
Abstract
An odorant screening by gas chromatography–olfactometry (GC–O) and a crude aroma extract dilution analysis (AEDA) applied to the volatiles isolated from a light and a dark liquid malt extract (LME) by solvent extraction and solvent-assisted flavour evaporation (SAFE) identified 28 odorants. Fifteen major odorants were subsequently quantitated and odour activity values (OAVs) were calculated as ratio of the concentration to the respective odour threshold value (OTV). Important odorants in the light LME included 3-(methylsulfanyl)propanal (OAV 1500), (E)-β-damascenone (OAV 430), and 4-ethenyl-2-methoxyphenol (OAV 91). In the dark LME, sotolon (OAV 780), 3-(methylsulfanyl)propanal (OAV 550), (E)-β-damascenone (OAV 410), acetic acid (OAV 160), and maltol (OAV 120) were of particular importance. To get an insight into the changes during malt extract production, the quantitations were extended to the malt used as the starting material for both LMEs. Addition of a minor amount of water to malt before volatile extraction was shown to be effective to cover the free as well as the bound malt odorants. Results showed that some LME odorants originated from the starting material whereas others were formed during processing. Important process-induced LME odorants included (E)-β-damascenone and 4-ethenyl-2-methoxyphenol in the light LME as well as maltol, sotolon, (E)-β-damascenone, and 2-methoxyphenol in the dark LME. In summary, the odorant formation during LME production was shown to be more important than the transfer of odorants from the malt.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Malts are germinated and re-dried grains, primarily of barley, but also of wheat, rye, and other cereals. The grains are first treated with water until the moisture content increases from ~ 12% to 43–48%. This initiates germination, during which enzymes start to break down starch to reducing sugars. After 4–6 days, the so-called green malt is dried in a kiln to obtain pale malt with a residual moisture content of 3.5–4% or dark malt with a residual moisture content of 1.5–2% [1, 2]. Malts can be further processed to malt extracts. For this purpose, the malt grains are milled and mashed with warm water. Application of a specific temperature program results in a substantial enzymatic degradation of biopolymers such as starch and proteins. After separation from the solid grain particles (malt draff), the aqueous phase is concentrated to yield a liquid malt extract (LME) with a syrup-like consistency. The LME can be further dried, e.g. by spray drying, to obtain a crystalline material marketed as dried malt extract (DME) [2, 3].
Malt extracts are mainly used in the baking industry. There is also a significant use of malt extracts for the production of confectionery, breakfast cereals, and other food products as well as for home brewing. In the baking industry, malt extracts are added to flours with low diastatic activity to provide fermentable sugars [4, 5], but also to enhance colour and aroma of bakery products. Particularly the contribution of malt extracts to the aroma of bakery products is yet poorly understood. The olfactory profile of malt extracts is characterized by malty, caramel-like, and honey-like odour notes. However, little is known on their molecular background.
In 1980, Farley and Nursten [6] applied gas chromatography–olfactometry (GC–O) and gas chromatography–mass spectrometry (GC–MS) to a volatile isolate obtained from an LME by simultaneous distillation/extraction (SDE) [7]. Forty-seven odour-active compounds were detected. GC–MS resulted in 38 structure proposals. However, structure assignments were not confirmed by GC–O of reference substances. Furthermore, the fact that SDE leads to extensive artefact formation [8] was not addressed. The compounds 2- and 3-methylbutanal were highlighted as odorants with malty characteristics and a high importance for the organoleptic properties of the LME. Przybylski and Kamiński [9] used GC to separate the volatiles isolated from a rye malt extract by liquid–liquid extraction into 39 fractions and evaluated their odour. GC–MS analysis led to 32 structure proposals, but no attempt was made to assess the role of the identified compounds for the odour of the fractions.
Unlike the scarce scientific literature on malt extract volatiles, numerous papers have been published on the volatiles in different malts, including green malt [10, 11], peated malt [12,13,14], dark malt [10, 15], crystal malt [11, 15, 16], caramel malt [10, 17,18,19], and roasted malt [15, 18, 20]. In summary, ~ 250 volatiles have been identified [21]. The odour contribution of individual malt volatiles was assessed in a study on caramel malt [10, 17]. Application of GC–O in combination with an aroma extract dilution analysis (AEDA) [22] to a volatile isolate obtained by solvent extraction and solvent-assisted flavour evaporation (SAFE) [23] revealed high flavour dilution (FD) factors for 3-methylbutanal, oct-1-en-3-one, 3-(methylsulfanyl)propanal, (2E,4E)-deca-2,4-dienal, 2- and 3-methylbutanoic acid, 4-hydroxy-2,5-dimethylfuran-3(2H)-one (HDMF), and vanillin. Quantitation experiments, the calculation of odour activity values (OAVs), aroma reconstitution, and omission tests finally showed that the malty smelling aldehydes 3-methylbutanal (OAV 235) and 2-methylpropanal (OAV 70) were key compounds in the aroma of the caramel malt.
In summary of the literature overview, a comprehensive study on the key odorants in malt extracts for the baking industry was still lacking. In particular, it was unknown, whether malt extract odorants mainly originate from a transfer of malt odorants or whether a de novo formation during the processing of the malt to the malt extract also makes a contribution. This knowledge, however, is crucial for a targeted optimization of the aroma of malt extracts. The objective of the present study was to elucidate the major odour-active compounds in a light LME and in a dark LME for the baking industry, both produced from the same malt, and relate their concentrations to the concentrations that were initially present in the malt.
Materials and methods
Malt and malt extract samples
Light LME (77.5–81.0% dry matter, 48.2% sugar), dark LME (59.0–65.0% dry matter, 22.1% sugar), and the Pilsner malt from which both malt extracts had been produced, were obtained from Ireks (Kulmbach, Germany). The light LME was obtained from the Pilsner malt after milling, mashing with water, and application of vacuum concentration to the aqueous extract. The dark LME was obtained from the light LME by an additional heating step followed by dilution with water. 1 kg Pilsner malt yielded 0.94 kg light LME and 1.20 kg dark LME.
Reference odorants
The following compounds were purchased from commercial sources: 1, 22 (Alfa Aesar, Karlsruhe, Germany), 2–5, 8–10, 12, 14, 16, 17, 19, 20, 23, 24, and 26–28 (Merck, Darmstadt, Germany). Compound 11 was a gift from Symrise (Holzminden, Germany). The following compounds were synthesised as detailed in the literature: 6 [24] and 18 [25].
