
Abstract
Dietary polyphenols like epigallocatechin-3-gallate (EGCG)—which represents the most abundant flavan-3-ol in green tea—are subject of several studies regarding their bioactivity and health-related properties. On many occasions, cell culture or in vitro experiments form the basis of published data. Although the stability of these compounds is observed to be low, many reported effects are directly related to the parent compounds whereas the impact of EGCG degradation and autoxidation products is not yet understood and merely studied. EGCG autoxidation products like its dimers theasinensin A and D, “P2” and oolongtheanin are yet to be characterized in the same extent as their parental polyphenol. However, to investigate the bioactivity of autoxidation products—which would minimize the discrepancy between in vitro and in vivo data—isolation and structure elucidation techniques are urgently needed. In this study, a new protocol to acquire the dimers theasinensin A and D as well as oolongtheanin is depicted, including a variety of spectroscopic and quadrupole time-of-flight high-resolution mass spectrometric (qTOF-HRMS) data to characterize and assign these isolates. Through nuclear magnetic resonance (NMR) spectroscopy, polarimetry, and especially circular dichroism (CD) spectroscopy after enzymatic hydrolysis the complementary atropisomeric stereochemistry of the isolated theasinensins is illuminated and elucidated. Lastly, a direct comparison between the isolated EGCG autoxidation products and the monomer itself is carried out regarding their antioxidant properties featuring Trolox equivalent antioxidant capacity (TEAC) values. These findings help to characterize these products regarding their cellular effects and—which is of special interest in the flavonoid group—their redox properties.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Green or black tea undergoes an increasing popularity all over the world. With billions of cups drunken daily, tea is the second most popular beverage following water [1]. Additionally, on-going food trends often feature tea or tea components. Several teas such as the Japanese Matcha tea can be used in so-called “superfoods” which are widely used in modern food recipes, for instance in green smoothies, home-made iced tea, or ice cream.
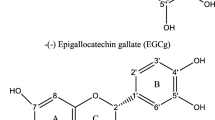
However, the high consumption of tea and its presence in new superfood-type edibles also carries emerging questions on tea ingredients and the use of single tea components [2]. These questions are concerning the chemical composition of these natural products, and of course, their bioactivity as well as toxicological properties [2, 3]. Emphasizing green or black tea-related foodstuff, polyphenolic compounds represent—besides caffeine—the most popular class of bioactive constituents [4]. Catechins were identified as secondary tea plant metabolites with a huge variety of bioactive but also toxic effects [5]. Epigallocatechin-3-gallate (EGCG, see Fig. 1) is one of the most abundant catechins in green tea [6], both in the leaves and freshly brewed. To study potential positive human health effects and to characterize human exposition, many in vivo and in vitro studies are published over the last decades [7]. For EGCG, an antioxidant effect scavenging reactive oxygen species [6, 8] or chemopreventive properties are found [9, 10]. Nevertheless, hepatic damages after the exposure with EGCG are also shown [2, 9, 11]. In 2018, the European Food Safety Authority published their recommendation of the maximal consumption of 800 mg EGCG per day for an average adult, to avoid hepatic toxicity [11]. To gather information on the mentioned bioactive and toxic mechanisms of EGCG, cell culture in vitro models are commonly used. Unfortunately, a low stability of EGCG in cell culture systems limits data interpretation [12,13,14,15,16]. In literature, several reasons for the lower stability of this catechin in cell culture compared to other models or even in vivo situations are given [7]. On the one hand, the main parameter influencing the stability seems to be the pH value of the cell culture medium or solvents [15], whereas non-enzymatic rearrangement reactions are supported at higher pH values [13]. Besides stereochemical rearrangements, oligo- and polymerization steps occur as well [16]. On the other hand, parameters like light exposure, temperature, double-charged metal ions, and salts also strongly influence the stability of this polyphenol. Some published studies include specific supplements to minimize EGCG degradation or autoxidation. For instance, the enzyme catalase is used to remove hydrogen peroxide which is also formed by EGCG in cell culture systems [12, 17,18,19,20]. While this helps to avoid hydrogen peroxide-induced side effects, catalase does not influence the formation of autoxidation products in vitro. Furthermore, stabilization of EGCG with antioxidants like ascorbic acid is described in literature [15]. Nonetheless, most published studies do not feature experimental conditions which guarantee EGCG stability over the whole incubation period. Therefore, the evaluation of observed effects after incubation with non-stabilized EGCG is difficult to achieve [16].

Chemical structure of epigallocatechin-3-gallate (EGCG), oolongtheanin digallate, P2, and the theasinensins A–H with their correspondent atom numbering, chemical sum formula, and exact neutral mass
One of the compound classes which are non-enzymatically formed under cell culture conditions after incubation of EGCG are the theasinensins (see Fig. 1) [12, 15]. These compounds represent dimeric, C–C linked flavan-3-ols [21]. The linkage between the two monomeric units is located at the B and B’ rings of the catechins for all of the compounds in this group [21,22,23]. The catechins EGCG, epicatechin-3-gallate (ECG), epigallocatechin (EGC), and epicatechin (EC) act as monomeric units, and within the scope of the dimeric nature of the theasinensins, the variety of binding possibilities of all these catechins leads to a wide structural diversity of this compound class [21]. An overview of the structural variability of the theasinensins is given in Fig. 1. A unique stereochemical feature of the theasinensins is the atropisomerism of its derivatives which is based on the B,B′ ring linkage—varying rotations of these two rings are not able to merge due to steric effects and hindrance. While EGCG dimerizes to the atropisomers theasinensin A and D, there also exist their hydrolyzed counterparts, which are combined dimers of EGCG and EGC (theasinensin B and H) or dimers of two EGC units (theasinensin C and E) [21, 23, 24]. Besides the theasinensins, there are various other autoxidation products which can be observed after incubation of EGCG in cell culture medium and its cultivation under in vitro conditions. In Fig. 1, the chemical structures of the in literature labeled compound “P2” and of oolongtheanin digallate, which is also called galloyl oolongtheanin [23], are presented. Compounds like P2 are being formed in a more complex way and their formation underlies specific parameters [14, 15, 25].
