
Abstract
The production of insects on an industrial scale has attracted the attention of the research and agricultural industry as novel protein sources. To detect the presence of Gryllodes sigillatus (GS) in feed and food, a real-time PCR method based on the mitochondrial cytochrome b (CYB) gene is proposed by this study. Forty DNA samples of animal and plant origin were used to confirm the specificity of the qPCR system. The detection method’s performance was evaluated on different processed GS matrices including native GS (UnGS) and different commercial products: crunchy roasted samples (RoGS), insect meal mixtures (ACGS) and energetic snacks containing GS (GSS). Data on sequencing were aligned with the reference gene to confirm the PCR products. The regression curve (y = −3.394 x + 42.521; R2 = 0.994, d.f. 14) between Ct values and Log DNA concentrations of Gryllodes sigillatus resulted in an efficiency of 96.4%. The severity of the technological processing treatments and the matrix structure affected the intensity of the PCR signal with the same amount of insect DNA as observed by different y-intercepts of the three-regression lines for RoGS, ACGS, and GSS. The real-time PCR method resulted in robust and sensitive outcomes able to detect low amounts of GS DNA (5 g/100 g) in a complex matrix, making it suitable for detecting the presence or absence of labeled Gryllodes sigillatus material both in feed and food.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Highlights
-
The method allows for the identification of Gryllodes sigillatus in a complex matrix.
-
Primers exhibited high sensitivity and accuracy.
-
PCR discrimination among insect species.
-
The qPCR system was successfully tested in complex matrices.
Introduction
Global welfare standards and the increasing world population [1] require/necessitate/demand/call for the consideration of protein sources derived from sustainable production systems capable of efficiently converting biomass [2,3,4,5]. The novel proteins from unconventional sources should be safe, nutritious, flexible and reliable as well as accepted by consumers. Insects may represent a more efficient way of producing animal protein and seem to meet this goal due to the use of cost-effective raw materials and environmental sustainability along with high nutritional value [6, 7]. Recently, the production of insects on an industrial scale has attracted the attention of the research and agricultural industry [8], because they are a good source of protein, vitamins, and energy [9] and are widely used for human consumption in many countries in Asia, Africa and South America [10]. Whole insects can be used raw, dried, crushed, textured, pulverized, ground, eaten after technological food processing or preserved by freeze-drying with or without the exoskeleton of chitin [11]. They can be consumed as snacks, used as ingredients of some typical preparations or undergo other technological treatments, depending on cultural habits [12]. In contrast, people in Western countries are not familiar with this kind of food even if some compounds derived from insects are already exploited by the food industry as coloring agents [11] that can be found in Smarties, yogurt, Campari, etc.[13]. Nevertheless, interest in including insects in the Western diet is increasing along with the number of restaurants serving insects as a delicacy and the number of recipe books dedicated to insect preparation [14, 15]. The expectation is that in the coming decades this novel protein source will represent 40% of the traditional meat consumption [16, 17], but estimated entry time into the market [18, 19] will vary depending on cultures and traditions. Recently, several studies have been carried out on the functionality, processing and industrial applications of this novel ingredient, trying to exploit by-products and waste streams obtained in other production processes. Moreover, research studies indicate that insects possess several features that make them well suited for use in animal feeds. The European Union identified seven insect species as suitable for use in aquafeed: Gryllodes sigillatus (GS) (tropical house cricket), Gryllus assimilis (Jamaican field cricket), Acheta domesticus (AC) (house cricket), Alphitobius diaperinus (lesser mealworm), Musca domestica (common housefly), Tenebrio molitor (yellow mealworm) and Hermetia illucens (black soldier fly) [20].