Stable isotopically substituted odorants
The following compounds were purchased from commercial sources: (2H3)-4 and (13C2)-26 (Merck), (2H5)-16 (CDN Isotopes, Quebec, Canada), and (13C2)-20 (aromaLAB, Planegg, Germany). The following compounds were synthesised as detailed in the literature: (2H3)-5 [26], (13C2)-8 [27], (2H2)-9 [28], (2H6)-11 [29], (2H3)-14 [30], (13C6)-22 [31], (13C2)-23 [32], and (2H3)-27 [33]. (13C2)-10 was prepared using the approach detailed in [31], but starting from (2E)-oct-2-enal instead of (2E)-(1,2-13C2)oct-2-enal. (13C2)-17 was synthesised as detailed below.
Miscellaneous chemicals
The following chemicals were purchased from commercial sources: (13C)ethanoic anhydride (Euriso-top, Saint-Aubin, France), furan, 2-methylpropan-2-ol, and sodium borohydride (Merck), nitromethane, aqueous sodium hypochlorite solution with 5% available chlorine, and tin(II) trifluoromethanesulfonate (VWR, Darmstadt, Germany). Diethyl ether, dichloromethane (CLN, Freising, Germany), and pentane (VWR) were freshly distilled through a column (120 cm × 5 cm) packed with Raschig rings.
Synthesis of (13C2)maltol [(13C2)-17]
1-(Furan-2-yl)(13C2)ethan-1-one
Following the approach published for the isotopically unmodified compound [34], Friedel–Crafts acylation of furan (75 mg, 1.1 mmol) with (13C)ethanoic anhydride (250 mg, 2.4 mmol) in dichloromethane (1 mL) using tin(II) trifluoromethanesulfonate (23 mg, 0.055 mmol) as Lewis acid resulted in 1-(furan-2-yl)(13C2)ethan-1-one.
1-(Furan-2-yl)(13C2)ethan-1-ol
The 1-(furan-2-yl)(13C2)ethan-1-one obtained above was dissolved in methanol (5 mL) and sodium borohydride (60 mg, 1.6 mmol) was slowly added, while maintaining the temperature below 40 °C. The solvent was removed in vacuo, water (20 mL) was added and the product was extracted with diethyl ether (3 × 50 mL). After drying over anhydrous sodium sulphate, the solvent was removed in vacuo to yield 1-(furan-2-yl)(13C2)ethan-1-ol [10].
Synthesis of (13C2)maltol [(13C2)-17]
Following the approach for the synthesis of the isotopically unmodified compound [35], the 1-(furan-2-yl)(13C2)ethan-1-ol obtained above was added to acetic acid (5 mL) and reacted with tert-butyl hypochlorite (1.3 g, 12 mmol) previously prepared as detailed in [36]. The resulting product was purified by silica gel chromatography with a pentane/diethyl ether gradient to afford 3.3 mg of 3-hydroxy-2-(13C)methyl(2-13C)-4H-pyran-4-one, that is (13C2)maltol, in 99.95% purity (GC–FID) equivalent to 2.4% overall yield. MS (EI): m/z (%) 71 (100), 128 (91), 45 (78), 43 (50), 57 (39), 55 (38), 99 (28), 54 (27), 44 (23), 42 (23), 56 (17), 53 (17), 41 (17), 69 (16), 58 (11), 46 (9), 39 (8), 72 (8), 40 (7), 126 (7), 52 (6), 70 (5). The mass spectrum in comparison with the mass spectrum of the isotopically unmodified compound (Supplementary file 1, Fig. S1 and Fig. S2) confirmed the incorporation of two 13C atoms and its suitability as internal standard in quantitation assays.
GC–O
A Trace Ultra series GC (Thermo Fisher Scientific, Dreieich, Germany) was equipped with a cold on-column injector, an FID (250 °C base temperature), and a custom-made sniffing port (230 °C base temperature) [13]. Two different fused silica columns were used, either a J&W DB-FFAP, 30 m × 0.32 mm i.d., 0.25 µm film thickness, or a J&W DB-5, 30 m × 0.25 mm i.d., 0.25 µm film thickness (both Agilent, Waldbronn, Germany). The end of the column was connected to a Y-shaped quick-seal glass connector (CHM, Fridolfing, Germany) that directed the effluent via two deactivated fused silica capillaries, 30 cm × 0.2 mm i.d. (Agilent) to the FID and the sniffing port, respectively. The carrier gas was helium at a constant pressure of 70 kPa for the DB-FFAP column and 95 kPa for the DB-5 column. The injection volume was 1 µL. The initial oven temperature of 40 °C was held for 2 min, followed by a gradient of 6 °C/min. The final temperatures were 230 °C for the DB-FFAP column and 240 °C for the DB-5 column. During GC–O analysis, a panellist placed his nose above the sniffing port and evaluated the effluent. The panellist marked odorous regions in the FID chromatogram plotted by a recorder and noted the odour quality [8]. A linear retention index (RI) was calculated for each odour-active compound from its retention time and the retention times of adjacent n-alkanes by linear interpolation [37].
GC–MS
A 7890B GC was equipped with a GC 80 autosampler, a multimode injector, and a J&W DB-FFAP column, 30 m × 0.25 mm i.d., 0.25 µm film thickness and was connected to a Saturn 220 ion trap mass spectrometer (Agilent). The carrier gas was helium at a constant flow of 1.0 mL/min. The injection volume was 1 µL. The injection temperature was 40 °C. The initial oven temperature of 40 °C was held for 2 min, followed by a gradient of 6 °C/min to a final temperature of 230 °C. Mass spectra were generated in the chemical ionisation (CI) mode with methanol as reagent gas. For data analysis the MS Workstation 7.0.2 software (Agilent) was used.
Heart-cut GC–GC–MS
A Trace GC Ultra (Thermo Fisher Scientific) was equipped with a PAL autosampler (CTC Analytics, Zwingen, Switzerland), a cold on-column injector, and a J&W DB-FFAP column, 30 m × 0.32 mm i.d., 0.25 μm film thickness (Agilent). The carrier gas was helium at a constant pressure of 110 kPa. The injection volume was 1–2 µL. The initial oven temperature of 40 °C was held for 2 min, followed by a gradient of 6 °C/min to a final temperature of 230 °C. The end of the column was connected to a moving column stream switching (MCSS) device (Thermo Fisher Scientific), which allowed for a time-programmed transfer of the eluate via deactivated fused silica capillaries (0.32 mm i.d.) either simultaneously to an FID (250 °C base temperature) and a sniffing port (230 °C base temperature) or via another deactivated fused silica capillary passed through a heated (250 °C) hose to a liquid nitrogen-cooled trap located in the oven of a CP 3800 GC (Agilent). Helium served as make-up gas for the MCSS device (50 kPa). The capillary in the second oven was a J&W DB-1701, 30 m × 0.25 mm i.d., 0.25 µm film thickness (Agilent). The initial oven temperature of 40 °C was held for 2 min, followed by a gradient of 6 °C/min to a final temperature of 240 °C. The end of this column was connected to a Saturn 2200 ion trap mass spectrometer (Agilent). Mass spectra were generated in the CI mode with methanol as reagent gas. For data analysis the MS Workstation 6.9.3 software (Agilent) was used.