Due to the instability of EGCG, it is not clear whether the observed effects can be traced back to this incubated catechin itself or its degradation and autoxidation products [16]. One possible way to characterize the contribution of the reaction products to the bioactivity of EGCG itself is the use of single autoxidation products in individual in vitro assays. Therefore, these compounds must be synthesized or isolated to obtain standards for cell culture experiments. To put the properties of these autoxidation products into perspective to EGCG, a characterization of their antioxidant capacity is crucial to identify possible sources of cellular effects [26]. It is hardly predictable if the high antioxidant effects of EGCG—whose (vicinal) hydroxy groups play an important role in its redox properties [8]—still remain after undergoing autoxidation reactions. The data availability on the antioxidant effects of green tea extract isolates is expandable, but shows high antioxidant activities for autoxidation products and theaflavins as well [27, 28]. Because of the low stability of these compounds [13, 15], the choice of technique is pivotal for the robustness of the analysis. The Trolox equivalent antioxidant capacity assay is an internationally standardized method whose protocol omits harsh reaction conditions for the educts [29].
In this work, a novel method for the semi-targeted autoxidation of EGCG to its dimers theasinensin A and D as well as to its autoxidation product oolongtheanin digallate is described and an isolation procedure to obtain these rather uncharacterized flavan-3-ols via semipreparative HPLC–UV is shown. To further describe the atropisomeric behavior of the theasinensins and to distinguish between theasinensin A and D, several structural elucidation methods are put into practice. Spectroscopic techniques as nuclear magnetic resonance (NMR) spectroscopy, polarimetry, photometry, and circular dichroism (CD) spectroscopy are utilized to achieve atropisomeric differentiation. Furthermore, the Trolox equivalent antioxidant capacity (TEAC) values are measured and calculated to perform a first characterization of the isolated autoxidation products regarding their antioxidant properties [29]. The described isolation method and these information on the spectroscopic and antioxidative properties help to characterize EGCG degradation and autoxidation product effects on in vitro experiments.
Materials and methods
Chemicals and reagents
All chemicals, reagents, and solvents are purchased at p. a. grade, if not stated otherwise. Solvents for chromatographic methods and dilution procedures are purchased at LC–MS purity from Fisher Scientific (Schwerte, Germany), acetonitrile-d3 (99.8% at-%D) for NMR is obtained from ARMAR Chemicals (Döttingen, Switzerland). Formic acid (98–100%, p. a.) for HPLC solvents is purchased from Merck KGaA (Darmstadt, Germany). Millipore water is obtained from a miniRO reverse osmosis station by Veolia Water Solutions & Technologies ELGA GmbH (Celle, Germany).
(−)-Epigallocatechin gallate (EGCG) is purchased at ≥ 96% purity from Cayman Chemical (Michigan, USA) for autoxidation product formation and isolation experiments. l-Ascorbic acid and PBS buffer salts (potassium hydrogen phosphate, sodium chloride, disodium phosphate) are obtained from Carl Roth GmbH & Co. KG (Karlsruhe, Germany). Tannase from Aspergillus ficuum is purchased at Sigma–Aldrich Chemie GmbH (Taufkirchen, Germany). For the determination of TEAC values, 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS, ≥ 99%) is purchased from Fluka Chemie GmbH (Buchs, Switzerland), potassium peroxodisulfate from Merck KGaA (Darmstadt, Germany), and trolox (( ±)-6-Hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid) from Sigma–Aldrich (Steinheim, Germany).
Isolation of EGCG autoxidation products
PBS buffer (4.2 g potassium hydrogen phosphate, 180 g sodium chloride, and 8.18 g disodium phosphate in 1 L Millipore water, dilution 1 + 19 (v/v) with Millipore water) is prepared and set to pH 8 using sodium hydroxide solution and hydrochloric acid. EGCG (0.1 mg/mL) is solved in this PBS buffer with a final volume of 10–20 mL. Solutions are stored in a water bath at 37 °C for 30 min. Thereafter, the solutions are immediately cooled down to 0 °C using a water/ice bath. Using a 200 mM ascorbic acid solution, the solutions are adjusted to an ascorbic acid concentration of 2 mM to stabilize the autoxidation products. The solutions are frozen at − 20 °C and subsequently freeze-dried. The resulting powders are reconstituted with 1–2 mL Millipore water (concentration of factor 10 in comparison to the initial solution), centrifugated at 20,000×g for 4 min at ambient temperature and directly used for preparative high-performance liquid chromatography (HPLC) coupled with an UV detector. A preparative Varian HPLC (Polaris solvent Delivery Module 210) and detector (ProStar 325) system is used, and the UV detector is set to a wavelength of 274 nm. A Macherey–Nagel (Düren, Germany) NUCLEODUR C18 HTec column (50 mm column length, 5 µm particle size, 21 mm inner diameter) is utilized running with acetonitrile (A) and water (B) with 0.1% formic acid, respectively. The gradient takes 25 min time per injection at a constant flow rate of 21 mL/min, with the following timetable for solvent A: 0 min 5%, 10 min 14.5%, 15 min 17%, 18 min 25%, 23 min 40%, 25 min 5%. The organic solvent of the combined fractions after the separation procedure is removed using rotary evaporation at water bath temperatures up to 35 °C. The remaining aqueous solution is again frozen and freeze-dried. The obtained powder is reconstituted in acetonitrile, centrifugated, and evaporated to dryness using constant nitrogen flow.
Compound identification and characterization via HPLC-qTOF-MS and purity check via HPLC-ELSD
The identification and MS/HRMS characterization of the isolated compounds is performed using a Bruker Daltonics GmbH & Co. KG (Bremen, Germany) Elute HPLC system coupled with a Bruker impact II qTOF mass spectrometer. In chapter 2.2 obtained compounds are dissolved in Millipore water and analyzed. This way, the isolated compounds can be assigned and matched with the expected autoxidation products theasinensin A/D and oolongtheanin digallate. The identification of the degradation and autoxidation products via mass spectrometry is performed by means of the exact mass and by comparison with fragmentation patterns from literature. A chromatographic separation ensures the differentiation between the two dimers theasinensin A and D. For the HPLC separation, a Macherey–Nagel NUCLEODUR phenyl-hexyl column (50 mm column length, 3 µm particle size, 2 mm inner diameter) is used, running a gradient with methanol (A) and water (B) with 0.1% formic acid, respectively, and the following timetable for solvent A: 0 min 5%, 1 min 5%, 8 min 40%, 10 min 100%, 12.5 min 100%, 12.51 min 5%, 15 min 5%. The solvent flow rate is constantly set to 400 µL/min. The qTOF mass spectrometer is set to Full MS mode and Auto MS/MS mode—to fragment the isolated compounds—with spectra rates of 1.6 and 3.2 Hz, respectively. The analysis is performed in negative ionization mode with a dry gas flow and temperature of 12.0 L/min and 250 °C, a nebulizer flow of 4.0 bar and a capillary voltage of − 3 kV.