The production and marketing of insects as food in Europe is regulated by the so-called ‘Novel Foods’ legislation—i.e. Regulation [14]. This regulation applies to all categories of foods that were not used for human consumption to a significant degree within the European Union before May 15, 1997, which is the case of insects. Since January 1, 2018, the Novel Food regulation has been in force; therefore, an authorization form must be submitted for each insect species intended to be considered as food and sold on the market. So far, Kreca Proti-Farm (NL), Micronutris (FR) and BiiF (BE) have applied for the inclusion of the following species in the European Union’s list: Alphitobius diaperinus, Gryllodes sigillatus, Tenebrio molitor, Locusta migratoria and Acheta domesticus. Among these, A. domesticus and G. sigillatus have increased their presence in the food market as a promising protein source for the human diet. Today, GS is commonly marketed as an alternative to AC, due to its ability to withstand cricket paralysis virus. GS, like other Gryllidae species, is susceptible to Acheta domesticus densovirus (AdDNV) with a variable degree, but without cases of severe mortality which instead occurs in AC, where AdDNV provokes high mortality and serious and irreparable damages to the production. For this reason, interest in rearing GS against AC increased after the bankruptcy of AC facilities [21].
In this context, controlling the composition of both feed and food is an important issue according to European rules, especially after outbreaks such as BSE (Bovine Spongiform Encephalopathy) or minced beef contaminated with horse meat; in any case, the safety and the control systems for insect components in food are still being developed [18, 22]. Today, ingredients of animal origin, used as raw materials, are identified based on typical and microscopically identifiable characteristics or by using polymerase chain reaction (PCR) following the provisions laid down in Annex VI of EU Commission Regulation No. 2019/152 [23]. Molecular biology techniques provide robust and valuable tools for the identification of both plant or animal species in feedstuffs [24]. DNA detection and identification of animal materials employing PCR, because of its high sensitivity, is considered the official analytical method to: (i) determine the species origin of processed animal proteins (PAPs), (ii) detect the presence of prohibited ingredients, (iii) confirm the presence of ingredients in animal feeds. Among the detection/identification of PAPs, insect species identification may represent a new challenge and the PCR methods may provide a possible solution. In particular, end-point PCR was developed to detect the presence of GS directly in foods [25], but a quantification was not possible with this technique. For this reason, an RT-PCR method is proposed in the present study.
Several methods have been applied for insect species identification, such as Enzyme-Linked Immunosorbent Assays (ELISA) for TM and nucleic acid-based approach applied to reliably detect the black soldier fly-specific DNA [26]. In more detail, DNA barcoding sequencing has been widely used in species identification and biodiversity research [27]. It has been shown that in many groups, including insects, interspecific variation in DNA sequences of some genes are higher than intraspecific variation thus providing an opportunity for DNA-based species identification. In any case, this technique is time-consuming in comparison with qPCR and it requires a pure sample and DNA from complex matrices can give unclear results [28]. An improved tool supporting molecular methods is Next-Generation Sequencing (NGS), which enables high-throughput and parallel taxonomic identification of multiple species, but a specialized working team and different statistical parameters should be employed to describe and evaluate the quality of the final genome assembly derived from NGS data output [26, 29]. In any case, the PCR method applied on specific DNA fragments is being advocated for species identification, because it is fast, cheap and has high sensitivity and specificity and has been employed to confirm morphological fragment identification [23, 30, 31].
The present research aims to develop a PCR-based method to detect Gryllodes sigillatus as a raw material in different processed feed and food preparations. The species-specific PCR primers and their accuracy are presented.
Materials and methods
Samples
The samples presented in the following list were used for method development and GS1 primer specificity test. Untreated insect samples of Gryllodes sigillatus (UnGS), Hermetia illucens, Acheta domesticus (AC), Gryllus assimilis, Musca domestica, Alphitobius diaperinus, Tenebrio molitor were purchased alive from Agripetgarden S.r.l. (PD)—Italy. Untreated insect samples of Bombus terrestris, Nezara viridula, Oecanthus pellucens, Sitophilus granaries, Sitophilus oryzae, Rhyzopertha dominica, Halyomorpha halys, Apis mellifera, Bombyx mori were kindly provided by the entomology division of the University of Udine, Italy. Species identification was confirmed by the examination of morphological features of adult individuals according to scientific standard literature [32, 33]. Muscle samples from selected animal species such as Sparus aurata (gilthead sea bream), Dicentrarchus labrax (European sea bass), Oncorhynchus mykiss (rainbow trout), Bos taurus (cattle), Ovis aries (sheep), Sepia officinalis (cuttlefish), Loligo vulgaris (squid), Mytilus edulis (mussel), Parapenaeus longirostris (pink shrimp), Penaeus kerathurus (striped prawn), Chamelea gallina (clam), Pleoticus muelleri (Argentine red Shrimp), Atherina boyeri, Pecten jacobaeus (scallop) and Aequipecten opercularis were purchased from GDO commerce, Italy.