Comprehensive two-dimensional gas chromatography–time of flight MS (GC × GC–TOFMS)
A 6890 GC (Agilent) was equipped with Combi PAL autosampler (CTC Analytics), a CIS 4 injector (Gerstel, Mülheim an der Ruhr, Germany), and a J&W DB-FFAP column, 30 m × 0.25 mm i.d., 0.25 µm film thickness (Agilent) used as first column. The carrier gas was helium at a constant flow of 2.0 mL/min. The injection volume was 1 µL. The initial oven temperature of 40 °C was held for 2 min, followed by a gradient of 6 °C/min to a final temperature of 230 °C. The end of the column was connected to a second column, J&W DB-1701, 2.7 m × 0.18 mm i.d., 0.18 µm film thickness (Agilent). A liquid nitrogen-cooled dual stage quad-jet thermal modulator was installed at the beginning of the second column and operated with a modulation period of 4 s. The major part of the second column was installed in a secondary oven mounted inside the primary GC oven. The initial oven temperature of the secondary oven was 45 °C and was held for 2 min, followed by a gradient of 6 °C/min to a final temperature of 240 °C. The end of the column was connected to a Pegasus II TOF mass spectrometer (Leco, Mönchengladbach, Germany) via a heated (250 °C) transfer line. Mass spectra were generated in the electron ionisation (EI) mode at 70 eV with a scan range of m/z 35 − 350 and a scan rate of 100 spectra/s. For data analysis, the GC Image (Lincoln, NE, USA) software was used.
Isolation of volatiles
LME samples (10 g) were diluted with water (20 mL) and stirred with diethyl ether (80 mL) at room temperature for 2 h. The aqueous phase was separated and stirred with a second portion of diethyl ether (80 mL). The combined organic phases were dried over anhydrous sodium sulphate and non-volatile matrix components were removed by SAFE at 40 °C [23]. The distillate was concentrated to a final volume of 1 mL using a Vigreux column (50 × 1 cm) and a Bemelmans microdistillation device [38].
AEDA
Volatile isolates prepared from light and dark LME as described above were repeatedly subjected to GC–O analysis (FFAP column) by three trained and experienced sniffers. After the results had become reproducible, the volatile isolates were stepwise diluted 1:10 with diethyl ether to obtain dilutions of 1:10, 1:100, 1:1000, and 1:10,000 of the initial solution. The diluted samples were also analysed by GC–O (three sniffers) and each odorant was assigned an FD factor defined as the dilution factor of the highest diluted sample in which the odorant was detected during GC–O analysis by any of the three panellists [8].
Odorant quantitation
LME samples (0.5–20 g) were diluted with water (4–40 mL). Diethyl ether (16–160 mL) and stable isotopically substituted odorants (cf. Supplementary file, Table S1; 0.03–400 µg) were added, and the mixture was stirred at room temperature overnight. The aqueous phase was separated, shaken with a second portion of diethyl ether (16–160 mL), and the organic phases were combined.
Commercial malt powder (2 g) was either added to diethyl ether (20 mL), or malt powder (0.5 g) was added to a mixture of diethyl ether (19 mL) and water (1 mL), or malt powder (0.5 g) was first mixed with water (2 mL), the mixture was allowed to stand for 30 s, 1 min, 2 min, 5 min, 15 min, or 180 min, and finally anhydrous sodium sulphate (10 g) suspended in diethyl ether (50 mL) was added. Stable isotopically substituted odorants (0.1–2 µg) were added and the mixture was stirred at room temperature overnight. The supernatant was decanted, the residue was stirred with a second portion of diethyl ether (20 mL), and the organic phases were combined.
Pilsner malt was frozen with liquid nitrogen and ground into a fine powder using a laboratory mill Grindomix GM 200 (Retsch, Haan, Germany) at 4000 rpm (10 s) and 10,000 rpm (10 s). Diethyl ether (19–190 mL) and water (1–10 mL) were added to the powder (0.5 − 20 g) followed by stable isotopically substituted odorants (0.01–20 µg), and the mixture was stirred at room temperature overnight. The supernatant was decanted, the residue was stirred with a second portion of diethyl ether (20–200 mL), and the organic phases were combined.
The ethereal extracts obtained from the different materials were dried over anhydrous sodium sulphate and non-volatiles were removed by SAFE at 40 °C. The SAFE distillates were separated into neutral and basic volatiles (NBV) and acidic volatiles (AV) as detailed in [39]. Fractions NBV and AV were concentrated to final volumes between 0.2 mL and 5 mL. Concentrates were analysed by using the GC–MS system (4), the heart-cut GC–GC–MS system (9), or the GC × GC–TOFMS system (5, 8, 10, 11, 14, 16, 17, 20, 22, 23, 26, and 27). Odorant concentrations were finally calculated from the area counts of the analyte peak and the internal standard peak as obtained from the extracted ion chromatograms of characteristic quantifier ions, the amount of LME or malt used, and the amount of standard added by employing a calibration line equation previously obtained from the analysis of analyte/standard mixtures in five different concentration ratios (5:1, 2:1, 1:1, 1:2, and 1:5). Quantifier ions and calibration line equations are available in the Supplementary file, Table S1. The individual concentrations of the two isomers of 9 were determined from the concentrations obtained for the sum of isomers and the ratios of isomers, which were determined by GC × GC–TOFMS using the approach detailed in [40].
Quantitative olfactory profiles
LME samples (5 g) were placed in cylindrical ground neck glasses (7 cm height, 3.5 cm i.d.) with lids (VWR, Darmstadt, Germany). A panel of 16–18 trained assessors (males and females, ages 21–49) orthonasally evaluated the intensities of pre-defined descriptors on a scale from 0 to 3 with 0.5 increments and 0 = not detectable, 1 = weak, 2 = moderate, and 3 = strong. Descriptors had previously been collected by free-choice profiling. Each descriptor was defined by the odour of a reference compound dissolved in water in a concentration ~ 100 times above its orthonasal odour threshold value (OTV). Reference compounds were selected on the basis of their odour and their occurrence in malt and other thermally treated foods [41, 42]. The twelve descriptors and the corresponding reference compounds were “smoky” (2-methoxyphenol; OTV 0.84 µg/kg [43]), “earthy” (2,3,5-trimethylpyrazine; OTV 11 µg/kg [44]), “roasty” (2-acetyl-2-thiazoline; OTV 0.079 µg/kg [44]), “seasoning-like” (sotolon; OTV 1.7 µg/kg [45]), “cooked potato-like” (3-(methylsulfanyl)propanal; OTV 0.43 µg/kg [43]), “cheesy” (3-methylbutanoic acid; OTV 490 µg/kg [43]), “fatty” ((2E,4E)-deca-2,4-dienal; OTV 0.027 µg/kg [43]), “malty” (3-methylbutanal; OTV 0.5 µg/kg [43]), “honey-like” (phenylacetic acid; OTV 68 µg/kg [46]), “caramel-like” (HDMF; OTV 87 µg/kg [47]), “vanilla-like” (vanillin; OTV 53 µg/kg [43]), and “clove-like” (4-allyl-2-methoxyphenol; OTV 1.8 µg/kg [48]). The results of the individual panellists were averaged by calculating the arithmetic mean.