After isolating and characterizing the autoxidation products, the purity of the isolated compounds is analyzed using HPLC coupled with an evaporative light-scattering detector (ELSD). An analytical Jasco HPLC system (Jasco Deutschland GmbH, Pfungstadt, Germany) and a Shimadzu (Shimadzu Deutschland GmbH, Duisburg, Germany) ELSD detector is used to measure possible impurities in the isolated fractions. A Macherey–Nagel NUCLEODUR phenyl-hexyl column (250 mm column length, 5 µm particle size, 4 mm inner diameter) is utilized, running a gradient with acetonitrile (A) and water (B) with 0.1% formic acid, respectively. The flow rate is constantly held at 1.3 mL/min. The gradient features the following timetable for solvent A: 0 min 5%, 2 min 5%, 16 min 40%, 21 min 100%, 25 min 100%, 25.1 min 5%, and 30 min 5%. The ELSD detector is set to a temperature of 50 °C. For the purity calculations, the peak area of the analyte is divided by the sum of all peaks measured during the analysis of the isolated compound solution minus the sum of all peaks during a blank measurement.
NMR spectroscopy
NMR spectra are obtained using a 600 MHz Agilent Technologies DD2 NMR spectrometer (Ratingen, Germany). 1H, 13C, gHSQC, gHMBC, COSY, and ROESY spectra are recorded. Deuterated acetonitrile with TMS as internal standard is chosen as NMR solvent for every compound to reduce compound degradation as seen in aqueous systems.
Polarimetry, photometry, circular dichroism spectroscopy, and enzymatic hydrolysis
Optical rotation values are recorded using a Schmidt + Haensch UniPol L1000 polarimeter (Berlin, Germany) with a sample light path length of 1 dm. Temperature, wavelength of the light, and used solvent are given with the calculated specific rotation values.
The UV absorption of theasinensin A, theasinensin D, and oolongtheanin digallate solutions in acetonitrile is measured at 274 nm using a Jasco V-750 spectrophotometer (Jasco Deutschland GmbH, Pfungstadt, Germany). Multiple solutions between 10 and 50 µg/mL are used and the extinction coefficient for each concentration and solution is calculated. After five measurements with different concentrations and extinctions between 0.1 and 1.0, the average extinction coefficient for each compound is calculated.
To measure the CD spectra, the compounds theasinensin A, D, and oolongtheanin digallate are dissolved in ACN. A solvent negative control is measured and subtracted from sample data. Different compound concentrations in the range 100–250 µg/mL are analyzed to get the most robust results. For the analysis, a Jasco J-600 CD spectropolarimeter (Jasco Deutschland GmbH, Pfungstadt, Germany) is used.
The enzymatic hydrolysis is carried out using tannase. 750 µg theasinensin A and D, respectively, are dissolved in 950 µL sodium acetate buffer (20 mM, pH 4.7), and 50 µL tannase solution (10 U/mL in the same buffer) is added. The solution is tempered at 30 °C for 60 min and subsequently, 4 mL of − 20 °C ACN is added to precipitate the enzyme. After centrifugation for 5 min at 3000×g, the solution is directly analyzed in the CD spectrometer. For the HPLC-qTOF-MS analysis, the centrifugated solution is diluted 1 + 9 (v/v) with water and analyzed using the same method as described in chapter 2.3.
Determination of TEAC values via Trolox assay
To analyze the antioxidative activity of the theasinensins and oolongtheanin digallate, the Trolox equivalent antioxidant capacity values are determined [29]. To obtain the ABTS radical cation solution, an aqueous solution of 7 mM ABTS and 2.45 mM potassium peroxodisulfate is stored at room temperature in the dark overnight for at least 12 h. The solution can be used for up to 48 h after addition of the radical initiator. The resulting, deep-blue solution is diluted 1 + 50 (v/v) with ethanol to an absorbance value A of A = 0.70 ± 0.20 at a wavelength of 734 nm and 30 °C. Trolox as well as the tested compounds are solved and diluted in ethanol and 2 µL/well of each solution is given in a 96 well plate. Blank wells of the same solvent are carried along as well. Each solution is measured in triplicates. For each well, 198 µL of the diluted ABTS radical cation solution is added, resulting in final Trolox concentrations between 0 and 30 µM. The well plate is sealed off and directly analyzed in a Tecan infinite 200 PRO microplate reader (Tecan Trading AG, Männedorf, Switzerland). After incubation in the plate reader for 6 min at 30 °C, the absorbance of each well at a wavelength of 734 nm is measured. The inhibition of absorbance relative to the blank of the same solvent after six minutes is plotted against the concentrations of Trolox or the tested compound, respectively. To calculate the TEAC value, the gradient of the linear regression of the testing compound plot is divided by the gradient of the linear regression of the Trolox plot. For the statistical analysis of the resulting TEAC values, a one-way analysis of variance (ANOVA) with a following Tukey post-hoc test is calculated. As level of significance, α = 0.05 is applied. The results of the calculated group division are given with asterisks.
Software
Microsoft Office 2019 is used to calculate, interpret, and plot analysis data. For statistical analysis, one-way ANOVA, and Tukey post hoc test, OriginPro 2021b (OriginLab Corporation, Northampton, USA) is used.
Results
To fulfill the aim of this project—the development of an isolation method of EGCG autoxidation products—information about the degradation of the compound itself is needed. This degradation is highly dependent on various factors. The pH value, temperature, solvent, light, ions in solution, and many other parameters have an effect on the stability of this particular polyphenol. To isolate degradation products, it is inevitable that the parameters and conditions of this autoxidation process are set up in a way that the concentration of the desired products is at its peak. Another contrary requirement for the most fitting parameters is the minimization of the parent compound concentration, in this case EGCG.
