
Abstract
The Maillard reaction is traditionally subdivided into three stages that start consecutively and run in parallel. Here, we show that N-ε-carboxymethyllysine (CML), a compound formed in the late stage of the reaction, can undergo a second glycation event at its secondary amino group leading to a new class of Amadori rearrangement products. When N-α-hippuryl-CML was incubated in the presence of reducing sugars such as glucose, galactose, ribose, xylose, maltose, or lactose in solution for 1 h at 75 °C, the compound was degraded by 6–21%, and N-ε-carboxymethyl-N-ε-deoxyketosyl lysine derivatives were formed. Under the same conditions, lysine was 5–10 times more reactive than CML. N-α-hippuryl-N-ε-carboxymethyl-N-ε-(1-deoxyfructosyl)-l-lysine (hippuryl-CMFL) and N-ε-carboxymethyl-N-ε-(1-deoxyfructosyl)-l-lysine (CMFL) were synthesized, isolated and characterized by MS/MS and NMR experiments. Depending on the reaction conditions, up to 21% of CMFL can be converted to the furosine analogue N-ε-carboxymethyl-N-ε-furoylmethyl-l-lysine (CM-Fur) during standard acid protein hydrolysis with hydrochloric acid. Incubation of bovine serum albumin (BSA) with glucose for up to 9 weeks at 37 °C revealed the formation of CMFL in the protein as assessed by HPLC–MS/MS in the MRM mode. Under these conditions, ca. 13% of lysine residues had been converted to fructosyllysine, and 0.03% had been converted to CMFL. The detection of glycation products of glycated amino acids (heterogeneous multiple glycation) reveals a novel pathway in the Maillard reaction.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
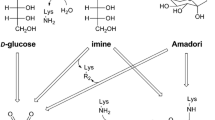
The Maillard reaction (glycation) was first described in 1912 as a complex reaction between amino acids and reducing sugars [1]. Since the classification of Hodge in 1953 [2], it has been generally accepted that individual reactions occur in parallel, but that the whole reaction develops in a linear fashion: Amadori rearrangement products (ARPs) are generated in the first stage of the reaction. They are degraded to 1,2-dicarbonyl compounds in the second stage of the reaction. In the late stage, dicarbonyl compounds react with nucleophilic side-chains and the N-termini of proteins, peptides and amino acids to stable glycation compounds sometimes also called “advanced glycation end products (AGEs)” [3,4,5]. One of these glycation compounds, N-ε-carboxymethyllysine (CML), can be formed by different pathways in food either by oxidative routes from the Schiff bases or from ARPs (routes A, B in Fig. 1) [6,7,8,9,10] or by reaction of the ε-amino group of lysine with glyoxal in an intramolecular Cannizzaro reaction (route C in Fig. 1), where glyoxal can originate from both glycation or lipid peroxidation reactions [8, 11]. In food, CML is analyzed mainly by liquid chromatography coupled to mass spectrometry [12,13,14]. Moderate CML concentrations were quantitated in milk (0.2–2.6 mg/kg) and in bread crumb (2.1–4.5 mg/kg) whereas boiled and fried meat products (1.1–28.2 mg/kg), evaporated milk (4.7–46.2 mg/kg) and bread crust (37.1–46.1 mg/kg) showed considerably higher values [12,13,14,15]. The daily intake of CML ranges between 2.1 and 11.3 mg [16,17,18]. The main issue in the analysis of CML is its possible formation as an artifact during acid hydrolysis of ARPs whose concentrations strongly exceed those of CML. Generally, acid hydrolysis of food samples is, therefore, preceded by reduction of ARPs with NaBH4 [12, 19]. The resulting glucitol amines can no longer react to CML. Even though a plethora of scientific articles describe adverse effects of Maillard reaction products—especially CML (for reviews, see [3] and [20])—no structure–activity relationship between dietary CML and adverse effects has been established yet [21].

Reactions leading to the formation and degradation of N-ε-carboxymethyllysine (CML). A Oxidation of the Schiff base, B oxidation of the Amadori product, C reaction of glyoxal with the ε-amino group of lysine, D bacterial degradation of CML by E. coli, E postulated reaction of the secondary amino group of CML with epicatechin
Glycated amino acids such as CML are not inert molecules, because these compounds can be degraded by microbes [22, 23]. Carboxymethylcadaverine (CM-CAD), a product resulting from degradation of CML by E. coli, is shown in Fig. 1 (route D). Moreover, the view of “advanced glycation end products” (AGEs) as being the final products of the Maillard reaction is more and more put into question. CML is formed during the baking process, but it is also degraded during this process [24]. The compound was postulated to be thermodynamically unstable, as it may also degrade in glycation reactions of sugar-casein mixtures [25]. A certain reactivity of CML in the presence of phenolic compounds such as epicatechin was suggested (route E in Fig. 1), but no reaction products have been isolated yet [26]. Moreover, it was not unequivocally shown that the ε-amino group of CML actually reacts, because CML was used as the free amino acid in that study with a free primary amino group that may react as well. To sum up, the reactivity of the secondary amino group of CML has not gained much attention yet.
In the present work, we show that a glycated functional group, namely the secondary amino group of CML, can be subject to a second glycation event. This secondary amino group is glycated with different reducing sugars. Moreover, the view of the Maillard reaction as a reaction with an inherent linearity is challenged by the preparation and detection in model systems of Amadori rearrangement products of CML. While homogeneous multiple glycation has been reported earlier, e.g., for difructosyl amino acids [27, 28], this is the first report on heterogeneous multiple glycation.
Materials and methods
Chemicals
Furosine [29] and N-α-benzoylglycyl-N-ε-fructosyllysine (N-α-hippuryl fructosyllysine) [30, 31] were synthesized according to the literature stated. Other chemicals were obtained from commercial suppliers: Ninhydrin, d-xylose, maltose monohydrate, d-mannose (Merck, Darmstadt, Germany). Acetic acid, N-α-Boc-lysine (Alfa Aesar, Karlsruhe, Germany). Benzoylglycyl-l-lysine (Bachem, Bubendorf, Switzerland). Glyoxylic acid monohydrate, bovine serum albumin, lactose (Fluka, Buchs, Switzerland). Palladium on activated charcoal, nonafluoropentanoic acid (NFPA), deuterium oxide, d-fructose, d-ribose (Sigma-Aldrich, Steinheim, Germany). DOWEX 50W-X8 ion exchange resin (200–400 mesh), silica gel (Acros, Geel, Belgium). d-Glucose (Roth, Karlsruhe, Germany). Lewatit S100 ion exchange resin (Bayer, Leverkusen, Germany). o-Dianisidine (Lancaster Synthesis, Morecambe, UK). d-Galactose (Serva, Heidelberg, Germany). Water for buffers and HPLC eluents was prepared by a Bi 18 E double distillation system (QCS, Maintal, Germany).