Results and discussion
Sensory characterization of light and dark LME
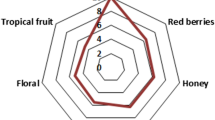
A light LME and a dark LME, both obtained from the same batch of malt, were orthonasally compared by a trained sensory panel in a quantitative olfactory profile analysis using 12 pre-defined descriptors. Results (Fig. 1) showed clear differences between the samples. The aroma of the dark LME was characterized by stronger smoky, earthy, roasty, seasoning-like, and malty notes and slightly more intense caramel-like and clove-like notes compared to the light LME. The profile of the light LME showed a higher intensity only in the honey-like odour note. The differences in the olfactory profiles corresponded to the different production protocols. In particular, the processing of the dark LME included an additional heating step, suggesting an increased formation of Maillard reaction products [49, 50].

Quantitative olfactory profiles of light and dark LME. Panellists rated the intensity of each descriptor on a scale from 0 to 3 in 0.5 increments with 0 = not detectable, 1 = weak, 2 = moderate, and 3 = strong
Screening for odour-active compounds in light and dark LME
To get a first insight into the odorants responsible for the different olfactory profiles of the light LME and the dark LME, the volatiles were isolated by solvent extraction and SAFE, and screened for odour-active compounds by GC–O and a crude AEDA using 1:10 dilutions. This resulted in a total of 28 odorants (Table 1). Preliminary structural assignments were achieved by comparing odour quality and RIs with data from literature [17] and from the Leibniz-LSB@TUM odorant database [42]. Assignments were confirmed by comparing the odour quality, the RIs on two columns of different polarity (DB-FFAP, DB-5), and the odour intensity at adequate concentration levels, as well as the mass spectra recorded by GC × GC–TOFMS to data of authentic reference substances analysed under the same conditions. Using this approach, 21 out of the 28 LME odorants were unequivocally identified. Due to low concentrations, no mass spectra were obtained for oct-1-en-3-one (3) and trans-4,5-epoxy-(2E)-dec-2-enal (18). Nevertheless, their identification was considered unambiguous due to their highly characteristic odour quality. As no reference compound was available, caramel-like smelling odorant 13 was only tentatively identified as dihydromaltol on the basis of a comparison of the odour quality, the RI, and the mass spectrum with literature data [17]. In summary, structures could be assigned to 24 out of the 28 malt extract odorants, all of which had already been reported as malt or malt extract components [6, 9, 17].
In both malt extract samples, a high FD factor of 1000 was determined for honey-like smelling phenylacetic acid (26) and vanilla-like smelling vanillin (27). Higher FD factors in the light LME than in the dark LME were in particular found for caramel-like smelling HDMF (20; FD 1000 vs. 10), metallic smelling trans-4,5-epoxy-(2E)-dec-2-enal (18; FD 100 vs. 1), clove-like smelling 4-ethenyl-2-methoxyphenol (22; FD 100 vs. < 1), and honey-like smelling phenylacetaldehyde (8; FD 100 vs. 10). Higher FD factors in the dark LME were obtained for the two caramel-like smelling compounds maltol (17; FD 1000 vs. 100) and dihydromaltol (13; FD 100 vs. 1), seasoning-like smelling sotolon (23; FD 1000 vs. 10), smoky smelling 2-methoxyphenol (14; FD 100 vs. 10), and the carboxylic acids acetic acid (4; FD 100 vs. 10) and 2-/3-methylbutanoic acid (9; FD 100 vs. 10). In both LMEs, the FD factors of highly volatile compounds such as 2- and 3-methylbutanal (1), and oct-1-en-3-one (3) were considerably lower than in malt [17], indicating losses in the concentration step during the malt extract production. Similar losses of such highly volatile malt compounds have been reported during wort boiling [51].
Odorant quantitation in light and dark LME and OAV calculation
To substantiate the differences in the odorants between the light LME and the dark LME and lift the investigations to a higher level of accuracy, 15 selected odorants were quantitated by GC–MS. Selection was based on the FD factors obtained in the screening experiments and according to literature on the relevance of the compounds for bread aroma [42, 52, 53]. Stable isotopically substituted odorants were employed as internal standards to compensate for losses during the sample workup. Results (Table 2) revealed concentrations in a range between 0.519 µg/kg for (2E,4E)-deca-2,4-dienal (11) and 869,000 µg/kg for acetic acid (4). To assess the odour potency of the odorants, OAVs were calculated by dividing the individual concentrations by the OTVs of the compounds in water.
In the light LME, 12 of 15 compounds showed OAVs ≥ 1. High OAVs were calculated for cooked potato-like smelling 3-(methylsulfanyl)propanal (5; OAV 1500), cooked apple-like smelling (E)-β-damascenone (11; OAV 430), clove-like smelling 4-ethenyl-2-methoxyphenol (22; OAV 91), and the two honey-like smelling odorants phenylacetaldehyde (8; OAV 70) and phenylacetic acid (26; OAV 58). In the dark LME, 11 of 15 compounds showed OAVs ≥ 1. High OAVs were calculated for seasoning-like smelling sotolon (23; OAV 780), cooked potato-like smelling 3-(methylsulfanyl)propanal (5; OAV 550), cooked apple-like smelling (E)-β-damascenone (11; OAV 410), vinegar-like smelling acetic acid (4; OAV 160), caramel-like smelling maltol (17; OAV 120), and honey-like smelling phenylacetic acid (26; OAV 62).
The differences found in the OAVs between the light LME and the dark LME very well corresponded to the differences in the quantitative olfactory profiles (cf. Fig. 1). For example, the stronger honey-like odour note in the light LME reflected the higher OAV of phenylacetaldehyde (8; OAV 70 vs. 20). With OAVs of 58 and 62, phenylacetic acid (26) did obviously not contribute to this sensory difference. In the dark LME, clearly higher OAVs were obtained for the well-known Maillard reaction products sotolon (23; OAV 780 vs. 7.4) and maltol (17; OAV 120 vs. 1.8) [49, 50] as well as for 2-methoxyphenol (14; OAV 26 vs. 2.6). This corresponded well to the higher intensities of the seasoning-like, the caramel-like, and the smoky odour notes in the quantitative olfactory profile. However, OAV data did not provide an explanation for the higher ratings obtained for the earthy, roasty, and malty notes in the dark LME.