Development of the isolation procedure
To obtain the autoxidation products of EGCG, an appropriate product-containing solution of these is needed. Possible materials would be tea due to the presence of several autoxidation products and EGCG dimers in fermented and semi-fermented tea variations, or aqueous in vitro systems because of the low EGCG stability and autoxidation product formation. The latter is chosen for this isolation method because of lower matrix interferences with other tea polyphenols which would also be present in tea or tea products. Therefore, a buffer system with PBS buffer is selected for the formation of EGCG autoxidation products. This buffer, which is set to a pH value of 8 and tempered to 37 °C, combines several factors influencing EGCG stability. A great advantage of the buffer systems is the simpler sample preparation, especially with preparative reverse-phase liquid chromatography. Due to information in literature indicating greater gallocatechin gallate and EGCG stability at higher concentrations, a concentration of 0.1 mg/mL in PBS buffer is chosen for an incubation time of 30 min [15]. This incubation time goes along with other observations made in cell culture systems. A stabilization of the newly formed autoxidation products with ascorbic acid is also described in literature and, therefore, included in our isolation method [15].
One main challenge while optimizing the preparative HPLC method is the chromatographic separation of the two atropisomers theasinensin A and D. The quality of this optimization defines the purity of the subsequently isolated compounds more than other parameters. The development finally leads to the utilization of a reverse-phase C18 HPLC column with acetonitrile and water with 0.1% formic acid as liquid phase, respectively. A preparative HPLC–UV chromatogram of a representative sample is shown in Fig. 2. The fractions, which are detected at the wavelength of maximum UV absorbance of EGCG and its degradation products at 274 nm, are collected and analyzed via HPLC-qTOF-MS to assign the collected fractions to the autoxidation products. The purity of the isolated compounds is checked via HPLC-ELSD analysis to achieve a detection of all possible impurities, also of these without chromophores, and is calculated to be < 95% for each isolated autoxidation product. For ELSD chromatograms, see Supplementary Material Fig. S1.

Semipreparative HPLC–UV chromatogram of a representative sample at 274 nm, processed and prepared after the described EGCG incubation method, run with the depicted HPLC method
Peak identification via HPLC-qTOF-MS and MS/HRMS characterization
The isolated fractions are directly analyzed via HPLC-qTOF-MS to assign the already known autoxidation products to the peaks measured via preparative HPLC–UV. For the assignment, the exact masses as well as fragmentation patterns are consulted. All analyses are recorded in negative ionization mode because of the high hydroxylation degree of the suspected compounds. Prior to the qTOF mass analyzer, a diode array detector is connected upstream to also detect UV light absorption of the autoxidation products. This further simplifies the assignment of the isolated compounds.
As a result of the HPLC-qTOF-MS analysis, the four marked compounds in the HPLC–UV chromatogram (see Fig. 2) are successfully identified as three autoxidation products—two different theasinensins, which are differentiated through chromatographic separation, and oolongtheanin digallate—and EGCG itself, which is not completely converted during the incubation in buffer before the isolation. Combined chromatograms of the qTOF fraction examination are given in Fig. 3. The assignment of the two EGCG dimers theasinensin A and D is accomplished later through other structural elucidation methods like nuclear magnetic resonance and circular dichroism spectroscopy. All compounds are matched to the suspected analytes using their particular exact mass and the arising calculated sum formulas. The mass error between calculated and detected m/z is below 3 ppm for EGCG and its autoxidation product which further assures a high accuracy of the qTOF fraction control: EGCG ([M−H]− m/z 457.0776) mass error 2.4 ppm; theasinensin A ([M−H]− m/z 913.1469, [M−2H]2− m/z 456.0698) mass error 1.6 ppm and 2.1 ppm, respectively; theasinensin D (same adduct m/z as theasinensin A) mass error 2.2 ppm and 3.0 ppm, respectively; and oolongtheanin digallate ([M−H]− m/z 883.1366) mass error 2.7 ppm. At higher concentrations—for example during the future fragmentation experiments—even lower mass error between 0.1 and 0.7 ppm are accomplished. While analyzing the theasinensins via qTOF mass spectrometry, it could be observed that both compounds have different ionization behavior: Both theasinensins show [M−H]− and [M−2H]2− ionization on the used instrument in negative polarity. The difference between the two atropisomers lies in the ratio between the intensity of the single negative ionization [M−H]− and the double negative ionization [M−2H]2−. The isotope pattern of the [M−2H]2− ion suggests a multiple charged ion based on the halved m/z gap of the individual isotope ions. While the first eluting theasinensin A has a higher intensity for the double-charged ion, it is the other way around for the second dimer, theasinensin D. More interestingly, the less intense adduct seems to feature one fifth of the Full MS ion intensity of the major adduct. This ratio is observable for both theasinensin A and D, as Fig. 4 shows.

Combined HPLC-qTOF-HRMS chromatograms of four isolated fractions after semipreparative HPLC–UV, mass width of each extracted ion chromatogram ± 0.01 Da, with spectrometric and spectroscopic techniques fractions could be identified as following: a Theasinensin A, b theasinensin D, c EGCG, and d oolongtheanin digallate

qTOF-HRMS spectra acquired in Full MS mode, normalized to the highest particular ion intensity. a Mass spectrum at the retention time of theasinensin A; b mass spectrum at the retention time of theasinensin D
MS/HRMS qTOF experiments are also performed to identify and characterize the autoxidation products. For these experiments, the impact II Auto MS/MS mode is used to fragment the parent ions theasinensin A and D as well as oolongtheanin digallate. The fragmentation patterns are in line to these described in literature. For all the analyzed autoxidation products, intense fragment ions are the parent ions with the loss of one and two gallic acid groups, as well as gallic acid itself ([M−H]− m/z 169.0142). The fragmentation patterns of the [M−H]− adduct and the [M−2H]2− adduct as parent ions, respectively, for each theasinensin A and theasinensin D are shown in Fig. 5a–d. The [M−H]− fragmentation pattern of oolongtheanin digallate can be found in Fig. 5e. There are observable differences in the fragmentation of the dimers depending on their three-dimensional, stereochemical properties. The main fragments for the EGCG dimers are represented by gallic acid itself and the hydrolyzed theasinensins, but there is an additional elimination of water during fragmentation of the gallic acid compared to theasinensin B/H and C/E (m/z 743.1253 and m/z 591.1144, respectively). In the case of the double negative charged ion, epigallocatechin (EGC, m/z 305.0667), and gallic acid are the most intense fragments. Looking at the parent ion theasinensin A [M−H]−, the intensities of the fragments theasinensin B-(H2O) and gallic acid in relation to the theasinensin C-(2 H2O) fragment intensity are higher than the relative intensities of the correspondent theasinensin D fragments. The fragmentation of the [M−2H]2− parent ion demonstrates a higher intensity of EGC—relative to gallic acid—for theasinensin D than for theasinensin A.

