, Gardner JC, Galas D, Schatzman RC, Beighton P, Papapoulos SE, Hamersma H, Brunkow ME (2002) A 52-kb deletion in the SOST-MEOX1 intergenic region on 17q12–q21 is associated with van Buchem disease in the Dutch population. Am J Med Genet 110:144–152
Loots GG, Kneissel M, Keller H, Baptist M, Chang J, Collette NM, Ovcharenko D, Plajzer-Frick I, Rubin EM (2005) Genomic deletion of a long-range bone enhancer misregulates sclerostin in van Buchem disease. Genome Res 15:928–935
Winkler DG, Sutherland MK, Geoghegan JC, Yu C, Hayes T, Skonier JE, Shpektor D, Jonas M, Kovacevich BR, Staehling-Hampton K, Appleby M, Brunkow ME, Latham JA (2003) Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J 22:6267–6276
Poole KE, van Bezooijen RL, Loveridge N, Hamersma H, Papapoulos SE, Löwik CWGM, Reeve J (2005) Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J 19:1842–1844
Ohyama Y, Nifuji A, Maeda Y, Amagasa T, Noda M (2004) Spaciotemporal association and bone morphogenetic protein regulation of sclerostin and osterix expression during embryonic osteogenesis. Endocrinology 145:4685–4692
Balemans W, van Hul W (2002) Extracellular regulation of BMP signaling in vertebrates: a cocktail of modulators. Dev Biol 250:231–250
Kusu N, Laurikkala J, Imanishi M, Usui H, Konishi M, Miyake A, Thesleff I, Itoh N (2003) Sclerostin is a novel secreted osteoclast-derived bone morphogenetic protein antagonist with unique ligand specificity. J Biol Chem 278:24113–24117
Bellido T, Ali AA, Gubrij I, Plotkin LI, Fu Q, O’Brien CA, Manolagas SC, Jilka RL (2005) Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology 146:4577–4583
Pockwinse SM, Wilming LG, Conlon DM, Stein GS, Lian JB (1992) Expression of cell growth and bone specific genes at single cell resolution during development of bone tissue-like organization in primary osteoblast cultures. J Cell Biochem 49:310–323
Sutherland MK, Geoghegan JC, Yu C, Winkler DG, Latham JA (2004) Unique regulation of SOST, the sclerosteosis gene, by BMPs and steroid hormones in human osteoblasts. Bone 35:448–454
Irie K, Ejiri S, Sakakura Y, Shibui T, Yajima T (2008) Matrix mineralization as a trigger for osteocyte maturation. J Histochem Cytochem 56:561–567
Keller H, Kneissel M (2005) SOST is a target gene for PTH in bone. Bone 37:148–158
Sutherland MK, Geoghegan JC, Yu C, Turcott E, Skonier JE, Winkler DG, Latham JA (2004) Sclerostin promotes the apoptosis of human osteoblastic cells: a novel regulation of bone formation. Bone 35:828–835
Li X, Ominsky MS, Niu QT, Sun N, Daugherty B, D’Agostin D, Kurahara C, Gao Y, Cao J, Gong J, Asuncion F, Barrero M, Warmington K, Dwyer D, Stolina M, Morony S, Sarosi I, Kostenuik PJ, Lacey DL, Simonet WS, Ke HZ, Paszty C (2008) Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res 23:860–869
Avsian-Kretchmer O, Hsueh AJ (2004) Comparative genomic analysis of the eight-membered ring cystine knot-containing bone morphogenetic protein antagonists. Mol Endocrinol 18:1–12
van Bezooijen RL, Papapoulos SE, Löwik CWGM (2005) Bone morphogenetic proteins and their antagonists: the sclerostin paradigm. J Endocrinol Invest 28:15–17
Ellies DL, Viviano B, McCarthy J, Rey JP, Itasaki N, Saunders S, Krumlauf R (2006) Bone density ligand, sclerostin, directly interacts with LRP5 but not LRP5G171V to modulate Wnt activity. J Bone Miner Res 21:1738–1749
Clevers H (2006) Wnt/beta-catenin signaling in development and disease. Cell 127:469–480
Boyden LM, Mao J, Belsky J, Mitzner L, Farhi A, Mitnick MA, Wu D, Insogna KL, Lifton RP (2002) High bone density due to a mutation in LDL-receptor-related protein 5. N Engl J Med 346:1513–1521
Little RD, Carulli JP, Del Mastro RG, Dupuis J, Osborne M, Folz C, Manning SP, Swain PM, Zhao SC, Eustace B, Lappe MM, Spitzer L, Zweier S, Braunschweiger K, Benchekroun Y, Hu X, Adair R, Chee L, FitzGerald MG, Tulig C, Caruso A, Tzellas N, Bawa A, Franklin B, McGuire S, Nogues X, Gong G, Allen KM, Anisowicz A, Morales AJ, Lomedico PT, Recker SM, van Eerdewegh P, Recker RR, Johnson ML (2002) A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am J Hum Genet 70:11–19