Sources of LME odorants
The differences in the concentrations of important odorants between the light and the dark LME already indicated a major impact of the processing steps from malt to extract, at least for the dark LME which faced a higher thermal impact. However, it was still unclear, whether a transfer of malt odorants or the odorant formation during malt extract processing is generally more important for the amount of odorants finally present in the LMEs. To clarify this, we aimed at quantifying all odorants previously quantitated in the light LME and the dark LME (cf. Table 2) also in the malt that served as starting material for both extracts.
It has been reported that in products with low moisture content such as chocolate, cornflakes, crackers, and malt, odour-active compounds are not only present in the free form, but to a major extent in a bound form from which the odorants are released by water contact [19, 54]. As during LME production the malt also gets into contact with water, we attempted to quantitate the free and bound odorants in the malt as sum. It was shown that in malt, the Strecker aldehydes 2- and 3-methylbutanal, phenylacetaldehyde, and 3-(methylsulfanyl)propanal increase 10–140-fold after water treatment [19]. 3-Oxazolines formed during Strecker degradation were suggested as the crucial hydrolabile precursors of these compounds [54]. Own preliminary experiments, however, showed that not only Strecker aldehydes, but also other odour-active compounds increase in malt upon water contact. Figure 2 exemplifies this for phenylacetaldehyde (8), phenylacetic acid (26), and vanillin (27). The substances were quantitated in a commercial malt powder by using stable isotopically substituted odorants as internal standards and two different workup procedures. To quantitate only the free compounds, pure diethyl ether was used as extraction solvent. By contrast, to cover additionally the bound compounds, a minor amount of water was added together with the diethyl ether. A similar approach had been suggested earlier for this purpose [10, 17].

Concentrations of phenylacetaldehyde, phenylacetic acid, and vanillin in malt powder determined without and with water addition before solvent extraction
Results showed clearly higher concentrations of all three odorants when water was added. Water addition increased the concentration of phenylacetaldehyde 33-fold from 60.6 µg/kg to 1990 µg/kg. The concentrations of phenylacetic acid and vanillin also significantly increased. With factors of 14 and 9, however, the increase was less pronounced. These observations suggested a combination of a general release mechanism applicable to volatiles in general and an additional formation of Strecker aldehydes as suggested before. Similar results were reported for oat products [55,56,57]. These authors suggested the cleavage of starch complexes as a general odorant release mechanism.
Although the experiments with water addition indicated a simultaneous quantitation of free and bound odorants in malt, it yet remained unclear whether the release of the bound odorants was exhaustive. To clarify this, we conducted a series of quantitation experiments during which water treatment and solvent extraction were temporally separated and the duration of the water treatment was varied. Experiments were again carried out with phenylacetaldehyde (8), phenylacetic acid (26), and vanillin (27). Results (Fig. 3) showed a very quick release of the bound odorants. The maximum value was already reached after 1 min of water contact and equalled the concentrations previously obtained after simultaneous addition of water and diethyl ether. Thus, the simultaneous addition of water and diethyl ether led to an exhaustive release of bound odorants and is an appropriate approach to quantitate the sum of free and bound volatiles in malt.

Concentrations of phenylacetaldehyde (a), phenylacetic acid (b), and vanillin (c) in malt powder after stirring with water for different periods (dots) in comparison to the concentrations determined after simultaneous addition of water and diethyl ether (straight line)
Application of this approach to the quantitation of important LME odorants in the malt that served as starting material for both, the light and the dark LMEs, resulted in concentrations ranging from 0.135 µg/kg for (E)-β-damascenone (11) to 404,000 µg/kg for acetic acid (4) (Table 3). High concentrations were additionally determined for phenylacetaldehyde (8; 7000 µg/kg), phenylacetic acid (26; 6250 µg/kg), 3-methylbutanoic acid (9a; 3810 µg/kg), 2-phenylethan-1-ol (16; 1360 µg/kg), and 3-(methylsulfanyl)propanal (5; 1190 µg/kg). To assess odorant loss and odorant formation on the way from the malt to the LMEs, the percentage of change was calculated from the odorant concentrations in the malt, the odorant concentrations in the LMEs, and the process yields. Results revealed huge differences: percentages ranged from 1.8% to 90,000% (Table 3).
Approximately half of the analysed compounds exhibited recoveries below 100% in both extracts. These included three compounds with comparatively high OAVs, namely the cooked potato-like smelling 3-(methylsulfanyl)propanal (5) and the honey-like smelling compounds phenylacetaldehyde (8) and phenylacetic acid (26). The recoveries for 3-(methylsulfanyl)propanal were 50% and 24% in the light LME and the dark LME, respectively. With 4.9% and 1.8%, phenylacetaldehyde showed the lowest recoveries of all compounds analysed. Higher recoveries were calculated for phenylacetic acid, namely 60% and 81%. Losses might be associated with thermal reactions and with evaporation during the vacuum concentration process.
Seven compounds showed an increase in at least one of the two LMEs, indicating a formation during malt extract production. Among them were vinegar-like smelling acetic acid (4), the phenolic compounds 2-methoxyphenol (14; smoky), 4-ethenyl-2-methoxyphenol (22; clove-like), and vanillin (27; vanilla-like), cooked apple-like smelling (E)-β-damascenone (11) as well as seasoning-like smelling sotolon (23) and caramel-like smelling maltol (17). Acetic acid showed a decrease from the malt to the light LME (43%) but an increase to the dark LME (260%). During the production of the dark LME, acetic acid might have been formed via sugar degradation and Strecker degradation of alanine [58,59,60,61]. Within the group of phenolic compounds, vanillin showed similar percentages in the light LME (150%) and the dark LME (270%), whereas 4-ethenyl-2-methoxyphenol showed an increase in the light LME (400%), but a decrease in the dark LME (3.8%), and 2-methoxyphenol showed a moderate increase in the light LME (140%), but a clearly higher increase in the dark LME (1800%). These observations are in line with the higher thermal impact associated with the additional heating step during the production of the dark LME. It has been demonstrated that the thermal decomposition of ferulic acid first results in 4-ethenyl-2-methoxyphenol [62,63,64,65] which presumably was accumulated in the light LME. A higher thermal impact converts 4-ethenyl-2-methoxyphenol to 2-methoxyphenol [62, 63], explaining the lower amount of 4-ethenyl-2-methoxyphenol and the higher amount of 2-methoxyphenol in the dark LME. With percentages of 1800% and 2200%, (E)-β-damascenone revealed a significant gain on the way from the malt to the LMEs, but only a small difference between the light and the dark LME. Thermal formation of (E)-β-damascenone is well known, but the compound also might have been formed from glycosides during mashing [66,67,68,69,70,71,72,73]. The highest increase of all compounds investigated was found for the Maillard reaction products maltol and sotolon in the dark LME, namely 90,000% and 39,000%. For both compounds, the percentages in the light LME were clearly lower (1100% and 290%) indicating that the additional heating step was crucial for their elevated formation in the dark LME. It has been demonstrated that sotolon can be formed by an aldol-reaction from butane-2,3-dione and hydroxyacetaldehyde, both being previously generated by retro-aldol cleavage of sugars [74]. The precursor of maltol is the disaccharide maltose [10, 75] resulting from the enzymatic breakdown of starch during germination and mashing.