qTOF-HRMS/MS fragmentation spectra and theasinensin ROESY couplings; MS spectra acquired in Auto MS/MS mode and normalized to the highest particular fragment ion intensity. a Fragment spectrum of the [M−H]− adduct of theasinensin A (m/z 913.1469), b fragment spectrum of the [M−2H]2− adduct of theasinensin A (m/z 456.0698), c fragment spectrum of the [M−H]− adduct of theasinensin D (m/z 913.1469), d fragment spectrum of the [M−2H]2− adduct of theasinensin D (m/z 456.0698), e fragment spectrum of the [M−H]− adduct of oolongtheanin digallate (m/z 883.1363), and f chemical structure of theasinensin A/D with all visible ROESY proton couplings
NMR spectroscopy
To further identify the isolated compounds, NMR spectra of the two EGCG dimers theasinensin A and D, as well as oolongtheanin digallate, are obtained. Coupling atoms are identified using gHMBC, gHSQC, COSY, and ROESY techniques. The chemical shifts of 13C and 1H atoms are displayed in Table 1 according to the atom numbering shown in Fig. 1. For the 1D NMR spectra, see Supplementary Material Figs. S2–S4.
There are differences in the analyzed chemical shifts of the atropisomers. The biggest discrepancy between theasinensin A and D occurs in the C and C′ ring, which share the same shifts due to the dimeric structure: The protons bound to C-3 and C-4 show the greatest span in chemical shift between the two compounds—5.06–5.51 ppm and 2.51–2.69 ppm, respectively. The differentiation between the dimers is possible based on the shift difference of these protons as well as the protons bound to C-8 in the A ring and C-6 at the B ring system. The discrepancy of the shift of the two lastly mentioned protons is of another special interest. A possible in- or decrease in their three-dimensional coupling depending on the B,B′ ring rotation would be detectable in NOESY or ROESY experiments. Unfortunately, there is no difference found in the coupling of these protons. All ROESY couplings of theasinensin A and theasinensin D are shown in Fig. 5f, the ROESY NMR spectra are shown in the Supplementary Material Fig. S5.
Polarimetry, photometry, CD spectroscopy, and enzymatic hydrolysis
To gather further information on the spectroscopic properties of the isolated compounds, the optical rotation of both theasinensin A/D and oolongtheanin digallate is measured and the extinction coefficients of these compounds at 274 nm are calculated. Based on the used concentration, the specific rotation and the extinction coefficients are calculated after solvent blank subtraction. All given optical rotation values and absorption values are measured using acetonitrile as solvent, the data are shown in Table 2.
CD spectra of the two theasinensins as well as oolongtheanin digallate are recorded. For the autoxidation products, a concentration of 150 µg/mL in acetonitrile is used to obtain the shown spectra in Fig. 6a. Both dimers show a negative Cotton effect, especially at wavelengths between 270 and 290 nm. Notably, there seems to be no difference in the CD spectra of the theasinensins A and D, despite their atropisomeric properties. The uniformity of the mass spectra and the fragment ions of the two theasinensins in addition to the chromatographic separation further assures that these compounds are isomeric, so there should be characteristic differences in CD absorbance. The gallic acid bound to the A and A′ ring might be the reason for possible interferences in the CD spectra. These groups have a great influence on the stereochemical characteristics of these compounds. An expectable difference in CD absorbance between theasinensin A and D might be covered by the gallic acid CD absorbance of higher intensity. To reduce this impact, tannase is used to expose the B,B′ ring system of the dimers. This enzyme hydrolyzes gallic acid from tannic acid or digallate in general. Fitting parameters for the incubation of the isolated theasinensins A and D with tannase are identified using gallotannin as positive control. After tannase incubation, the hydrolysis products are analyzed using HPLC-qTOF-MS to control the success of the enzymatic reaction, see Fig. 7. For the dimers, two new signals at m/z 609.1250 could be identified as hydrolysis products from theasinensin A and D—named theasinensin C and theasinensin E, see Fig. 1—with one signal for each theasinensin atropisomer, respectively. These products also differ in their retention times, as it is expected because these compounds have different isomeric properties as well. After the qTOF-HRMS control, the CD spectra of the hydrolyzed theasinensins are obtained and shown in Fig. 6b. After blank solvent correction, the two hydrolyzed theasinensins differ in their CD absorbance at their UV absorbance maximum around 270–290 nm. While the hydrolyzed theasinensin A shows a positive Cotton effect relative to the negative control, it is the other way around for the hydrolyzed theasinensin D. These data further verify the isomeric and atropisomeric characteristics and differences between the theasinensins A and D and help to develop isolation and structure elucidation methods for EGCG autoxidation products.

Blank value corrected circular dichroism spectra of the autoxidation products before and after the enzymatic hydrolysis. a CD spectra of theasinensin A, theasinensin D, and oolongtheanin digallate before the hydrolysis; b CD spectra of theasinensin C and theasinensin E as hydrolysis products after enzymatic hydrolysis

Combined HPLC-qTOF-HRMS chromatograms of theasinensin A and theasinensin D before and after tannase hydrolysis, mass width of each extracted ion chromatogram ± 0.01 Da. a Theasinensin A, b theasinensin A after tannase hydrolysis to theasinensin C, c theasinensin D, and d theasinensin D after tannase hydrolysis to theasinensin E
Trolox equivalent antioxidant capacity of EGCG and its autoxidation products
To characterize the antioxidant capacity of the autoxidation products, EGCG as well as theasinensin A, D and oolongtheanin digallate is analyzed regarding their trolox equivalent antioxidant capacity. This way, a direct comparison between the polyphenolic monomer and its dimers is possible. For all compounds, a concentration range of 0.5–5 µM is tested and narrowed down to their linear range. These concentrations induced absorbance inhibition values up to 80% relative to the negative solvent control. The inhibition of absorption at 734 nm after 6 min incubation at 30 °C is shown for both compounds in Fig. 8, with the correspondent inhibition by Trolox on the same well plate, respectively. The TEAC values are calculated based on the linear regression gradient of relative absorbance inhibition by EGCG and its autoxidation products and their particular correspondent trolox point of reference. Furthermore, an EGCG equivalent antioxidant capacity (EEAC) value is calculated in the same manner for theasinensin A, D, and oolongtheanin digallate. For all compounds, the TEAC and EEAC values are given in Table 3. For all linear regressions, coefficients of determination R2 > 0.97 are obtained.