Gong Y, Slee RB, Fukai N, Rawadi G, Roman-Roman S, Reginato AM, Wang H, Cundy T, Glorieux FH, Lev D, Zacharin M, Oexle K, Marcelino J, Suwairi W, Heeger S, Sabatakos G, Apte S, Adkins WN, Allgrove J, Arslan-Kirchner M, Batch JA, Beighton P, Black GC, Boles RG, Boon LM, Borrone C, Brunner HG, Carle GF, Dallapiccola B, de Paepe A, Floege B, Halfhide ML, Hall B, Hennekam RC, Hirose T, Jans A, Juppner H, Kim CA, Keppler-Noreuil K, Kohlschuetter A, LaCombe D, Lambert M, Lemyre E, Letteboer T, Peltonen L, Ramesar RS, Romanengo M, Somer H, Steichen-Gersdorf E, Steinmann B, Sullivan B, Superti-Furga A, Swoboda W, van den Boogaard MJ, van Hul W, Vikkula M, Votruba M, Zabel B, Garcia T, Baron R, Olsen BR, Warman ML (2001) LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 107:513–523
Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang J, Harris SE, Wu D (2005) Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem 280:19883–19887
Semenov M, Tamai K, He X (2005) SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J Biol Chem 280:26770–26775
van Bezooijen RL, Svensson JP, Eefting D, Visser A, van der Horst G, Karperien M, Quax PH, Vrieling H, Papapoulos SE, ten Dijke P, Löwik CWGM (2007) Wnt but not BMP signaling is involved in the inhibitory action of sclerostin on BMP-stimulated bone formation. J Bone Miner Res 22:19–28
Veverka V, Henry AJ, Slocombe PM, Ventom A, Mulloy B, Muskett FW, Muzylak M, Greenslade K, Moore AR, Zhang L, Gong J, Qian X, Paszty C, Taylor RJ, Robinson MK, Carr MD (2009) Characterization of the structural features and interactions of sclerostin: molecular insight into a key regulator of Wnt-mediated bone formation. J Biol Chem 284:10890–10900
Weidauer SE, Schmieder P, Beerbaum M, Schmitz W, Oschkinat H, Mueller TD (2009) NMR structure of the Wnt modulator protein Sclerostin. Biochem Biophys Res Commun 380:160–165
Hens JR, Wilson KM, Dann P, Chen X, Horowitz MC, Wysolmerski JJ (2005) TOPGAL mice show that the canonical Wnt signaling pathway is active during bone development and growth and is activated by mechanical loading in vitro. J Bone Miner Res 20:1103–1113
Bonewald LF, Johnson ML (2008) Osteocytes, mechanosensing and Wnt signaling. Bone 42:606–615
Yadav VK, Ryu JH, Suda N, Tanaka KF, Gingrich JA, Schutz G, Glorieux FH, Chiang CY, Zajac JD, Insogna KL, Mann JJ, Hen R, Ducy P, Karsenty G (2008) Lrp5 controls bone formation by inhibiting serotonin synthesis in the duodenum. Cell 135:825–837
Knothe Tate ML, Adamson JR, Tami AE, Bauer TW (2004) The osteocyte. Int J Biochem Cell Biol 36:1–8
Han Y, Cowin SC, Schaffler MB, Weinbaum S (2004) Mechanotransduction and strain amplification in osteocyte cell processes. Proc Natl Acad Sci USA 101:16689–16694
Tatsumi S, Ishii K, Amizuka N, Li M, Kobayashi T, Kohno K, Ito M, Takeshita S, Ikeda K (2007) Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab 5:464–475
Robinson JA, Chatterjee-Kishore M, Yaworsky PJ, Cullen DM, Zhao W, Li C, Kharode Y, Sauter L, Babij P, Brown EL, Hill AA, Akhter MP, Johnson ML, Recker RR, Komm BS, Bex FJ (2006) Wnt/beta-catenin signaling is a normal physiological response to mechanical loading in bone. J Biol Chem 281:31720–31728
Lin C, Jiang X, Dai Z, Guo X, Weng T, Wang J, Li Y, Feng G, Gao X, He L (2009) Sclerostin mediates bone response to mechanical unloading via antagonizing Wnt/beta-catenin signaling. J Bone Miner Res 24:1651–1661
Sawakami K, Robling AG, Ai M, Pitner ND, Liu D, Warden SJ, Li J, Maye P, Rowe DW, Duncan RL, Warman ML, Turner CH (2006) The Wnt co-receptor LRP5 is essential for skeletal mechanotransduction but not for the anabolic bone response to parathyroid hormone treatment. J Biol Chem 281:23698–23711
Robling AG, Niziolek PJ, Baldridge LA, Condon KW, Allen MR, Alam I, Mantila SM, Gluhak-Heinrich J, Bellido TM, Harris SE, Turner CH (2008) Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem 283:5866–5875
Moustafa A, Sugiyama T, Saxon LK, Zaman G, Sunters A, Armstrong VJ, Javaheri B, Lanyon LE, Price JS (2009) The mouse fibula as a suitable bone for the study of functional adaptation to mechanical loading. Bone 44:930–935
Pleiner-Duxneuner J, Zwettler E, Paschalis E, Roschger P, Nell-Duxneuner V, Klaushofer K (2009) Treatment of osteoporosis with parathyroid hormone and teriparatide. Calcif Tissue Int 84:159–170