Synthesis of N-α-hippuryl-N-ε-carboxymethyl-l-lysine (N-α-hippuryl-CML)
The compound was synthesized based on previous protocols [31, 32]. Benzoylglycyllysine (461.0 mg, 1.5 mmol) and glyoxylic acid monohydrate (166.4 mg, 1.81 mmol) were dissolved in 30 mL of double-distilled water, and the pH was adjusted to 8.75 with 1 M sodium hydroxide. Then, 50 mg of palladium on charcoal was added, and the mixture was incubated under a hydrogen atmosphere overnight. The catalyst was filtered off, and the clear filtrate was evaporated to dryness, drawn on silica gel and subjected to column chromatography on 40 g of silica gel using the solvents methanol/ethyl acetate 20/80 (350 mL), methanol/ethyl acetate 40/60 (250 mL), methanol/ethyl acetate 50/50 (250 mL), methanol/ethyl acetate 60/40 (350 mL), and methanol/ethyl acetate 70/30 (150 mL). Spots were detected after chlorination using a modified Reindel Hoppe reagent [22]. The target compound was found to elute with the last two solvents. The fractions were pooled, evaporated to dryness and then lyophilized yielding 549.8 mg of the target compound as a white foam, which was stored at − 18 °C.
Analytical data: HPLC–UV–MS/MS (gradient 1): tR, 7.7 min; [M + H]+ (m/z 366), UV, λmax 219 nm. Content = 82.8%, based on comparison with an authentic standard [31]. Yield: 549.6 mg (83%).
Synthesis of N-α-hippuryl-N-ε-carboxymethyl-N-ε-(1-deoxyfructosyl)-l-lysine (N-α-hippuryl-CMFL)
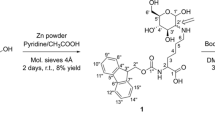
N-α-hippuryl-CML (21.5 mg, 0.05 mmol) and glucose (90.0 mg, 0.5 mmol) were dissolved in 41.5 mL methanol, and 2.1 mL of acetic acid was added. The mixture was heated under reflux and stirred for 4 h at 75 °C. After cooling, the mixture was evaporated to dryness using a rotary evaporator, and the residue was dissolved in 30 mL water. Semi-preparative HPLC was then performed with a high-pressure gradient system consisting of two pumps K-1001, an online degasser, a UV detector K-2501, and a fraction collector K-16 (all from Knauer, Berlin, Germany). On a stainless steel column (RP-18, Eurospher 100, 300 mm × 8 mm, 5 µm, Knauer) with a guard column (30 mm × 8 mm) filled with the same material, separation was performed at room temperature using 0.1% trifluoroacetic acid (TFA) in double-distilled water (solvent A), and 0.1% TFA in a mixture of acetonitrile and water (90/10, v/v) as the eluents. Between 0 and 30.5 min, a linear gradient was formed starting from 0% B to 38% B, and the absorbance was monitored at 254 nm. Prior to injection, 1 mL of sample solution was diluted with 1 mL of solvent A, and the whole sample was injected. The effluent was fractionated between 29 and 32 min. The combined fractions were evaporated to dryness and once more fractionated with the same method. Finally, the combined eluate fractions were evaporated to dryness and lyophilized to yield the target compound as a white powder, which was stored at − 18 °C.
Analytical data: 1H-NMR, see Table 1. HPLC–UV-MS/MS (gradient 1): tR, 6.7 min; [M + H]+ (m/z 528), UV, λmax 218 nm. Content = 67.4%, based on comparison with N-α-hippuryl-CML as a standard. Yield: 20.3 mg (77%).
Synthesis of N-ε-carboxymethyl-N-ε-(1-deoxyfructosyl)-l-lysine (CMFL)
As generally applied for synthesis of CML [31, 32], N-α-Boc-lysine (1000 mg, 4.1 mmol) and glyoxylic acid monohydrate (478.4 mg, 5.2 mmol) were dissolved in 30 mL of water, and the pH was adjusted to 8.75 with 3 M sodium hydroxide. After addition of 50 mg of palladium on charcoal, the mixture was stirred overnight at room temperature under a hydrogen atmosphere. Then, the mixture was filtered, water was evaporated in vacuo, and 7.2 g (40 mmol) glucose, 100 mL methanol, and 500 µL of glacial acetic acid were added. The mixture was heated at 70 °C for 4 h, and after cooling and vacuum evaporation of methanol, the residue was taken up in 75 mL of water. The pH was adjusted to 3.0, and the solution was applied to a column (15 × 2.5 cm) filled with the strongly acidic cation exchange resin Lewatit S100 (20–50 mesh), previously equilibrated with 300 mL of 6 M HCl and 300 mL of water. After rinsing the column with 300 mL of water, the compounds were left bound to the exchanger overnight to remove the Boc protecting group. Elution was performed with 250 mL 2 M pyridine, whose pH had been adjusted to 6.0 with glacial acetic acid. The eluate was evaporated to dryness in vacuo, the residue was dissolved in 30 mL of water, and the pH was adjusted to 2.0 with HCl. The solution was applied to a glass column (1.5 × 50 cm) filled with the strongly acidic cation exchange resin DOWEX 50W-X8 (100–200 mesh), which had previously been equilibrated with 300 mL of 6 M HCl, 300 mL of water, and 300 mL of 0.01 M HCl. The elution was performed first with 40 mL of 0.01 M HCl, then with 200 mL of 1 M HCl, and lastly with 700 mL of 1.5 M HCl. Fractions (10 mL) were collected with a fraction collector (RediFrac, Pharmacia Biotech, Uppsala, Sweden). Target compounds were detected by spotting 1 µL of each fraction on TLC plates and spraying with a solution of 0.1% ninhydrin in ethanol. CMFL was found to elute between 350 and 430 mL of 1.5 M HCl, well separated from the following CML band. The respective fractions were pooled, filtered, and evaporated to dryness in vacuo. After redissolving in water, the solution was lyophilized to yield CMFL hydrochloride as an off-white powder, which was stored at − 18 °C.