Conclusions
The odour-active compounds in LMEs clearly differ from the odour-active compounds in the malt used for their production. Whereas some malt odorants decrease during extract production, others show an enormous increase. Thus, the formation of odorants during LME production is much more important than the mere transfer of odorants from malt to LMEs.
Abbreviations
- AEDA:
-
Aroma extract dilution analysis
- AV:
-
Acidic volatiles
- CI:
-
Chemical ionisation
- DME:
-
Dry malt extract
- EI:
-
Electron ionisation
- FD factor:
-
Flavour dilution factor
- FFAP:
-
Free fatty acid phase
- FID:
-
Flame ionisation detector
- GC:
-
Gas chromatography
- GC × GC:
-
Comprehensive two-dimensional gas chromatography
- GC–O:
-
Gas chromatography–olfactometry
- HDMF:
-
4-Hydroxy-2,5-dimethylfuran-3(2H)-one
- i.d.:
-
Inner diameter
- LME:
-
Liquid malt extract
- MCSS:
-
Moving column stream switching
- MS:
-
Mass spectrometry
- NBV:
-
Neutral and basic volatiles
- TOF:
-
Time of flight
- OAV:
-
Odour activity value
- OTV:
-
Odour threshold value
- RI:
-
Retention index
- SAFE:
-
Solvent-assisted flavour evaporation
- SDE:
-
Simultaneous distillation/extraction
- Cyclotene:
-
2-Hydroxy-3-methylcyclopent-2-en-1-one
- (E)-β-Damascenone:
-
(2E)-1-(2,6,6-Trimethylcyclohexa-1,3-dien-1-yl)but-2-en-1-one
- Dihydromaltol:
-
5-Hydroxy-6-methyl-2,3-dihydro-4H-pyran-4-one
- Maltol:
-
3-Hydroxy-2-methyl-4H-pyran-4-one
- γ-Nonalactone:
-
5-Pentyloxolan-2-one
- Sotolon:
-
3-Hydroxy-4,5-dimethylfuran-2(5H)-one
- Vanillin:
-
4-Hydroxy-3-methoxybenzaldehyde
References
Narziß L, Back W, Gastl M, Zarnkow M (2017) Die Technologie der Malzbereitung. In: Narziß L, Back W, Gastl M, Zarnkow M (eds) Abriss der Bierbrauerei. Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
Narziß L (2004) Malz. In: Heiss R (ed) Lebensmitteltechnologie: Biotechnologische, chemische, mechanische und thermische Verfahren der Lebensmittelverarbeitung. Springer, Berlin, Heidelberg
Kunze W (2016) Technologie Brauer und Mälzer. Versuchs- u. Lehranstalt f. Brauerei, Berlin
Poiana MA, Dogaru D, Mateescu C, Mucete D, Parvulescu L (2008) Researches regarding the influence of malt extract addition on the bread quality. Bull Univ Agric Sci Vet Med Cluj-Napoca, Agric 62:332–337
Man S, Paucean A, Muste S, Damian M (2012) Preliminary study of the malt extract addition on the wholemeal bread quality. Bull Univ Agric Sci Vet Med Cluj-Napoca, Agric 69:505–507
Farley DR, Nursten HE (1980) Volatile flavor components of malt extract. J Sci Food Agric 31:386–396
Nickerson GB, Likens ST (1966) Gas chromatography evidence for the occurrence of hop oil components in beer. J Chromatogr A 21:1–5
Steinhaus M (2020) Gas chromatography–olfactometry: principles, practical aspects and applications in food analysis. In: Gas A (ed) Tranchida PQ. Chromatography in Food Analysis. The Royal Society of Chemistry, Cambridge, UK, pp 337–399
Przybylski R, Kamiński E (1983) Thermal degradation of precursors and formation of flavour compounds during heating of cereal products. Part II. The formation and changes of volatile flavour compounds in thermally treated malt extracts at different temperature and pH. Nahrung 27:487–496
Fickert B (1999) Untersuchungen zur Bildung von Aromastoffen bei der Mälzung von Getreide. Verlag Dr. Hut, München
Dong L, Piao Y, Zhang X, Zhao C, Hou Y, Shi Z (2013) Analysis of volatile compounds from a malting process using headspace solid-phase micro-extraction and GC–MS. Food Res Int 51:783–789
Deki M, Yoshimura M (1974) Volatile components of peated malt. III. Identification of acidic and basic components. Chem Pharm Bull 22:1760–1764
Deki M, Yoshimura M (1974) Volatile components of peated malt. II. Identification of neutral components. Chem Pharm Bull 22:1754–1759
Deki M, Yoshimura M (1974) Volatile components of peated malt. I. Identification of phenolic compounds. Chem Pharm Bull 22:1748–1753
Yahya H, Linforth RST, Cook DJ (2014) Flavour generation during commercial barley and malt roasting operations: a time course study. Food Chem 145:378–387
Beal AD, Mottram DS (1994) Compounds contributing to the characteristic aroma of malted barley. J Agric Food Chem 42:2880–2884
Fickert B, Schieberle P (1998) Identification of the key odorants in barley malt (caramalt) using GC/MS techniques and odour dilution analyses. Nahrung 42:371–375
Vandecan SMG, Saison D, Schouppe N, Delvaux F, Delvaux FR (2010) Optimisation of specialty malt volatile analysis by headspace solid-phase microextraction in combination with gas chromatography and mass spectrometry. Anal Chim Acta 671:55–60
Buhr K, Pammer C, Schieberle P (2010) Influence of water on the generation of Strecker aldehydes from dry processed foods. Eur Food Res Technol 230:375–381
Parr H, Bolat I, Cook D (2021) Modelling flavour formation in roasted malt substrates under controlled conditions of time and temperature. Food Chem 337:127641
VCF volatile compounds in food, database, version 16.7 (1963–2020) Van Dongen WD, Donders, JJH (eds) BeWiDo B.V.: Reeuwijk, The Netherlands. https://www.vcf-online.nl. Accessed 04 December 2020
Schieberle P, Grosch W (1987) Evaluation of the flavor of wheat and rye bread crusts by aroma extract dilution analysis. Z Lebensm Unters Forsch 185:111–113
Engel W, Bahr W, Schieberle P (1999) Solvent assisted flavour evaporation - a new and versatile technique for the careful and direct isolation of aroma compounds from complex food matrices. Eur Food Res Technol 209:237–241
Ullrich F, Grosch W (1988) Flavour deterioration of soya-bean oil: identification of intense odour compounds formed during flavour reversion. Lipid/Fett 90:332–336
Schieberle P, Grosch W (1991) Potent odorants of the wheat bread crumb - differences to the crust and effect of a longer dough fermentation. Z Lebensm Unters Forsch 192:130–135