Acheta domesticus (mAC) and Locusta migratoria flour were obtained from Crunchy Critters (Ilkeston, United Kingdom).
Gryllodes sigillatus meal (mGS), Cruncky Roasted Gryllodes sigillatus (RoGS) and energetic bars for human consumption (GSS) with different GS inclusion level (0, 5.2, 6.3, 10.6 g/100 g in GSS0, GSS1, GSS2 and GSS3, respectively) were purchased from Eat Grub Ltd, London, United Kingdom.
Finally, a serial dilution of commercial GS meal (mGS) from Eat Grub, and AC meal (mAC) from Crunchy Critters were also prepared as mixtures (approximately 100 g) at the laboratories of the University of Udine (Table 1) to be used as calibration material.
Sample preparation and DNA extraction
All the samples were stored at − 20 °C; if needed tissues were cut using a scalpel, put into a Lysing matrix A tube (MP biomedicals, Santa Ana, California, USA) and disrupted by a TissueLyser II bead mill (Qiagen, Hilden, Germany).
DNA was extracted from 20–25 mg of whole insect/pupae and samples of animals and feeds and food with the DNeasy Blood & Tissue® (Qiagen, Hilden, Germany) according to the manufacturer’s instruction. The DNA concentration and purity were evaluated by measuring the absorbance at 260 nm and the 260/280 nm ratio, respectively, using a NanoDrop One Micro-UV/Vis spectrophotometer (Thermo Fisher Scientific, Wilmington, U.S.A.). DNA integrity was also tested by denaturing 1.2% agarose gel electrophoresis in 1X TBE buffer and ethidium bromide staining.
DNA primers
All samples were analyzed using unspecific insect primers, Ins3.2F (ATAATTGGTGGATTTGGAAATTGA) and Ins3.2R (GGTGGATAA ACAGTT CATCCTGT), designed according to Koppel et al. [31]. A specific pair of primers for Gryllodes sigillatus was designed by retrieving the sequence of the mitochondrial cytochrome b (CYB) gene (accession no. KR903358.1) from GenBank. The pair of primers GS1Fw (GATCAAACAATCCCCTAGGTGTC) and GS1re (CTGGGTCTCCAAGTAT-TAAGGATTAG) were designed using the online software Primer3 (https://primer3.ut.ee) with Lucia algorithm. The expected GS1 amplified sequence was blasted in BLASTn to evaluate the specificity of the sequence against the deposited sequences in GenBank. The end-point PCR technique was performed to screen the primers specific amplification using the GoTaq DNA polymerase Kit (Promega, Milano, Italy). Each PCR reaction was conducted in a final volume of 20 µl, using 4 µl of PCR Green or colorless GoTaq Reaction Buffer (1.5 mMol MgCl2), 13.5 µl of distilled water, 0.4 µl of PCR nucleotide Mix at 10 mM, 0.1 µl of GoTaq DNA polymerase (5 µ/µl), 0.5 µl of 10 µM of each primer and 1 µl of DNA template (< 0.5 µg/50 µl) were used. The PCR thermal protocol consisted of one cycle of 3 min at 98 °C, then 1 min at 95 °C, 1 min at 60 °C and 1 min at 72 °C for 35 cycles and a final cycle of 5 min at 72 °C. Each PCR product was subsequently gel verified for the expected product length (bp).