Resulting plots of the Trolox equivalent antioxidant capacity assay. a Relative absorbance inhibition through trolox (circle), EGCG (grey diamond), theasinensin A (triangle), and theasinensin D (black square) with each linear regression; b relative absorbance inhibition through trolox (circle), EGCG (grey diamond) and oolongtheanin digallate (cross) with each linear regression
Discussion
In this study, an isolation method of the EGCG degradation and autoxidation products theasinensin A, theasinensin D and oolongtheanin digallate is presented as well as structure elucidation techniques to characterize these compounds. The chromatographic separation which is shown in this study does not mark the first analytical method to separate the described compounds, but it is the first preparative isolation procedure published in literature to yield multiple autoxidation products. The degradation and autoxidation of EGCG and flavonoids similar to EGCG in buffer to theasinensins and oolongtheanin digallate is known to occur [15, 23], but it is not used as point of origin for theasinensin isolation with these specific incubation parameters and a dedicated sample preparation method before. The conversion of EGCG into theasinensin A via dehydrotheasinensin A using copper chloride and ascorbic acid is described in literature [23, 30]. While this approach is similar to the one described here, it is further from in vitro conditions. The advantage of a realistic in vitro buffer system is the easier possibility to modify the method to isolate other, non-abundant by-products that are also present in cell culture systems. Other released analytical separation methods usually base upon reversed phase C18 columns with ACN and water as mobile phase. While the preparative HPLC isolation method that is shown here rests upon the same column material and eluent, the developed analytical method with the purpose of peak identification via HPLC-qTOF-HRMS operates on phenyl-hexyl column material with methanol and water as mobile phase. This enables the separating capacity of π–π stacking between the phenyl part of the stationary phase and the gallic acid groups of the reviewed compounds, leading to an improved separation of the theasinensin atropisomers. This separation also helps to identify and assign theasinensin A and D to the isolated compounds.
The obtained data on HRMS and MS/HRMS characteristics of the theasinensins and oolongtheanin digallate also enable a comparison with previous information published in literature. While the fragmentation patterns of the isolated polyphenols fit to the spectra of earlier studies [14], differences in multiple charged ion intensities of the parental compounds have not yet been described. In this regard, it could be demonstrated that theasinensin A shows a factor 5 higher intensity for the [M−2H]2− adduct relative to the single charged ion in Full MS mode, and vice versa concerning theasinensin D. A possible explanation for this observation could be the different three-dimensional, stereoscopic orientation of the two atropisomers. Due to the varying alignment of the bridged B,B′ rings and the two gallate groups of the two dimers, a chelating effect of double-positive charged ions is imaginable, with different distinct tendencies based on the steric orientation. Different cation chelating properties could explain the varying stability and intensity of the correspondent parent ions among the theasinensins.
Although theasinensin A and D and oolongtheanin digallate have been described in literature before, a new spectroscopic evaluation of these compounds is necessary and inevitable to distinguish between these highly similar isolated compounds. While NMR data are available, the difference in the chemical shift between several carbon atoms and protons of the theasinensin atropisomers is too small to tell these compounds apart, at least without another structural elucidation methods to confirm and reassure the findings. The NMR data of the atropisomers differs in a few signals, which can be traced back on different three-dimensional coupling of atoms due to the inversed B,B′ ring rotation. Starting with the information on the NMR chemical shifts of theasinensin A and D atoms, publications in literature indicate different chemical shifts for the methylene group protons bound to C-4 at the C ring of theasinensin A. While Hashimoto et al. could not differentiate between these protons of theasinensin D [21], a splitting could be observed in this present study. The chemical shift of the methylene protons of theasinensin D could be numbered to 2.69 ppm and 2.75 ppm. While most of the chemical shifts measured via NMR accord and chime with previous results, there are only a few protons with different tendencies between earlier and this present study [21,22,23]. For example, the proton bound to C-6 at the B ring shows a chemical shift of 6.88 ppm and 6.76 ppm for theasinensin A and D, respectively, with a downfield shift at theasinensin D. In literature, a high field shift for this proton is described [21]. Nevertheless, the obtained data fit to these displayed in earlier studies in nearly all aspects but expand the structural information on these compounds with further NMR techniques like NOESY and ROESY elucidation. Differences in individual shifts might be traced back on different NMR solvents which do not match in every published study [21, 22].
Besides qTOF-HRMS and NMR spectroscopy, the specific rotation of the isolated compounds is analyzed and calculated. While there are observable differences to previous publications, the optical rotation still clearly indicates whether the first and second eluting theasinensin is equal to theasinensin A or D, respectively. Calculated specific optical rotation values may be prone to imprecisely adjusted concentrations or impurities. Nonaka et al. as well as Hashimoto et al. represent the only source of specific optical rotation values for these EGCG dimers and they do not give any information about the purity of their theasinensin A and D solutions [21, 22]. Additionally, their optical rotation values are given for slightly differing temperatures at 15 °C and 28 °C and different solvents as well. Although the values do not fit precisely—for theasinensin A \(\left[ {\upalpha } \right]_{{\text{D}}}^{{{20}}}\) − 218.6° vs. \(\left[ {\upalpha } \right]_{{\text{D}}}^{{{15}}}\) − 226.8° [22] and for theasinensin D \(\left[ {\upalpha } \right]_{{\text{D}}}^{{{20}}}\) − 116.0° vs. \(\left[ {\upalpha } \right]_{{\text{D}}}^{{{28}}}\) − 158.6° [21]—, the difference in the optical rotation between theasinensin A and D is great enough to help the atropisomeric assignment, especially in combination with the other spectrometric and spectroscopic methods performed in this study.
While there are no CD data available for theasinensin A and D, the obtained CD spectra of the hydrolysis products theasinensin C and E fit to these described in literature at the UV absorbance maximum between 270 and 290 nm. Hashimoto et al. also show a positive Cotton effect for theasinensin C and—vice versa—a negative Cotton effect for theasinensin E [21]. A reason for the missing CD information in literature on theasinensins with bound gallic acid groups might be the overlaying effects of these substituents on this spectroscopic analysis. Nevertheless, the structure elucidation via CD spectroscopy was executed successfully through tannase hydrolysis and helps to further assure the atropisomeric differentiation. For oolongtheanin digallate, there is no comparable CD information given in literature. The additional CD absorbance maximum of this compound at 350 nm can be used as supplementary structural elucidation technique to distinguish between EGCG autoxidation products.