Analytical data: 1H-NMR, see Table 1. HPLC–MS/MS (gradient 1): tR, 3.1 min; fragmentation (100 V, 10 eV) of [M + H]+ (m/z 367): 331 (100), 349 (83), 367 (63), 205 (51), 313 (30), 130 (17), 84 (8), 283 (8). Yield: 130.6 mg (7.9% from lysine).
Synthesis of N-ε-carboxymethyl-N-ε-furoylmethyl-l-lysine (carboxymethylfurosine, CM-Fur)
N-α-hippuryl-CML (250 mg, 0.57 mmol) and glucose (1.2330 g, 6.9 mmol) were dissolved in a mixture of 482 mL methanol and 24 mL glacial acetic acid. The mixture was heated to 75 °C and stirred for 6 h in an oil bath. Afterwards, the solvents were evaporated and the residue was subjected to flash column chromatography on 50 g of silica gel using a mixture of methanol and ethyl acetate (70/30, v/v) as the eluent. Fractions of 10 mL were collected manually and spotted on silica gel plates. Detection with chlorine/dianisidine [33] revealed positive spots eluting between 100 and 500 mL of the solvents. The respective fractions were combined, evaporated in vacuo, taken up in 250 mL of 10 M HCl and hydrolysed for 23 h at 110 °C. Then, HCl was evaporated, and the residue was taken up in 30 mL of water and subjected to cation-exchange chromatography on a column of DOWEX 50W-X8 (100–200 mesh, 2.5 × 25 cm) previously equilibrated with 250 mL of 6 M HCl, 250 mL of water, and 250 mL of 0.01 M HCl. After the addition of the sample (30 mL), the column was rinsed first with 50 mL of 0.01 M HCl, and then with 300 mL of 1.5 M HCl, 300 mL of 2 M HCl, 300 mL of 3 M HCl, and 300 mL of 6 M HCl. Spotting test and HPLC–MS/MS revealed that the target compound had eluted with 6 M HCl. After evaporation of the solvent, the residue was taken up in 5 mL of water. HPLC with UV-detection at 280 nm by comparison with a furosine standard assuming similar molar absorptivity revealed that the concentration of CM-FUR in this solution was 1.3 µM, equivalent to a yield of 2.0 mg (6.4 µmol) of the compound (molar yield from N-α-hippuryl-CML, 1.1%).
Analytical data: HPLC–MS/MS (gradient 1): tR, 8.0 min; UV, λmax 281 nm. Fragmentation (100 V, 10 eV) of [M + H]+ (m/z 313): 130 (100), 313 (56), 84 (17), 184 (13).
Characterization of synthesized compounds
NMR spectra were recorded on an Avance III instrument (1H, 600 MHz; 13C, 150 MHz) from Bruker (Rheinstetten, Germany). Chemical shifts are given in parts per million (ppm), relative to the internal HOD signal (4.70 ppm). Coupling constants (J) are reported in Hz. Beyond one-dimensional spectra, also two-dimensional spectra such as DEPT-135 (distortionless enhancement by polarization transfer), COSY (1H,1H-correlation spectroscopy), HSQC (heteronuclear single quantum coherence), and HMBC (heteronuclear multiple bond correlation) were recorded. The chromatographic purity, UV maxima, molecular mass, and fragmentation behavior of the substances were assessed by HPLC–MS/MS using gradient 1 (see below). The content of target substances in a preparation was determined by HPLC–UV by comparison with either authentic standard or substances with identical chromophores (e.g., N-α-hippuryl lysine or furosine).
Stability of N-α-hippuryl-CML in the presence of reducing sugars
An amount of 16.5 µmol of different sugars (ribose, xylose, 2.5 mg; glucose, mannose, galactose, 3.0 mg; maltose, lactose, 5.9 mg) was weighed into a screw-cap tube, and 1 mL of either 3 mM N-α-hippuryl-Lys in a mixture of methanol and DMF (30/70, v/v) or 1 mL of 3 mM N-α-hippuryl-CML in a mixture of methanol and DMF (30/70, v/v) was added. After addition of 50 µL of glacial acetic acid, the tubes were closed and heated at 75 °C for 1 h in an oil bath during constant stirring. Afterwards, the mixtures were completely evaporated to dryness and taken up in 1 mL of 10 mM NFPA. After centrifugation, the supernatants were transferred into HPLC vials and analyzed by HPLC–UV and HPLC–MS/MS. All incubations were performed in triplicate.
Model incubations regarding the formation of protein-bound CMFL
Based on various protocols for the preparation of so-called “AGE-BSA” [34,35,36], bovine serum albumin (BSA) was incubated at a concentration of 50 mg/mL in phosphate-buffered saline (PBS) in the presence of 0.5 M glucose at 37 °C for up to 9 weeks. In a parallel set of samples, glucose was replaced by 0.25 M glucose and 0.25 M fructose. In a further set of samples, no sugar was added. All incubations were performed in triplicate. Samples were withdrawn at the beginning of the experiment, and after 1, 3 days, 1, 3, 6, and 9 weeks. The samples were deep-frozen immediately. Prior to analysis, the samples were dialyzed against water for 72 h using tubings with an MWCO of 12 kDa. Water was changed twice daily. After dialysis, the samples were lyophilized and subjected to acid hydrolysis.
Acid hydrolysis for determination of CMFL by conversion to CM-Fur was performed by adding 10 mL of 10 M HCl to 10 mg of protein in a screw-cap tube. The tubes were incubated in a drying oven for 23 h at 110 °C. Then, the hydrolysates were completely evaporated to dryness first by rotary evaporation, and then in a vacuum centrifuge. The residue was reconstituted in 1000 µL of 10 mM NFPA and centrifuged (10,000×g, RT, 10 min) prior to LC–MS/MS and amino acid analysis.
High-pressure liquid chromatography with UV-detection (HPLC–UV)
Analysis of hippuryl amino acids was performed with a gradient system consisting of a solvent organizer (K-1500; Knauer, Berlin, Germany), an autosampler (Basic Marathon; Spark Holland, Emmen, Netherlands) a pump (Smartline 1000, Knauer), an online degasser (Knauer), a column oven, and a diode array detector (DAD 2.1L, Knauer). For peak evaluation, the software ClarityChrom (Chrom Tech Inc., Apple Valley, MN) was used.