The pair of primers amplified the target sequence and was tested for amplification efficiency and sensitivity in qPCR. These parameters were determined by six serial tenfold dilutions of pure GS DNA in three replicates. Real-time PCR was performed in a total volume of 20 µl, 10 µl of EvaGreen Buffer mix, 8.2 µl of molecular biology grade water, 0.4 µl of each primer, Forward and Reverse at a final concentration of 200 nM, and 1 µl of DNA. qPCR was carried out on a CX-96 (Biorad Srl, Milano, Italy) with the following standard program: initial denaturation steps of 3 min at 98 °C, followed by 45 cycles of 15 s at 95 °C and 30 s at 60 °C. Besides, a melting curve was determined using the following parameters: starting at 65 °C for 90 s temperature increment 1 °C, 5 s each step, 95 °C was reached. In all runs, a No Template Control (NTC) with pure water instead of DNA was added. All tested samples were diluted to obtain a concentration of 10 ng/µl and evaluated using a NanoDrop One Micro-UV/Vis spectrophotometer (Thermo Fisher Scientific, Wilmington, U.S.A.). When results of different runs were compared (e.g. in specificity testing) the threshold was always set manually to the same level. The baseline setting was done automatically.
The specificity of the primer pairs was estimated using 10 ng of DNA extracted from 40 samples including animals and ingredients. The GS1 primer sensitivity was evaluated by 6 serial tenfold dilutions in the concentration range from 10 ng to 10 fg, of DNA isolated from the target insect species (UnGS) and the processed products (RoGS, ACGS).
Sequencing
The samples, that in PCR have been amplified for both Ins3.2 and GS1 were purified using the QIAquick® PCR Purification Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The purified products from GS1 PCR were subsequently sequenced (Eurofins Genomics, Milano, Italy). The forward and reversed sequences were clustered and aligned with the reference gene sequence (accession no. KR903358.1) using the multiple sequence alignment program CLUSTAL OMEGA (https://www.ebi.ac.uk/) (data shown in supplementary material).
Data analysis
The limit of detection (LOD) for GS1 was defined as the lowest dilution level for which all replicates still gave a positive result. When appropriate, data were subjected to the linear regression model. The efficiency \({\text{E}}\, = \,\left( {{1}0^{{ - {1}/{\text{s}}}} - {1}} \right)*{1}00\) of each qPCR system was calculated based on the slope(s) of the regression curve obtained by plotting Ct values against log DNA concentrations. The regression lines were compared and tested for parallelism. The following model was used:
where ɛ is the error associated with the model and z is an “independent” dummy variable as described by Andrade & Estévez-Pérez (2014). The Student’s t test was used to evaluate the null hypothesis \(H_{0} :\beta_{{3}} = \, 0\).
Results and discussion
In silico analysis
The GS PCR system was developed based on the mitochondrial cytochrome b (CYB). The sequence from GenBank Accession No. KR903358.1 (346 bp) was chosen as Gryllodes sigillatus reference for method development and alignments with all other species. The GS1Fw and GS1re primers amplified a sequence of 149 bp included in this region. The specificity of the whole amplicon and the primer-binding sites checked in silico by BLASTn database revealed 85% of identity with Lepidogryllus comparatus and 83% of identity with Gryllus bimaculatus for the whole amplicon. A comparison between the Gryllodes-specific primers and the sequences from other Arthropods demonstrated an encouraging number of mismatches.
The genus Lepidogryllus sp. is of minor interest regarding the intended scope of the system developed primarily for feed and food on the European market, where those species are not allowed. In the data set, only Acheta domesticus revealed close identities (82%) with the targeted Gryllodes sigillatus sequence; in any case, the AC samples gave negative results with the GS primers.
To achieve 100% specificity for an insect, targeted PCR system seems to be unrealistic taking into consideration the biodiversity of insects [35]. The in silico analysis confirmed the application of the reported primers in the qPCR test and the results obtained in the present study revealed good specificity for GS against the species allowed for feed and the ones proposed for human consumption.
Specificity testing with biological samples
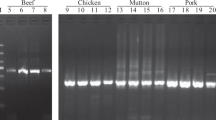
The primers specificity was tested in triplicate on 40 different DNA samples of animal and plant origin with the starting concentration of DNA at 10 ng/µl for each pair of primers. Data are reported in Table 2. The Ins3.2 primers amplified only the samples belonging to an insect family, as previously observed in Koppel et al. (2019). On the contrary, the GS1 primers gave clear positive signals in qPCR (Ct = 21.5) only for the target species, Gryllodes sigillatus, thus showing the specificity of the GS1 primers compared to the unspecific insect detection of Ins3.2. These results allow the use of GS1 primers to discriminate GS DNA in a complex matrix without the risk of unspecific amplification sequences.