Sen A, Grosch W (1991) Synthesis of six deuterated sulfur containing odorants to be used as internal standards in quantification assays. Z Lebensm Unters Forsch 192:541–547
Schuh C, Schieberle P (2006) Characterization of the key aroma compounds in the beverage prepared from darjeeling black tea: quantitative differences between tea leaves and infusion. J Agric Food Chem 54:916–924
Steinhaus M, Schieberle P (2005) Characterization of odorants causing an atypical aroma in white pepper powder (Piper nigrum L.) based on quantitative measurements and orthonasal breakthrough thresholds. J Agric Food Chem 53:6049–6055
Sen A, Laskawy G, Schieberle P, Grosch W (1991) Quantitative determination of beta-damascenone in foods using a stable isotope dilution assay. J Agric Food Chem 39:757–759
Cerny C, Grosch W (1993) Quantification of character-impact odour compounds of roasted beef. Z Lebensm Unters Forsch 196:417–422
Kiefl J, Pollner G, Schieberle P (2013) Sensomics analysis of key hazelnut odorants (Corylus avellana L. ‘Tonda Gentile’) using comprehensive two-dimensional gas chromatography in combination with time-of-flight mass spectrometry (GC×GC-TOF-MS). J Agric Food Chem 61:5226–5235
Blank I, Schieberle P, Grosch W (1993) Quantification of the flavour compounds 3-hydroxy-4,5-dimethyl-2(5H)-furanone and 5-ethyl-3-hydroxy-4-methyl-2(5H)-furanone by stable isotope dilution assay. In: Schreier P, Winterhalter P (eds) Progress in flavour precursor studies. Allured Publishing, Carol Stream, pp 103–109
Semmelroch P, Laskawy G, Blank I, Grosch W (1995) Determination of potent odorants in roasted coffee by stable isotope dilution assays. Flavour Fragrance J 10:1–7
Komoto I, Matsuo J, Kobayashi S (2002) Catalytic Friedel-Crafts acylation of heteroaromatics. Top Catal 19:43–47
Harada R, Iwasaki M (1983) Syntheses of maltol and ethylmaltol. Agric Biol Chem 47:2921–2922
Bentzinger G, Souza W, Mullie C, Agnamey P, Dassonville-Klimpt A, Sonnet P (2016) Asymmetric synthesis of new antimalarial aminoquinolines through Sharpless aminohydroxylation. Tetrahedron Asymmetry 27:1–11
Van den Dool H, Kratz PD (1963) A generalization of the retention index system including linear temperature programmed gas–liquid partition chromatography. J Chromatogr A 11:463–471
Bemelmans JMH (1979) Review of isolation and concentration techniques. In: Land GG, Nursten HE (eds) Progress in flavour research. Applied Science Publishers, London, pp 79–98
Tatsu S, Matsuo Y, Nakahara K, Hofmann T, Steinhaus M (2020) Key odorants in japanese roasted barley tea (Mugi-Cha)—differences between roasted barley tea prepared from naked barley and roasted barley tea prepared from hulled barley. J Agric Food Chem 68:2728–2737
Steinhaus M, Sinuco D, Polster J, Osorio C, Schieberle P (2008) Characterization of the aroma-active compounds in pink guava (Psidium guajava, L.) by application of the aroma extract dilution analysis. J Agric Food Chem 56:4120–4127
Dunkel A, Steinhaus M, Kotthoff M, Nowak B, Krautwurst D, Schieberle P, Hofmann T (2014) Nature’s chemical signatures in human olfaction: a foodborne perspective for future biotechnology. Angew Chem, Int Ed 53:7124–7143
Kreissl J, Mall V, Steinhaus P, Steinhaus M (2019) Leibniz-LSB@TUM odorant database, version 1.0. Leibniz-Institute for Food Systems Biology at the Technical University of Munich, Freising, Germany. https://www.leibniz-lsb.de/en/databases/leibniz-lsbtum-odorant-database. Accessed 04 December 2020
Czerny M, Christlbauer M, Christlbauer M, Fischer A, Granvogl M, Hammer M, Hartl C, Hernandez N, Schieberle P (2008) Re-investigation on odour thresholds of key food aroma compounds and development of an aroma language based on odour qualities of defined aqueous odorant solutions. Eur Food Res Technol 228:265–273
Mall V, Schieberle P (2017) Evaluation of key aroma compounds in processed prawns (whiteleg shrimp) by quantitation and aroma recombination experiments. J Agric Food Chem 65:2776–2783
Steinhaus M, Sinuco D, Polster J, Osorio C, Schieberle P (2009) Characterization of the key aroma compounds in pink guava (Psidium guajava L.) by means of aroma re-engineering experiments and mission tests. J Agric Food Chem 57:2882–2888
Wagner J, Granvogl M, Schieberle P (2016) Characterization of the key aroma compounds in raw licorice (Glycyrrhiza glabra L.) by means of molecular sensory science. J Agric Food Chem 64:8388–8396
Munafo JP, Didzbalis J, Schnell RJ, Steinhaus M (2016) Insights into the key aroma compounds in mango (Mangifera indica L. ‘Haden’) fruits by stable isotope dilution quantitation and aroma simulation experiments. J Agric Food Chem 64:4312–4318
Kreissl J, Schieberle P (2017) Characterization of aroma-active compounds in italian tomatoes with emphasis on new odorants. J Agric Food Chem 65:5198–5208
Cerny C (2008) The aroma side of the Maillard reaction. Ann N Y Acad Sci 1126:66–71
Starowicz M, Zieliński H (2019) How Maillard reaction influences sensorial properties (color, flavor and texture) of food products? Food Rev Int 35:707–725
De Schutter DP, Saison D, Delvaux F, Derdelinckx G, Rock J-M, Neven H, Delvaux FR (2008) Release and evaporation of volatiles during boiling of unhopped wort. J Agric Food Chem 56:5172–5180
Birch AN, Petersen MA, Hansen AS (2014) Aroma of wheat bread crumb. Cereal Chem 91:105–114
Pico J, Bernal J, Gómez M (2015) Wheat bread aroma compounds in crumb and crust: a review. Food Res Int 75:200–215
Granvogl M, Beksan E, Schieberle P (2012) New insights into the formation of aroma-active Strecker aldehydes from 3-oxazolines as transient intermediates. J Agric Food Chem 60:6312–6322
Guth H, Grosch W (1993) Odorants of extrusion products of oat meal—changes during storage. Z Lebensm Unters Forsch 196:22–28