Hippuryl amino acids were analyzed at room temperature on a stainless-steel column (250 mm × 4.6 mm, 5 µm) filled with Eurospher-100 RP-18 material with an integrated guard column (5 mm × 4 mm) of the same material (Knauer, Berlin, Germany). As solvent A, 10 mM nonafluoropentanoic acid (NFPA) in water was used, whereas solvent B was 10 mM NFPA in acetonitrile. The gradient started at 10% B, which was increased to 50% during 29 min. The injection volume was 20 µL. The absorbance was read at 228 nm and 254 nm, simultaneously, and UV spectra were recorded between 190 and 400 nm throughout the analytical run.
High-pressure liquid chromatography with mass-spectrometric detection (HPLC–MS/MS)
These analyses were performed on the high pressure gradient system 1200 Series (Agilent Technologies, Böblingen, Germany), consisting of a binary pump, an online degasser, a column oven and an autosampler. The stainless steel column Zorbax 300 SB-C18 (50 mm × 2.1 mm, 3.5 µm, Agilent) was used. Solutions of 10 mM NFPA in double distilled water (solvent A) and of 10 mM NFPA in acetonitrile (solvent B) were used in the gradient mode at a flow rate of 0.25 mL/min (gradient 1; 0 min, 10% B; 15 min, 66% B; 19 min, 66% B; 20 min, 10% B; 28 min, 10% B) [22]. The column temperature was maintained at 35 °C. The injection volume was 5 µL. The absorbance was measured at 220 nm, and 280 nm, respectively, while UV spectra were recorded simultaneously (200–350 nm; step size, 1 nm). For mass-spectrometric detection, the mass spectrometer 6410 Triple Quad (Agilent) was employed working in the positive mode. The capillary voltage was set at 4000 V, and the source temperature at 350 °C. The HPLC effluent was transferred to the mass spectrometer between 2 and 20 min. In the scan mode, mass spectra were recorded in the m/z range 100–800 (scan time, 500 ms; fragmentor voltage, 100 V). In the product ion scan mode, chromatograms were recorded between 3 and 20 min for expected [M + H]+ ions after adjustment of the fragmentor voltage and collision energy.
In the MRM mode, the gradient was optimized for analysis of furosine and CM-Fur (gradient 2). The proportion of B was increased from 5 to 45% during 9 min. UV-detection was performed at 220 and 280 nm, and the transitions given in Table 2 were recorded between 3 and 14 min.
Results and discussion
Stability of CML in the presence of reducing sugars
Emerging evidence suggests that the so-called “advanced glycation end products”, an expression widely used for glycated amino acids, are indeed not end products of the Maillard reaction, but that they can be degraded in chemical and biochemical reactions [22, 23, 26]. While it is established that CML is utilized by microorganisms [22], the chemical fate of the compound has not been studied until now. We hypothesized that, being a secondary amine, CML can undergo glycation reactions as described also for other secondary amines such as dibenzylamine and morpholine [37].
Therefore, we performed a stability study by subjecting CML to glycation conditions [30]. The primary amino group of CML was protected by a hippuryl residue to impede side-reactions at this group. Due to its UV activity, this group also enables direct analysis of the reaction products without the need for sample work-up that could lead to the formation of by-products. First, N-α-hippuryl-CML was incubated with a ten-fold molar excess of glucose for 4 h, and HPLC coupled to DAD and mass-spectrometry was applied for structural elucidation of the reaction products. A new peak appeared in the chromatogram eluting after 6.8 min (Peak 1, Fig. 2), shortly before N-α-hippuryl-CML (Peak 2). This peak had the same UV spectrum as N-α-hippuryl-CML, but a different mass spectrum. The main peak in the mass spectrum of peak 1 had an m/z ratio of 528, which is equivalent to the attachment of a sugar molecule to N-α-hippuryl-CML and condensation of one molecule of water. The structure of the assumed reaction product is also depicted in Fig. 2. Beneath this peak, a sodium adduct (m/z 550), a potassium adduct (m/z 566), and an ion representing the loss of a molecule of water (m/z 510) were detected. Reaction products of N-α-hippuryl-CML that can be assigned to the corresponding Amadori products were detected in all co-incubations of N-α-hippuryl-CML with different sugars (Table 3). Even though CML is more stable under these reaction conditions than lysine, the results show that up to 21% are degraded during heating with ribose. The extent of degradation of N-α-hippuryllysine was proportional to the amount of sugar that is present in the open-chain form. It was reported that the yield of Amadori products increases with increasing acyclic form [38, 39]. The effect was observed similarly for degradation of N-α-hippuryl-CML, but it was less pronounced than during incubation of N-α-hippuryllysine. The reaction products that have been assigned via LC–MS/MS according to their m/z values are listed in Table 3. Comparison of the stability of both hippuryl amino acids showed that lysine is 5–10 times more reactive than CML.

A RP-HPLC with UV and MS detections of a mixture of N-α-hippuryl-CML and glucose after heating in boiling methanol for 4 h. B MS spectrum of peak (1) with the proposed structure of a glucose-derived Amadori product incorporating the secondary amino group of N-α-hippuryl-CML
Synthesis, isolation and characterization of hippuryl-CMFL and CMFL
Based on different literature methods on the synthesis of ARPs [30, 37] the synthesis of N-α-hippuryl-CMFL (Fig. 3) was optimized. An increase in the excess of glucose form six-fold to ten-fold did not lead to an increase in the formation of N-α-hippuryl-CMFL. Other solvents than methanol such as water, propanol and dioxan were also not leading to higher yields. The best yields were obtained by synthesis in boiling methanol and a synthesis time of 4 h. After this time, the yields decreased, possibly because of side-reactions such as methyl ester formation in N-α-hippuryl-CML. Such a product was detected qualitatively in the LC–MS chromatograms (peak 3, Fig. 3) with an m/z ratio of 380, equivalent to the addition of the CH3 group to and removal of a proton from one of the carboxy groups of N-α-hippuryl-CML.