Efficiency and limit of detection (LOD)
The efficiency of the GS1 qPCR system evaluated by serial tenfold dilutions of UnGS, RoGS, and ACGS100 DNA is presented in Fig. 1. Positive results were observed until 10–5 dilutions. Such values reflect the higher presence of mitochondrial target genomes relative to genomic DNA copies in the cell. Linear regression relationship was observed within the dilution range between Ct values and Log DNA concentrations. An efficiency of 96.4% was calculated from the regression curve of UnGS (y = − 3.394 x + 42.521; R2 = 0.994, d.f. 14) and RoGS (y = − 3.412 x + 43.023; R2 = 0.999, d.f. 14) while an efficiency of 95.4% resulted from the regression curve of ACGS100 (y = − 3.436 x + 47.010; R2 = 0.998, d.f. 8). The slope value, the amplification efficiency and the determination coefficient of the linear relationships resulted within the range of the requested parameters reported in the guidelines of the European Network of GMO laboratories for the verification of a quantitative real-time PCR method [36]. The values of the slope of the standard curves were in the range of − 3.6 ≤ slope ≤ − 3.1 and the amplification efficiencies were in the range 90—110%. Those results show the specificity of the primers designed for the target DNA and the melting curve underlines the amplification of a single fragment, confirmed by gel electrophoresis run.

PCR Linear regression relationship from five tenfold dilutions of Untreated GS (UnGS), roasted GS (RoGS) and ACGS mix (ACGS) DNA
qPCR validation
All the samples and complex matrices were tested in qPCR using the Ins3.2 primers; the samples that gave a positive result were subsequently tested in qPCR with GS1 primers. Only the samples UnGS, RoGS, ACGS meal mix and GSS (1–3) resulted in a positive signal and the amplified products were afterward sequenced to verify the amplified sequences that were blasted against the reference gene (CYB accession no. KR903358.1). The alignment (supplementary materials) shows no differences between the UnGS (reference DNA) and the other five sequenced samples, thus confirming that the qPCR analyses amplified selectively only the target G. sigillatus DNA; on the contrary, the six sequenced sequences of the qPCR amplification resulted in four different bases when compared with the reference CYB sequence (accession no. KR903358.1). Such discrepancy could be due to low accuracy in the deposited sequence in GenBank or punctiform intraspecies variation of the selected gene; anyway, these variations did not prevent the specific identification of the species.
Effect of technological treatment
The effect of GS matrix processing was evaluated on the detection method performance by utilizing the native G. sigillatus (UnGS) and the different commercial products: crunchy roasted samples (RoGS), the mixture of AC and GS commercial meals (ACGS) and energetic bars containing GS (GSS).
The linear regression of six ½ dilutions of 10 ng of DNA extract from UnGS and RoGS samples are shown in Fig. 2. The 10 ng/µl of UnGS DNA sample resulted in a Ct value 21.5 and the normalized curve of its six ½ dilutions is expressed by the relationship y = − 3.344 (± 0.090) x + 28.170 (± 0.118) (R2 = 0.995, d.f. = 17). The 10 ng/µl of RoGS DNA sample resulted in a figure of Ct value of 22.5 and the normalized curve of six ½ dilution is expressed by the relationship: y = − 3.728 (± 0.157) x + 30.001 (± 0.205) (R2 = 0.984, d.f. = 17). Testing the two regression lines for parallelism, the following relationship was obtained for the overall data y = 42.521 (± 0.288) − 3.394 (± 0.064) x− 0.018 (± 0.090) z + 0.502 (± 0.408) xz where the ± indicate the standard error associated to each coefficient. The Student’s t test for the coefficient of the interaction x × z (t = 1.231) was not significant (P = 0.234) so the lines are parallel with no significant difference between slope values (− 3.344 and − 3.727 for UnGS and RoGS, respectively). It should be noted that the dilutions from RoGS DNA samples resulted in approximately two additional cycles to be detected. The PCR results on the DNA extracted from the snacks for human consumption characterized by different GS inclusion (GSS) and from the mixture of AC and GS commercial meal samples (ACGS) are presented in Fig. 3. A clear positive signal was detected for the 10 ng DNA/PCR samples. No template control (NTC) and negative controls were tested and resulted in negative or weak signals. The samples with different levels of GS inclusion in the complex matrix of the snacks samples (GSS) resulted in a normalized relationship expressed by y = − 10.867 (± 1.094) x + 39.201 (± 1.021) (R2 = 0.936, d.f. = 8). The linear relationship obtained with the ACGS mixture is expressed by y = − 4.5572 (± 0.244) x + 35.67 (± 0.395) (R2 = 0.965, d.f. = 20).