The analysis of the antioxidant capacity of the autoxidation products in comparison to their parental monomer EGCG reflects a first characterization of the isolated compounds acquired by this protocol using the Trolox assay. While EGCG has been intensively characterized in literature, the antioxidant properties of other tea ingredients like the theasinensins have only been analyzed using more specialized photometric methods [27]. The calculated EGCG TEAC value of 3.50 is slightly lower than equivalent published values [31]. The functionality of the assay has been tested with other flavonoids as well, like quercetin, where published TEAC values are also replicable. Although another method for the determination of antioxidant effects show comparable properties of EGCG and its autoxidation products [27], the Trolox assay indicates a higher capacity of the theasinensins, which reaches almost double with theasinensin D. An explanation for this discrepancy lies in the different methodical approach, which might not take all challenges regarding these compounds into account. For example, other assays often feature an incubation with other oxidable agents like linoleic acid over a prolonged period of time at higher temperatures [27], not considering the low stability of EGCG and its autoxidation products in these conditions [13]. Altogether, the comparison between the monomer and the autoxidation products is possible and successful nevertheless, showing an even greater antioxidant capacity for theasinensin A, D and oolongtheanin digallate than for EGCG itself. This is also observable in the EGCG equivalent antioxidant capacity value. The statistical analysis shows a clear group division of EGCG and its autoxidation products, with theasinensin A and oolongtheanin digallate being in the same statistical group. The even higher antioxidant capacity of theasinensin D could be statistically supported using one-way ANOVA. This finding further indicates that these rather uncharacterized compounds might be the source of many in vitro, in vivo or cell culture-related effects—which might be traced back to EGCG with the current state of knowledge.
All in all, this study presents a novel generation and isolation protocol of EGCG dimers and autoxidation products, namely theasinensin A, theasinensin D, and oolongtheanin digallate. Besides the isolation procedure, several spectroscopic structure elucidation techniques, as well as high-resolution mass spectrometric information are featured and shown. To perform a characterization of the isolated products, these compounds are compared to the monomer EGCG regarding their antioxidant capacity using the Trolox assay. All this information should help to characterize the effects of polyphenolic degradation and reaction products like these which are present in food-based material as well as in cell culture and in vitro situations. To fully understand the characteristics of such compounds and their impact on cell culture systems and the human health, there are numerous aspects that must be further investigated in upcoming experiments—which base on protocols concerning the isolation and methods for the structure elucidation of these compounds.
Availability of data and material
Not applicable.
Code availability
Not applicable.
References
Brody H (2019) Tea. Nature 566(7742):S1. https://doi.org/10.1038/d41586-019-00394-5
Sergi CM (2020) Epigallocatechin-3-gallate toxicity in children: a potential and current toxicological event in the differential diagnosis with virus-triggered fulminant hepatic failure. Frontiers 10:1563. https://doi.org/10.3389/fphar.2019.01563
Hu J, Webster D, Cao J, Shao A (2018) The safety of green tea and green tea extract consumption in adults—results of a systematic review. Regul Toxicol Pharm 95:412–433. https://doi.org/10.1016/j.yrtph.2018.03.019
Lin Y-L, Juan I-M, Chen Y-L, Liang Y-C, Lin J-K (1996) Composition of polyphenols in fresh tea leaves and associations of their oxygen-radical-absorbing capacity with antiproliferative actions in fibroblast cells. J Agric Food Chem 44(6):1387–1394. https://doi.org/10.1021/jf950652k
Marais JPJ, Deavours B, Dixon RA, Ferreira D (2008) The stereochemistry of flavonoids. In: Grotewold E (ed) The science of flavonoids. Springer, New York. https://doi.org/10.1007/978-0-387-28822-2_1
Du G-J, Zhang Z, Wen X-D, Yu C, Calway T, Yuan C-S, Wang C-Z (2012) Epigallocatechin gallate (EGCG) is the most effective cancer chemopreventive polyphenol in green tea. Nutrients 4(11):1679–1691. https://doi.org/10.3390/nu4111679
Song X, Du J, Zhao W, Guo Z (2017) Epigallocatechin-3-gallate (EGCG): mechanisms and the combined applications. Comb Chem High Throughput Scr 20:872–885. https://doi.org/10.2174/1386207321666171218115850
Duthie GG, Duthie SJ, Kyle JAM (2000) Plant polyphenols in cancer and heart disease: implications as nutritional antioxidants. Nutr Res Rev 13(1):79–106. https://doi.org/10.1079/095442200108729016
Gupta S, Ahmad N, Nieminen A-L, Mukhtar H (2000) Growth inhibition, cell-cycle dysregulation, and induction of apoptosis by green tea constituent (−)-epigallocatechin-3-gallate in androgen-sensitive and androgen-insensitive human prostate carcinoma cells. Toxicol Appl Pharm 164(1):82–90. https://doi.org/10.1006/taap.1999.8885
Shim J-H, Su Z-Y, Chae J-I, Kim DJ, Zhu F, Ma W-Y, Bode AM, Yang CS, Dong Z (2010) Epigallocatechin gallate suppresses lung cancer cell growth through Ras-GTPase-activating protein SH3 domain-binding protein 1. Cancer Prev Res 3(5):670–679. https://doi.org/10.1158/1940-6207.CAPR-09-0185
EFSA Panel on Food Additives and Nutrient Sources added to Food (ANS), Younes M, Aggett P, Aguilar F, Crebelli R, Dusemund B, Filipič M, Frutos MJ, Galtier P, Gott D, Gundert-Ry U, Lambré C, Leblanc J-C, Lillegaard IT, Moldeus P, Mortensen A, Oskarsson A, Stankovic I, Waalkens-Berendsen I, Woutersen RA, Andrade RJ, Fortes C, Mosesso P, Restani P, Arcella D, Pizzo F, Smeraldi C, Wright M (2018) Scientific opinion on the safety of green tea catechins. EFSA J 16(4):5239. https://doi.org/10.2903/j.efsa.2018.5239