Scheme of preparative procedures carried out in the present work. R1 = Benzoylglycyl. (i) Glyoxylic acid, Pd/C, RT, pH 8.75, 16 h, (ii) glucose, H+, 75 °C, 4 h, (iii) 10 M HCl, 23 h, 110 °C
After optimization of the reaction conditions in small scale, the synthesis was upscaled, and the compound N-α-hippuryl-CMFL was then isolated by semi-preparative HPLC. The synthesis of free CMFL was performed starting from N-α-Boc-lysine. The intermediate product N-α-Boc-CML was not isolated but directly converted to the ARP N-α-Boc-CMFL by incubation with glucose in boiling methanol. The formation of N-α-Boc-fructosyllysine as a by-product from residual N-α-Boc-lysine was accepted, because CMFL and N-ε-fructosyllysine strongly differ in their charges and were easily separated by ion-exchange chromatography.
Both products were characterized by one- and two-dimensional NMR experiments (Table 1). Data for the hippuryl residue were in good agreement with literature data [30]. When compared with either N-ε-fructosyllysine or CML, certain differences in the chemical shifts were recognized for the 13C and 1H signals of atoms in close vicinity to the tertiary amine nitrogen of CMFL. In CMFL, the protons of the carboxymethyl group are more strongly deshielded than in “normal” CML or N-α-hippuryl-CML, because there are more electron-withdrawing groups in immediate vicinity of these protons. In the present study, these protons were shifted to 3.83–3.90 ppm in N-α-hippuryl-CMFL and CMFL, while literature reports shifts between 3.55 and 3.81 ppm for the carboxymethyl protons in CML and N-α-hippuryl-CML [30, 32]. The same applies to the C-1 and H-1 signals at the fructopyranosyl residue which are also shifted downfield. These protons have chemical shifts between 3.40 and 3.52 ppm in CMFL and N-α-hippuryl-CMFL, compared to literature data of 3.17–3.18 ppm for the same protons in ARPs of different mono- and disaccharides. The 13C signal of the C-1 of the fructopyranosyl residue is at 59.7 ppm in N-α-hippuryl-CMFL and CMFL, while it is upfield shifted (52.3–52.7 ppm) in common ARPs [30, 32]. For CMFL, all signals of the fructopyranosyl residue were detected as in N-α-hippuryl-CMFL, but the signals in the lysyl chain were slightly shifted. The signals of the protons at the C-2 were found upfield by 0.4 ppm in CMFL. This is due to the incorporation of the amino group in a peptide bond, while the carboxy group is still free. Similar shifts were observed at the C-2 between free and dipeptide-bound glycated amino acids [32]. Even though the α-amino group of lysine was protected during synthesis, a side-reaction at this group may not be excluded, and this would have led to a compound isobaric to CMFL. However, this would have led to stronger differences in the chemical shifts of the 13C atoms and protons in vicinity to the α-amino group. Moreover, cross peaks between (i) the protons at the carboxymethyl group and the C-1 of the fructosyl residue and (ii) the protons at C-6 of lysine and the C-1 of the fructosyl residue were clearly visible in the HMBC spectrum. This excludes an N-α modified CML species and proves the existence of a lysine derivative modified at the ε-amino group both by a carboxymethyl group and a fructosyl group.
The product ion spectrum of CMFL (Fig. 4) shows fragment ions of the ion m/z 367 ([M + H]+ of CMFL) that result from the lysyl (m/z 84, 130) and CML residues (m/z 205). Moreover, three successive eliminations of water are visible at higher m/z values which are due to fragmentations in the fructosyl residue. Successive water eliminations, eliminations of three molecules of water and HCHO (m/z 283) as well as the loss of the fructosyl moiety (m/z 205) are common fragmentations observed during tandem MS of ARPs [40].

Product ion spectrum obtained during measurement of a standard solution of N-ε-carboxymethyl-N-ε-(1-deoxyfructosyl)-l-lysine (CMFL) by HPLC–MS/MS. Operating conditions: Fragmentor voltage, 100 V, collision energy, 10 eV
Stability of CMFL during acid hydrolysis
As CMFL can theoretically also be formed during food processing, further studies focused on chemical reactions during analysis of the compound. ARPs such as N-ε-fructosyllysine are converted to the stable marker furosine during acid hydrolysis used for the determination of the amino acid composition of proteins [30, 41]. Furosine is widely applied as a marker for heat treatment of food and blockage of lysine [42]. N-α-hippurylfructosyllysine is converted to 56.2% lysine, 32.4% furosine, 15.9% pyridosine, and 1.6% CML during hydrolysis with 6 M HCl [30]. When N-α-hippuryl-CMFL was subjected to acid hydrolysis under the same conditions, the compound was mainly degraded to CML (98%), while ca. 2% accounted for a new UV-active peak which was different from furosine. Furosine itself was not detected following acid hydrolysis of N-α-hippuryl-CMFL. The peak had a similar UV spectrum as furosine with a UV maximum at 281 nm (Fig. 5). The m/z ratio of the protonated pseudomolecular ion of the new UV-active peak was 313. N-α-fructosyllysine loses three molecules of water during formation of furosine, and the same chemical reaction may be expected for CMFL, leading to a molecular mass of 312 Da. Thus, we postulated that the new peak should be N-ε-carboxymethyl-N-ε-furoylmethyl-l-lysine (carboxymethylfurosine, CM-Fur). An independently synthesized standard of CM-Fur showed the same fragmentation pattern (Fig. 5). It is known that the yield of furosine from fructosyllysine increases with increasing concentration of HCl used for hydrolysis [30]. Therefore, the yields of CM-Fur and furosine were compared for different concentrations of HCl (Fig. 6). As for FL [29], a linear relationship between the concentration of hydrochloric acid and the yield of CM-Fur could be established for hydrolysis of N-α-hippuryl-CMFL. At a concentration of 4 M HCl, formation of CM-Fur from CMFL is nearly negligible. Ca. 16 times more furosine is formed from fructosyllysine than CM-Fur is formed from N-α-hippuryl-CMFL on a mass basis when 6 M HCl is used. Regarding the different molar masses of the educts, the excess is ca. 9.4-fold on a molar basis. The excess decreases with increasing HCl concentration. Maximum conversion was reached at 10 M HCl, which is an applicable concentration also for food analysis. The conversion to CM-Fur may be expected to rise further in 12 M HCl, but this would not be a convenient concentration for the analysis of liquid samples. Compared to the published conversion factors of ARPs to furosine [30], a molar conversion of 11% of CMFL to CM-Fur can be estimated for hydrolysis with 8 M HCl which rises to ca. 21% for hydrolysis with 10 M HCl.