GS PCR regression curve resulting from six-fold dilutions of Untreated GS (UnGS) and Roasted GS (RoGS) DNA. Data expressed as mean value ± standard deviation of triplicate measurements

GS PCR regression curve of different percentages of GS derivatives inclusion in the snack (GSS) and the mixture of commercial AC and GS meals (ACGS) compared with the UnGS regression curve. Data expressed as mean value ± standard deviation of triplicate measurements
When the regression lines of UnGS and ACGS were tested for parallelism, the following relationship was obtained for the overall data y = 34.834 (± 0.450) − 3.908 (± 0.314) x− 6.665 (± 0.475) z + 0.564 (± 0.335) xz. The Student’s t test for the coefficient of the interaction x × z (t = 1.681) was not significant (P = 0.105) so the lines resulted parallel with no significant difference between slopes (− 3.344 and − 4.557 for UnGS and ACGS, respectively).
It was worthwhile to note that the comparison between the UnGS regression line and the ACGS regression line reveals that additional PCR cycles are needed to detect the samples from the mixtures (ACGS) in comparison with the samples not subjected to technological treatments (UnGS).
In contrast, the GSS regression line presents a significantly different slope if compared to the UnGS linear relationship (P = 0.001). Testing the regression lines of UnGS and GSS for parallelism, the following relationship was obtained for the overall data y = 28.170 (± 0.175) − 3.344 (± 0.134) x + 11.030 (± 0.703) z− 7.523 (± 0.742) xz. The Student’s t test for the coefficient of the interaction x × z (t = -10.133) indicates that the lines are not parallel. The relationships presented in Fig. 3 underline the different characteristics of the matrices considered in the test. As observed by Hrnčírová, Bergerová, & Siekel (2008), the technological processing could be one of the factors affecting the integrity of the DNA extracted from food matrices that influences the CT needed for the qPCR detection. In fact, the nature/intensity of the processing treatments determined weak or not negligible degrading effect, as also suggested by the different y-intercepts of the regression lines for UnGS, RoGS, and ACGS. In addition, the possible presence of inhibitors, that could not be eliminated during the DNA extraction process, could affect the CT response of a complex matrix.
Moreover, in the case of GSS, the different particle size of the bars ingredients represents another factor that should be taken into account; this might have made the DNA extraction from fine components, like insect meal, easier with respect to the other ingredients. Consequently, such factor could have affected the sample expected ingredients ratio.
All these factors should be taken into account and specific calibration curves based on the sample characteristics should be considered to quantify the target DNA in different processed samples.
Conclusions
The real-time PCR method described in the present study resulted in robust and sensitive outcomes making it suitable for the purpose for which it was designed.
The use of the two pairs of primers enables a clear insect and GS discrimination and the sequencing of the GS1 qPCR products confirms the accuracy of the Gryllodes sigillatus detection and the robustness of the technique.
In addition, the results of the present study indicate that with the same initial amount of insect DNA, different signals are obtained depending on the severity of the technological processing treatment which the matrix is subjected to and the possible presence of inhibitors.
In conclusion, the complexity of the matrix, possible inhibitors, and the integrity of the extracted DNA can affect the quantification of GS in the sample, but the method is always capable of detecting the GS target DNA and the sequencing of the PCR product confirms the specificity of the amplified product. Moreover, the combination of the two pairs of primers is able to certify the univocal identification. However, if the method must be applied for relative quantification, specific calibration curves should be developed and validated for different technological processes.