Leupin O, Kramer I, Collette NM, Loots GG, Natt F, Kneissel M, Keller H (2007) Control of the SOST bone enhancer by PTH using MEF2 transcription factors. J Bone Miner Res 22:1957–1967
Silvestrini G, Ballanti P, Leopizzi M, Sebastiani M, Berni S, di Vito M, Bonucci E (2007) Effects of intermittent parathyroid hormone (PTH) administration on SOST mRNA and protein in rat bone. J Mol Histol 38:261–269
O’Brien CA, Plotkin LI, Galli C, Goellner JJ, Gortazar AR, Allen MR, Robling AG, Bouxsein M, Schipani E, Turner CH, Jilka RL, Weinstein RS, Manolagas SC, Bellido T (2008) Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS One 3:e2942
Kramer I, Loots GG, Studer A, Keller H, Kneissel M (2009) Parathyroid hormone (PTH) induced bone gain is blunted in SOST overexpressing and deficient mice. J Bone Miner Res 25:178–189
Sevetson B, Taylor S, Pan Y (2004) Cbfa1/RUNX2 directs specific expression of the sclerosteosis gene (SOST). J Biol Chem 279:13849–13858
Yao W, Cheng Z, Pham A, Busse C, Zimmermann EA, Ritchie RO, Lane NE (2008) Glucocorticoid-induced bone loss in mice can be reversed by the actions of parathyroid hormone and risedronate on different pathways for bone formation and mineralization. Arthritis Rheum 58:3485–3497
Kamiya N, Ye L, Kobayashi T, Mochida Y, Yamauchi M, Kronenberg HM, Feng JQ, Mishina Y (2008) BMP signaling negatively regulates bone mass through sclerostin by inhibiting the canonical Wnt pathway. Development 135:3801–3811
Goldring SR, Goldring MB (2007) Eating bone or adding it: the Wnt pathway decides. Nat Med 13:133–134
Li X, Ominsky MS, Warmington KS, Morony S, Gong J, Cao J, Gao Y, Shalhoub V, Tipton B, Haldankar R, Chen Q, Winters A, Boone T, Geng Z, Niu QT, Ke HZ, Kostenuik PJ, Simonet WS, Lacey DL, Paszty C (2009) Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis. J Bone Miner Res 24:578–588
Ominsky M, Stouch B, Doellgast G, Gong J, Cao J, Gao Y, Tipton B, Haldankar R, Winters A, Chen Q, Graham K, Zhou L, Hale M, Henry A, Lightwood D, Moore A, Popplewell A, Robinson M, Vlasseros F, Jolette J, Smith SY, Kostenuik PJ, Simonet WS, Lacey DL, Paszty C (2006) Administration of sclerostin monoclonal antibodies to female cynomolgus monkeys results in increased bone formation, bone mineral density and bone strength. Read at the Annual Meeting of the American Society for Bone and Mineral Research, 2006 Sept 18, Philadelphia, PA
Eddleston A, Marenzana M, Moore AR, Stephens P, Muzylak M, Marshall D, Robinson MK (2009) A short treatment with an antibody to sclerostin can inhibit bone loss in an ongoing model of colitis. J Bone Miner Res 24:1662–1671
Padhi D, Stouch B, Jang G, Fang L, Darling M, Glise H, Robinson M, Harris SE, Posvar E (2007) Anti-sclerostin antibody increases markers of bone formation in healthy postmenopausal women. J Bone Miner Res 21 Suppl 1:S37
Whyte MP, Reinus WH, Mumm S (2004) High-bone-mass disease and LRP5. N Engl J Med 350:2096–2099
Rickels MR, Zhang X, Mumm S, Whyte MP (2005) Oropharyngeal skeletal disease accompanying high bone mass and novel LRP5 mutation. J Bone Miner Res 20:878–885
Kansara M, Tsang M, Kodjabachian L, Sims NA, Trivett MK, Ehrich M, Dobrovic A, Slavin J, Choong PF, Simmons PJ, Dawid IB, Thomas DM (2009) Wnt inhibitory factor 1 is epigenetically silenced in human osteosarcoma, and targeted disruption accelerates osteosarcomagenesis in mice. J Clin Invest 119:837–851
Acknowledgements
The authors thank Dr. H. Hamersma for providing clinical information and pictures of the patient with sclerosteosis presented in Fig. 1. This work was supported by grants from the European Commission (HEALTH-F2-2008-201099, TALOS) and Agentschap NL/IOP Genomics (IGE07001A).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
The authors have stated that they have no conflict of interest.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Moester, M.J.C., Papapoulos, S.E., Löwik, C.W.G.M. et al. Sclerostin: Current Knowledge and Future Perspectives. Calcif Tissue Int 87, 99–107 (2010). https://doi.org/10.1007/s00223-010-9372-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-010-9372-1