A RP-HPLC with UV and MS detections of a mixture of furosine and N-ε-carboxymethylfurosine (CM-Fur). B UV spectra of furosine (grey line) and CM-Fur (black line). C Product ion spectrum of CM-Fur. Operating conditions: Fragmentor voltage, 100 V, collision energy, 10 eV

Peak areas of furosine (closed boxes) and CM-Fur (open boxes) after hydrolysis of 50 µg/mL solutions of FL and hippuryl-CMFL in hydrochloric acid of increasing molarity. Data are means ± S.D. (n = 3)
Beneath standard acid hydrolysis (6 M HCl, 23 h, 110 °C), acid hydrolysis with prior reduction using NaBH4, and also enzymatic hydrolysis would be probable methods for sample preparation. CML can be formed as a byproduct from ARPs during acid hydrolysis due to oxidation. Therefore, reduction of ARPs by NaBH4 is often implemented as a step in CML analysis [12, 19]. This type of sample pretreatment was also tested in the present study. During amino acid analysis, a new peak, probably identical to N-ε-glucitol-N-ε-carboxymethyllysine or N-ε-mannitol-N-ε-carboxymethyllysine was detectable in considerable amounts (Fig. 7), however, as the identity of the peak(s) could not yet be definitely proven by mass spectrometry, this analytical route was not further followed. Provided that such a compound is equally well ionizable as CM-Fur, this could turn out to be a better way for CMFL analysis.

Amino acid analysis with VIS detection after post-column derivatization with ninhydrin of solutions of N-α-hippuryl-CML (grey line) and N-α-hippuryl-CMFL (black line) after reduction with NaBH4 and subsequent acid hydrolysis (6 M HCl, 23 h, 110 °C) and proposed structure of the reaction product that results from reduction of CMFL
Formation of CMFL during protein glycation
A multitude of approaches is available in the literature for the targeted modification of bovine serum albumin (BSA) to derivatives such as “CML-BSA”, or “AGE-BSA” often simply using glucose or fructose as the starting sugar [34,35,36]. Owing to the ease of formation of CMFL in model systems, we decided to elucidate if the formation of CMFL may play a role during incubation of glucose with BSA under physiological conditions. BSA was incubated with a tenfold molar excess of glucose with respect to the lysine residues for up to 9 months, and samples were taken regularly, dialyzed against water, and lyophilized. A mixture of fructose and glucose was also used, because it is known that fructose may lead to a higher degree of CML formation. Acid hydrolysis using 10 M HCl was performed on the lyophilized samples to convert fructosyllysine to furosine and CMFL to CM-Fur. The concentrations of furosine and CM-Fur were determined by HPLC with UV and MS/MS detections and standard addition (Fig. 8). From the data (Table 4) and the lysine concentration in BSA [43], we estimate that ca. 13% of the lysine residues of BSA had been converted to fructosyllysine after co-incubation of BSA with glucose, whereas only 0.015% had been converted to CMFL. Thus, the ratio between fructosyllysine and CMFL is ca. 1000:1. If this ratio is the same in other food items such as bread, a concentration of ca. 5–10 mg/kg CMFL may be expected, when a concentration of 6–7 g/kg ARPs is present [3]. It is important to consider that in many food items, the ratio between ARPs and CML is about 100 to 1 [3, 44], so a significant part of about 10% of CML may be fructosylated. The conversion rates were slightly smaller when glucose was replaced by a mixture of glucose and fructose. When N-ε-fructosyllysine as the free amino acid was subjected to hydrolysis in the presence of 10 M HCl, no CM-Fur could be detected.

RP-HPLC with MS/MS detection of carboxymethylfurosine (CM-Fur) in the MRM mode. a BSA sample incubated in phosphate buffer for 9 weeks, then isolated and hydrolyzed in the presence of 10 M HCl for 23 h at 110 °C. b BSA sample incubated with glucose in phosphate buffer for 9 weeks and worked up as sample a. c, d Addition of different concentrations of CM-Fur standard
Taken together, glycation of the ε-amino group of CML is possible with different reducing mono- and disaccharides leading to heterogeneous multiply glycated species constituting a new class of compounds in the Maillard reaction. CMFL, and possibly also other ARPs of CML, can be converted to CM-Fur analogously to fructosyllysine for analysis by HPLC–UV or MS. However, as the biggest part of CMFL is converted to CML during acid hydrolysis, there is an urgent need to clarify as to how much the currently published data for CML are biased owing to artificial neoformation of the compound from products of multiple glycation. Exposure assessment both for CML and CMFL will be improved in this way. Lastly, two further pathways of formation of CMFL are possible: one would be the carboxymethylation of FL, e.g., by glyoxal, during advanced glycation, and the other would be the oxidation of bis(fructosyl)-lysine. Some other lysine-derived glycated amino acids such as CEL may react in the same way, whereas amide-AGEs [5] should not. In CEL, however, due to steric hindrance of the additional methyl group, glycation by sugars is supposed to be even less pronounced. In general, the chemical reactivity of glycated amino acids during glycation deserves further attention. In the case of CML, this may explain, at least in part, its instability during glycation shown in different works [24, 25].