For these reasons, the method is suitable for detecting the presence/absence of Gryllodes sigillatus DNA both in feed and food and GS1 primers will be used to implement a genosensor biochip [38].
References
FAO (Food and Agriculture Organization) (2011) World livestock 2011 livestock in food security. FAO (Food and Agriculture Organization)
Halloran A, Flore R, Vantomme P, Roos N (2018) Edible insects in sustainable food systems. Springer, London
Boland MJ, Rae AN, Vereijken JM et al (2013) The future supply of animal-derived protein for human consumption. Trends Food Sci Technol 29(1):62–73
FAO (2013) Edible insects. Future prospects for food and feed security. FAO
Remans R (2020) The changing nature of our food systems. Nat Food 1:21–21. https://doi.org/10.1038/s43016-019-0015-z
Smetana SM, Mathys A, Heinz V (2016) Life Cycle Assessment perspectives of insect proteins for feed and food. In: Paper presented to Putting LCA into Practice—10th International Conference on LCA of Food 2016 Dublin, Ireland, 19–21 October 2016
Dobermann D, Swift JA, Field LM (2017) Opportunities and hurdles of edible insects for food and feed. Nutr Bull 42(4):293–308
van Huis A, Tomberlin JK (2017) Insects as food and feed: from production to consumption (free online e-module). Wageningen Academic Publishers, Wageningen
Ramos-Elorduy J, Moreno JMP, Prado EE et al (1997) Nutritional value of edible insects from the state of Oaxaca, Mexico. J Food Compos Anal. https://doi.org/10.1006/jfca.1997.0530
Gahukar RT (2016) Edible Insects Farming: Efficiency and Impact on Family Livelihood, Food Security, and Environment Compared With Livestock and Crops. In: Insects as Sustainable Food Ingredients.
Schabel HG (2010) Forest insects as food: a global review. In: Forest insects as food: humans bite back.
DeFoliart GR (1992) Insects as human food Gene DeFoliart discusses some nutritional and economic aspects. Crop Prot. https://doi.org/10.1016/0261-2194(92)90020-6
DeFoliart GR (1999) INSECTS AS FOOD: Why the Western Attitude Is Important. Annu Rev Entomol. https://doi.org/10.1146/annurev.ento.44.1.21
EU 2015/2283 (2015) Regulation (Eu) 2015/2283 of the European Parliament. Off J Eur Union
La Barbera F, Verneau F, Videbæk PN et al (2020) A self-report measure of attitudes toward the eating of insects: construction and validation of the Entomophagy Attitude Questionnaire. Food Qual Prefer 79:103757. https://doi.org/10.1016/j.foodqual.2019.103757
de Kuijer O, Wielenga DK (1999) Een vergelijking van de milieubelasting van vlees en vleesalternatieven en de aantrekkelijkheid van de alternatieven voor consumenten
Sogari G, Amato M, Biasato I et al (2019) The potential role of insects as feed: A multi-perspective review. Animals 9(4):119
van der Spiegel M, Noordam MY, van der Fels-Klerx HJ (2013) Safety of novel protein sources (insects, microalgae, seaweed, duckweed, and rapeseed) and legislative aspects for their application in food and feed production. Compr Rev Food Sci Food Saf. https://doi.org/10.1111/1541-4337.12032
Alexandratos N, Bruinsma J (2012) World agriculture towards 2015/2030: the 2012 revision. ESA Working paper (Vol. 12 No. 3)
European Commission (2017) Commision Regulation 2017/893.. as regards to the provisions on processed animal proteins. Off J Eur Union 138:92–116
Weissman DB, Gray DA, Thi Pham H, Tijssen P (2012) Billions and billions sold: Pet-feeder crickets (Orthoptera: Gryllidae), commercial cricket farms, an epizootic densovirus, and government regulations make for a potential disaster. Zootaxa. https://doi.org/10.11646/zootaxa.3504.1.3
Vandeweyer D, Wynants E, Crauwels S et al (2018) Microbial dynamics during industrial rearing, processing, and storage of tropical house crickets ( Gryllodes sigillatus ) for Human Consumption. Appl Environ Microbiol. https://doi.org/10.1128/aem.00255-18