Klensporf D, Jelen HH (2005) Analysis of volatile aldehydes in oat flakes by SPME-GC/MS. Pol J Food Nutr Sci 14:389–395
Dach AM (2015) Characterization of key aroma compounds in oat pastry and their generation during processing. Verlag Deutsche Forschungsanstalt für Lebensmittelchemie, Freising
Hofmann T, Münch P, Schieberle P (2000) Quantitative model studies on the formation of aroma-active aldehydes and acids by Strecker-type reactions. J Agric Food Chem 48:434–440
Davídek T, Devaud S, Robert F, Blank I (2006) Sugar fragmentation in the Maillard reaction cascade: isotope labeling studies on the formation of acetic acid by a hydrolytic β-dicarbonyl cleavage mechanism. J Agric Food Chem 54:6667–6676
Smuda M, Glomb MA (2013) Fragmentation pathways during Maillard-induced carbohydrate degradation. J Agric Food Chem 61:10198–10208
Davídek T, Robert F, Devaud S, Vera FA, Blank I (2006) Sugar fragmentation in the Maillard reaction cascade: formation of short-chain carboxylic acids by a new oxidative α-dicarbonyl cleavage pathway. J Agric Food Chem 54:6677–6684
Tressl R, Kossa T, Renner R, Koeppler H (1976) Gas chromatographic−mass spectrometric investigations on the formation of phenolic and aromatic hydrocarbons in food. Z Lebensm Unters Forsch 162:123–130
Fiddler W, Parker WE, Wasserman AE, Doerr RC (1967) Thermal decomposition of ferulic acid. J Agric Food Chem 15:757–761
Langos D, Granvogl M (2016) Studies on the simultaneous formation of aroma-active and toxicologically relevant vinyl aromatics from free phenolic acids during wheat beer brewing. J Agric Food Chem 64:2325–2332
Coghe S, Benoot K, Delvaux F, Vanderhaegen B, Delvaux FR (2004) Ferulic acid release and 4-vinylguaiacol formation during brewing and fermentation: indications for feruloyl esterase activity in saccharomyces cerevisiae. J Agric Food Chem 52:602–608
Averbeck M, Schieberle PH (2009) Characterisation of the key aroma compounds in a freshly reconstituted orange juice from concentrate. Eur Food Res Technol 229:611–622
Sefton MA, Skouroumounis GK, Elsey GM, Taylor DK (2011) Occurrence, sensory impact, formation, and fate of damascenone in grapes, wines, and other foods and beverages. J Agric Food Chem 59:9717–9746
Czerny M, Grosch W (2000) Potent odorants of raw arabica coffee. Their changes during roasting. J Agric Food Chem 48:868–872
Isoe S, Katsumura S, Sakan T (1973) The synthesis of damascenone and β-damascone and the possible mechanism of their formation from carotenoids. Helv Chim Acta 56:1514–1516
Bezman Y, Bilkis I, Winterhalter P, Fleischmann P, Rouseff RL, Baldermann S, Naim M (2005) Thermal oxidation of 9‘-cis-neoxanthin in a model system containing peroxyacetic acid leads to the potent odorant β-damascenone. J Agric Food Chem 53:9199–9206
Roberts DD, Mordehai AP, Acree TE (1994) Detection and partial characterization of eight β-damascenone precursors in apples (Malus domestica Borkh. Cv. Empire). J Agric Food Chem 42:345–349
Liang Z, Fang Z, Pai A, Luo J, Gan R, Gao Y, Lu J, Zhang P (2020) Glycosidically bound aroma precursors in fruits: a comprehensive review. Crit Rev Food Sci Nutr. https://doi.org/10.1080/10408398.2020.1813684
Jin H, Rogers P (2000) Novel recovery of malt flavours from their glycosidically bound precursors. Tech Q - Master Brew Assoc Am 37:79–83
Hofmann T, Schieberle P (1996) Studies on intermediates generating the flavour compounds 2-methyl-3-furanthiol, 2-acetyl-2-thiazoline and sotolon by Maillard-type reactions. In: Taylor AJ, Mottram DS (eds) Flavour science. Woodhead Publishing, UK, pp 182–187
Yaylayan VA, Mandeville S (1994) Stereochemical control of maltol formation in Maillard reaction. J Agric Food Chem 42:771–775
Wagner J, Schieberle P, Granvogl M (2017) Characterization of the key aroma compounds in heat-processed licorice (succus liquiritiae) by means of molecular sensory science. J Agric Food Chem 65:132–138
Sellami I, Mall V, Schieberle P (2018) Changes in the key odorants and aroma profiles of hamlin and valencia orange juices not from concentrate (NFC) during chilled storage. J Agric Food Chem 66:7428–7440
Grosshauser S, Schieberle P (2013) Characterization of the key odorants in pan-fried white mushrooms (Agaricus bisporus L.) by means of molecular sensory science: comparison with the raw mushroom tissue. J Agric Food Chem 61:3804–3813
American Society of Testing and Materials (2005) Standard E679–04. Standard practice for determination of odor and taste thresholds by a forced-choice ascending concentration series method of limits, vol 15.08. ASTM Book of Standards, West Conshohocken, PA, pp 38–44
Acknowledgments
The authors thank Inge Kirchmann for her skilful technical assistance and research intern Mirijam Adams for her help during sample workups. Special thanks go to Dr. Martin Erdmann and Dr. Thomas Kunte, Ireks GmbH, Kulmbach, Germany, for providing the malt and LME samples and for helpful discussions during the project.
Funding
Open Access funding enabled and organized by Projekt DEAL. This IGF Project of the FEI was supported via AiF within the program for promoting the Industrial Collective Research (IGF) of the German Ministry of Economics and Energy (BMWi), based on a resolution of the German Parliament. Project No. 18669 N.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Compliance with ethics requirements
This article does not contain any studies with human or animal subjects.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rögner, N.S., Mall, V. & Steinhaus, M. Odour-active compounds in liquid malt extracts for the baking industry. Eur Food Res Technol 247, 1263–1275 (2021). https://doi.org/10.1007/s00217-021-03707-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-021-03707-z