Abbreviations
- AGE:
-
Advanced glycation end product
- ARP:
-
Amadori rearrangement product
- BSA:
-
Bovine serum albumin
- CM-CAD:
-
Carboxymethylcadaverine
- CMFL:
-
N-ε-Carboxymethyl-N-ε-fructosyllysine
- CM-Fur:
-
N-ε-Carboxymethylfurosine
- CML:
-
N-ε-Carboxymethyllysine
- COSY:
-
Correlation spectroscopy
- DAD:
-
Diode array detection
- DEPT:
-
Distortionless enhancement by polarization transfer
- DMF:
-
Dimethyl formamide
- HMBC:
-
Heteronuclear multiple bond correlation
- HSQC:
-
Heteronuclear single quantum coherence
- MRM:
-
Multiple reaction monitoring
- MWCO:
-
Molecular weight cut-off
- NFPA:
-
Nonafluoropentanoic acid
- NMR:
-
Nuclear magnetic resonance
- PBS:
-
Phosphate-buffered saline
- RT:
-
Room temperature
- TFA:
-
Trifluoroacetic acid
- TLC:
-
Thin layer chromatography
References
Maillard LC (1912) Action des acides aminés sur les sucres. Formation des mélanoïdines par voie méthodique. CR Acad Sci 145:66–68
Hodge JE (1953) Dehydrated foods. Chemistry of browning reactions in model systems. J Agric Food Chem 1:928–943. https://doi.org/10.1021/jf60015a004
Hellwig M, Henle T (2014) Baking, ageing, diabetes: a short history of the Maillard reaction. Angew Chem Int Ed Engl 53:10316–10329. https://doi.org/10.1002/anie.201308808
Brownlee M, Vlassara H, Cerami A (1984) Nonenzymatic glycosylation and the pathogenesis of diabetic complications. Ann Intern Med 101:527–537. https://doi.org/10.7326/0003-4819-101-4-527
Baldensperger T, Jost T, Zipprich A, Glomb MA (2018) Novel α-oxoamide advanced-glycation endproducts within the N6-carboxymethyl lysine and N6-carboxyethyl lysine reaction cascades. J Agric Food Chem 66:1898–1906. https://doi.org/10.1021/acs.jafc.7b05813
Ahmed MU, Thorpe SR, Baynes JW (1986) Identification of Nε-carboxymethyllysine as a degradation product of fructoselysine in glycated protein. J Biol Chem 261:4889–4894. https://doi.org/10.1016/S0021-9258(19)89188-3
Namiki M, Hayashi T, Ohta Y (1977) Novel free radicals formed by the amino-carbonyl reactions of sugars with amino acids, amines, and proteins. Adv Exp Med Biol 86B:471–501. https://doi.org/10.1007/978-1-4757-9113-6_28
Glomb MA, Monnier VM (1995) Mechanism of protein modification by glyoxal and glycolaldehyde, reactive intermediates of the Maillard reaction. J Biol Chem 270:10017–10026. https://doi.org/10.1074/jbc.270.17.10017
Cho S-J, Roman G, Yeboah F, Konishi Y (2007) The road to advanced glycation end products: a mechanistic perspective. Curr Med Chem 14:1653–1671. https://doi.org/10.2174/092986707780830989
Kasper M, Schieberle P (2005) Labeling studies on the formation pathway of Nε-carboxymethyllysine in Maillard-type reactions. Ann NY Acad Sci 1043:59–62. https://doi.org/10.1196/annals.1333.007
Fu M-X, Requena JR, Jenkins AJ, Lyons TJ, Baynes JW, Thorpe SR (1996) The advanced glycation end product, Nε-(carboxymethyl)lysine, is a product of both lipid peroxidation and glycoxidation reactions. J Biol Chem 271:9982–9986. https://doi.org/10.1074/jbc.271.17.9982
Assar SH, Moloney C, Lima M, Magee R, Ames JM (2009) Determination of Nε-(carboxymethyl)lysine in food systems by ultra performance liquid chromatography-mass spectrometry. Amino Acids 36:317–326. https://doi.org/10.1007/s00726-008-0071-4
Hull GLJ, Woodside JV, Ames JM, Cuskelly GJ (2012) Nε-(carboxymethyl)lysine content of foods commonly consumed in a Western style diet. Food Chem 131:170–174. https://doi.org/10.1016/j.foodchem.2011.08.055
Scheijen JLJM, Clevers E, Engelen L, Dagnelie PC, Brouns F, Stehouwer CDA, Schalkwijk CG (2016) Analysis of advanced glycation endproducts in selected food items by ultra-performance liquid chromatography tandem mass spectrometry: presentation of a dietary AGE database. Food Chem 190:1145–1150. https://doi.org/10.1016/j.foodchem.2015.06.049
Chen G, Smith JS (2015) Determination of advanced glycation endproducts in cooked meat products. Food Chem 168:190–195. https://doi.org/10.1016/j.foodchem.2014.06.081
Birlouez-Aragon I, Saavedra G, Tessier FJ, Galinier A, Ait-Ameur L, Lacoste F, Niamba C-N, Alt N, Somoza V, Lecerf J-M (2010) A diet based on high-heat-treated foods promotes risk factors for diabetes mellitus and cardiovascular diseases. Am J Clin Nutr 91:1220–1226. https://doi.org/10.3945/ajcn.2009.28737
Delgado-Andrade C, Tessier FJ, Niquet-Léridon C, Seiquer I, Navarro MP (2012) Study of the urinary and faecal excretion on Nε-carboxymethyllysine in young human volunteers. Amino Acids 43:595–602. https://doi.org/10.1007/s00726-011-1107-8
Scheijen JLJM, Hanssen NMJ, Van Greevenbroek MM, Van der Kallen C, Feskens EJM, Stehouwer CDA, Schalkwijk CG (2018) Dietary intake of advanced glycation endproducts is associated with higher levels of advanced glycation endproducts in plasma and urine: the CODAM study. Clin Nutr 37:919–925. https://doi.org/10.1016/j.clnu.2017.03.019
Niquet-Léridon C, Tessier FJ (2011) Quantification of Nε-carboxymethyl-lysine in selected chocolate-flavoured drink mixes using high-performance liquid chromatography-linear ion trap tandem mass spectrometry. Food Chem 126:655–663. https://doi.org/10.1016/j.foodchem.2010.10.111
Delgado-Andrade C (2016) Carboxymethyl-lysine: thirty years of investigation in the field of AGE formation. Food Funct 7:46–57. https://doi.org/10.1039/c5fo00918a
Hellwig M, Humpf H-U, Hengstler J, Mally A, Vieths S, Henle T (2019) Quality criteria for studies on dietary glycation compounds and human health. J Agric Food Chem 67:11307–11311. https://doi.org/10.1021/acs.jafc.9b04172
Hellwig M, Auerbach C, Müller N, Samuel P, Kammann S, Beer F, Gunzer F, Henle T (2019) Metabolization of the advanced glycation end product N-ε-carboxymethyllysine (CML) by different probiotic E. coli strains. J Agric Food Chem 67:1963–1972. https://doi.org/10.1021/acs.jafc.8b06748
Bui TPN, Troise AD, Fogliano V, De Vos WM (2019) Anaerobic degradation of N-ε-carboxymethyllysine, a major glycation end-product, by human intestinal bacteria. J Agric Food Chem 67:6594–6602. https://doi.org/10.1021/acs.jafc.9b02208
Cheng L, Jin C, Zhang Y (2014) Investigation of variations in the acrylamide and Nε-(carboxymethyl) lysine contents in cookies during baking. J Food Sci 79:T1030–T1038. https://doi.org/10.1111/1750-3841.12450
Nguyen HT, Van der Fels-Klerx HJ, Van Boekel MAJS (2016) Kinetics of Nε-(carboxymethyl)lysine formation in aqueous model systems of sugars and casein. Food Chem 192:125–133. https://doi.org/10.1016/j.foodchem.2015.06.110
Li Y, Li L, Lund MN, Li B, Zhang X (2018) Reduction of Nε-(carboxymethyl) lysine by (−)-epicatechin and (−)-epigallocatechin gallate: the involvement of a possible trapping mechanism by catechin quinones. Food Chem 266:427–434. https://doi.org/10.1016/j.foodchem.2018.06.009
Anet EFLJ (1960) Degradation of carbohydrates 1. Isolation of 3-deoxyhexosones. Austr J Chem 13:396–403
Feather MS, Mossine VV (1998) Correlations between structure and reactivity of Amadori compounds: the reactivity of acyclic forms. In: O’Brien J, Crabbe MJC, Nursten HE, Ames JM (eds) The Maillard reaction in foods and medicine. Woodhead Publishing Ltd.