EC ( 2009) Commission regulation (EC) No 152/2009 of 27 January 2009 laying down the methods of sampling and analysis for the official control of feed. Off J Eur Union 36:1–130
Peres B, Barlet N, Loiseau G, Montet D (2007) Review of the current methods of analytical traceability allowing determination of the origin of foodstuffs. Food Control. https://doi.org/10.1016/j.foodcont.2005.09.018
Tramuta C, Gallina S, Bellio A et al (2018) A set of multiplex polymerase chain reactions for genomic detection of nine edible insect species in foods. J Insect Sci 18:3. https://doi.org/10.1093/jisesa/iey087
Zagon J, di Rienzo V, Potkura J et al (2018) A real-time PCR method for the detection of black soldier fly (Hermetia illucens) in feedstuff. Food Control 91:440–448. https://doi.org/10.1016/j.foodcont.2018.04.032
Liu ZF, Ci XQ, Li L et al (2017) DNA barcoding evaluation and implications for phylogenetic relationships in Lauraceae from China. PLoS ONE. https://doi.org/10.1371/journal.pone.0175788
Kim D-W, Yoo WG, Park HC et al (2012) DNA Barcoding of Fish, Insects, and Shellfish in Korea. Genomics Inform. https://doi.org/10.5808/gi.2012.10.3.206
Chun J, Oren A, Ventosa A et al (2018) Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int J Syst Evol Microbiol. https://doi.org/10.1099/ijsem.0.002516
Calvo JH, Zaragoza P, Osta R (2001) Technical note: A quick and more sensitive method to identify pork in processed and unprocessed food by PCR amplification of a new specific DNA fragment. J Anim Sci 79(8):2108–2112
Köppel R, Schum R, Habermacher M et al (2019) Multiplex real-time PCR for the detection of insect DNA and determination of contents of Tenebrio molitor, Locusta migratoria and Achaeta domestica in food. Eur Food Res Technol 245:559–567. https://doi.org/10.1007/s00217-018-03225-5
Capinera JL (2008) Encyclopedia of entomology, vol 4. Springer Science & Business Media, Berlin
Klausnitzer B (2011) Exkursionsfauna von Deutschland
Andrade JM, Estévez-Pérez MG (2014) Statistical comparison of the slopes of two regression lines: A tutorial. Anal Chim Acta 838:1–2
Hebert PDN, Cywinska A, Ball SL, DeWaard JR (2003) Biological identifications through DNA barcodes. Proc R Soc B Biol Sci 270:313–321. https://doi.org/10.1098/rspb.2002.2218
Goerlich O, Grohmann L, Mazzara M et al (2017) Verification of analytical methods for GMO testing when implementing interlaboratory validated methods. Version 2. Test Anal GMO-containing Foods Feed 8:245
Hrnčírová Z, Bergerová E, Siekel P (2008) Effects of technological treatment on DNA degradation in selected food matrices of plant origin. J Food Nutr Res 47(1):23–28
Daniso E, Melpignano P, Tulli F (2020) An OLED-based genosensor for the detection of Hermetia illucens in feeds. Food Control 113:107179. https://doi.org/10.1016/j.foodcont.2020.107179
Acknowledgements
This work was supported by project Ager SUSHIN cod. 2016-0112. The authors wish to thank prof. P. Zandigiacomo for kindly confirming insect species identification and Dr. Dianne Cannizzo for accurate language revision.
Funding
Open access funding provided by Università degli Studi di Udine within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Contributions
E.D. and F.T. have participated in conception and design; E.D., R.C. and G.C. performed analysis and interpretation of the data; E.D. and F.T. have drafted the article and revised it critically for important intellectual content; E.T. provided funds for activity. All authors had approved the final version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Daniso, E., Tulli, F., Cardinaletti, G. et al. Molecular approach for insect detection in feed and food: the case of Gryllodes sigillatus. Eur Food Res Technol 246, 2373–2381 (2020). https://doi.org/10.1007/s00217-020-03573-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-020-03573-1