Hong J, Lu H, Meng X, Ryu J-H, Hara Y, Yang CS (2002) Stability, cellular uptake, biotransformation, and efflux of tea polyphenol (−)-epigallocatechin-3-gallate in HT-29 human colon adenocarcinoma cells. Cancer Res 62(24):7241–7246
Krupkova O, Ferguson SJ, Wuertz-Kozak K (2016) Stability of (−)-epigallocatechin gallate and its activity in liquid formulations and delivery systems. J Nutr Biochem 37:1–12. https://doi.org/10.1016/j.jnutbio.2016.01.002
Sang S, Lee M-J, Hou Z, Ho C-T, Yang CS (2005) Stability of tea polyphenol (−)-epigallocatechin-3-gallate and formation of dimers and epimers under common experimental conditions. J Agric Food Chem 53:9478–9484. https://doi.org/10.1021/jf0519055
Wu Q-Q, Liang Y-F, Ma S-B, Li H, Gao W-Y (2019) Stability and stabilization of (−)-gallocatechin gallate under various experimental conditions and analyses of its epimerization, auto-oxidation, and degradation by LC-MS. J Sci Food Agric 99(13):5984–5993. https://doi.org/10.1002/jsfa.9873
Xiao J, Högger P (2015) Stability of dietary polyphenols under the cell culture conditions: avoiding erroneous conclusions. J Agric Food Chem 63(5):1547–1557. https://doi.org/10.1021/jf505514d
Long LH, Clement MV, Halliwell B (2000) Artifacts in cell culture: rapid generation of hydrogen peroxide on addition of (−)-epigallocatechin, (−)-epigallocatechin gallate, (+)-catechin, and quercetin to commonly used cell culture media. Biochem Biophys Res Commun 273(1):50–53. https://doi.org/10.1006/bbrc.2000.2895
Nakagawa H, Wachi M, Woo JT, Kato M, Kasai S, Takahashi F, Lee I-S, Nagai K (2002) Fenton reaction is primarily involved in a mechanism of (−)-epigallocatechin-3-gallate to induce osteoclastic cell death. Biochem Biophys Res Commun 292(1):94–101
Nakazato T, Ito K, Ikeda Y, Kizaki M (2005) Green tea component, catechin, induces apoptosis of human malignant B cells via production of reactive oxygen species. Clin Cancer Res 11(16):6040–6049. https://doi.org/10.1006/bbrc.2002.6622
Simon H-U, Hak-Yehia A, Levi-Schaffer F (2000) Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 5(5):415–418. https://doi.org/10.1023/A:1009616228304
Hashimoto F, Nonaka G-I, Nishioka I (1988) Isolation and structure elucidation of B, B′-linked bisflavanoids, theasinensins D-G and oolongtheanin from oolong tea. Chem Pharm Bull 36(5):1676–1684. https://doi.org/10.1248/cpb.36.1676
Nonaka G-I, Kawahara O, Nishioka I (1983) A new class of dimeric flavan-3-ol gallates, theasinensins A and B, and proanthocyanidin gallates from green tea leaf. Chem Pharm Bull 31(11):3906–3914. https://doi.org/10.1248/cpb.31.3906
Tanaka T, Watarumi S, Matsuo Y, Kamei M, Kouno I (2003) Production of theasinensins A and D, epigallocatechin gallate dimers of black tea, by oxidation–reduction dismutation of dehydrotheasinensin A. Tetrahedron 59(40):7939–7947. https://doi.org/10.1016/j.tet.2003.08.025
Shii T, Tanaka T, Watarumi S, Matsuo Y, Miyata Y, Tamaya K, Tamaru S, Tanaka K, Matsui T, Kouno I (2011) Polyphenol composition of a functional fermented tea obtained by tea-rolling processing of green tea and loquat leaves. J Agric Food Chem 59(13):7253–7260. https://doi.org/10.1021/jf201499n
Yoshino K, Suzuki M, Sasaki K, Miyase T, Sano M (1999) Formation of antioxidants from (−)-epigallocatechin gallate in mild alkaline fluids, such as authentic intestinal juice and mouse plasma. J Nutr Biochem 10(4):223–229. https://doi.org/10.1016/S0955-2863(98)00103-X
Blokhina O, Virolainen E, Fagerstedt KV (2003) Antioxidants, oxidative damage and oxygen deprivation stress: a review. Ann Bot Lond 91:179–194. https://doi.org/10.1093/aob/mcf118
Hashimoto F, Masateru O, Masuoka C, Ito Y, Sakata Y, Shimizu K, Nonaka G-I, Nishioka I, Nohara T (2003) Evaluation of the anti-oxidative effect (in vitro) of tea polyphenols. Biosci Biotechnol Biochem 67(2):396–401. https://doi.org/10.1271/bbb.67.396
Shibamoto T, Junji T, Toshihiko O (1998) Functional foods for disease prevention I: fruits, vegetables, and teas. American Chemical Society, Washington DC
Re R, Pellegrini N, Proteggente A, Pannala A, Yang M, Rice-Evans C (1999) Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic Biol Med 26(9):1231–1237. https://doi.org/10.1016/S0891-5849(98)00315-3
Shii T, Miyamoto M, Matsuo Y, Tanaka T, Kouno I (2011) Biomimetic one-pot preparation of a black tea polyphenol theasinensin A from epigallocatechin gallate by treatment with copper(II) chloride and ascorbic acid. Chem Pharm Bull 59(9):1183–1185. https://doi.org/10.1248/cpb.59.1183
Muzolf-Panek M, Gliszczyńska-Świgło A, Szymusiak H, Tyrakowska B (2012) The influence of stereochemistry on the antioxidant properties of catechin epimers. Eur Food Res Technol 235(6):1001–1009. https://doi.org/10.1007/s00217-012-1826-4
Acknowledgements
We thank Dr. Jens Köhler from the Institute for Pharmaceutical and Medicinal Chemistry of the WWU Münster for the measurement of the NMR spectra.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Not applicable.
Corresponding author
Ethics declarations
Conflict of interest
No conflicts of interest are declared.
Compliance with ethics requirements
This publication does not contain any human or animal experimental data and features only experiments without human participants or animal subjects.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Alfke, J., Kampermann, U., Kalinina, S. et al. Isolation and structural elucidation of dimeric epigallocatechin-3-gallate autoxidation products and their antioxidant capacity. Eur Food Res Technol 247, 2961–2975 (2021). https://doi.org/10.1007/s00217-021-03846-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-021-03846-3