Henle T, Zehetner G, Klostermeyer H (1995) Fast and sensitive determination of furosine. Z Lebensm-Unters Forsch 200:235–237. https://doi.org/10.1007/BF01190503
Krause R, Knoll K, Henle T (2003) Studies on the formation of furosine and pyridosine during acid hydrolysis of different Amadori products of lysine. Eur Food Res Technol 216:277–283. https://doi.org/10.1007/s00217-002-0649-0
Grunwald S, Krause R, Bruch M, Henle T, Brandsch M (2006) Transepithelial flux of early and advanced glycation compounds across Caco-2 cell monolayers and their interaction with intestinal amino acid and peptide transport systems. Br J Nutr 95:1221–1228. https://doi.org/10.1079/bjn20061793
Hellwig M, Geissler S, Matthes R, Peto A, Silow C, Brandsch M, Henle T (2011) Transport of free and peptide-bound glycated amino acids: Synthesis, transepithelial flux at Caco-2 cell monolayers, and interaction with apical membrane transport proteins. ChemBioChem 12:1270–1279. https://doi.org/10.1002/cbic.201000759
Sajapin J, Hellwig M (2020) Studies on the synthesis and stability of α-ketoacyl peptides. Amino Acids 52:1425–1438. https://doi.org/10.1007/s00726-020-02902-8
Vlassara H, Brownlee M, Cerami A (1985) High-affinity-receptor-mediated uptake and degradation of glucose-modified proteins: a potential mechanism for the removal of senescent macromolecules. Proc Natl Acad Sci USA 82:5588–5592. https://doi.org/10.1073/pnas.82.17.5588
Yamagchi S, Yonekura H, Yamamoto Y, Katsuno K, Sato F, Mita I, Ooka H, Satozawa N, Kawakami T, Nomura M, Yamamoto H (1997) Advanced glycation end products-driven angiogenesis in vitro. J Biol Chem 272:8723–8730. https://doi.org/10.1074/jbc.272.13.8723
Valencia JV, Weldon SC, Quinn D, Kiers GH, DeGroot J, TeKoppele J, Highes TE (2004) Advanced glycation end product ligands for the receptor for advanced glycation end products: biochemical characterization and formation kinetics. Anal Biochem 324:68–78. https://doi.org/10.1016/j.ab.2003.09.013
Hodge JE, Fisher BE (1963) Amadori rearrangement products. Methods Carbohydr Chem 2:99–107
Chanda D, Harohally NV (2018) Revisiting Amadori and Heyns synthesis: critical percentage of acyclic form play the trick in addition to catalyst. Tetrahedron Lett 59:2983–2988. https://doi.org/10.1016/j.tetlet.2018.06.050
Bunn HF, Higgins PJ (1981) Reaction of monosaccharides with proteins: possible evolutionary significance. Science 213:222–224. https://doi.org/10.1126/science.12192669
Ruan ED, Wang H, Ruan Y, Juárez M (2013) Study of fragmentation behavior of Amadori rearrangement products in lysine-containing peptide model by tandem mass spectrometry. Eur J Mass Spectrom 19:295–303. https://doi.org/10.1255/ejms.1237
Finot PA, Bricout J, Viani R, Mauron J (1986) Identification of a new lysine derivative obtained upon acid hydrolysis of heated milk. Experientia 24:1097–1099. https://doi.org/10.1007/BF02147778
Erbersdobler HF, Somoza V (2007) Forty years of furosine—forty years of using Maillard reaction products as indicators of the nutritional quality of foods. Mol Nutr Food Res 51:423–430. https://doi.org/10.1002/mnfr.200600154
Stein WH, Moore S (1949) Amino acid composition of β-lactoglobulin and bovine serum albumin. J Biol Chem 178:79–91
Troise AD, Vitiello D, Tsang C, Fiore A (2016) Encapsulation of ascorbic acid promotes the reduction of Maillard reaction products in UHT milk. Food Funct 7:2591–2602. https://doi.org/10.1039/c6fo00151c
Acknowledgements
We thank Dr. Tilo Lübken and Anett Rudolph (Chair of Organic Chemistry, TU Dresden) for recording the NMR spectra. We thank Karla Schlosser and Dr. Anke Förster (Chair of Food Chemistry, TU Dresden) for performing the amino acid analyses.
Funding
Open Access funding enabled and organized by Projekt DEAL. The study was not supported by any external funding sources.
Author information
Authors and Affiliations
Contributions
Conceptualization, MH, TH; investigations, JN, MH; writing, MH, JN, TH.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Compliance with ethics requirements
This article does not contain any studies with human or animal subjects.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hellwig, M., Nitschke, J. & Henle, T. Glycation of N-ε-carboxymethyllysine. Eur Food Res Technol 248, 825–837 (2022). https://doi.org/10.1007/s00217-021-03931-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-021-03931-7