
Abstract
Identification of crops present in food and/or feed matrices represents an important step in the screening strategies targeting genetically modified organisms (GMO). Soybean, maize, oilseed rape, rice, cotton, sugar beet and potato are to date the most important sources of genetically modified materials imported in the European Union (EU). In order to allow detection of their presence in an integrated screening approach, a set of SYBR®Green real-time polymerase chain reaction (qPCR) methods has been developed which can be used under the same assay conditions and at similar efficiency for each of the abovementioned crops. Each qPCR method is shown to meet the performance criteria (i.e. specificity, limit of detection and PCR efficiency) set by the European Network of GMO Laboratories (ENGL). When combined with the equivalent qPCR methods targeting GMO elements, these crop-specific SYBR®Green qPCR methods can aid the development of an efficient tool for determining GMO presence in food and/or feed products.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
To date, about 130 genetically modified (GM) plants have received an authorization for the commercial usage as food and/or feed in the world [1]. Most of these GM plants are derived from crops of critical importance to the EU food and feed producers: soybean, maize, oilseed rape, rice, sugar beet, cotton [2]. EU consumers are reluctant to the introduction of GM materials in the food/feed chain. For this, traceability of GM products on the EU market is regulated by the means of mandatory labelling rules (EC/1829/2003 [3] and EC/1830/2003 [4]). Considering the diversity of GMOs authorized for food and feed use, enforcement activities to verify compliance with these rules have become increasingly complex, time-consuming and expensive. To date, most GMO detection strategies are PCR-based approaches [5]. A state of the art overview on the different techniques currently applied is presented in Querci et al. [6]. Several of these approaches consist of a two-step process: (1) a generic screening for the presence of crop(s) and of GM material using common genetic elements present in authorized GMOs [such as the CaMV 35S promoter, Agrobacterium tumefaciens nopaline synthase terminator (tNOS)] [5] and (2) a GMO identification step wherein so-called event-specific methods are used to univocally identify which GMOs are present [7].
Several GMO detection approaches introduce hierarchal logic into the screening approach. Leimanis et al. [8] used a micro-array platform for a combination of crop, trait-, GM element-, construct- and event-specific GMO screening targets. Morriset et al. [9] used NAIMA, a nucleic acid sequence-based amplification (NASBA [10]) approach adapted on an array platform to develop a multi-target GMO screening tool. Both technologies require specific equipment, and the validation of array devices remains challenging. Waiblinger et al. [11] have presented a tiered-approach-based Taqman single PCR-based approach, while Van den Bulcke et al. [12] have developed an integrated GMO screening approach based on hierarchal logic, the so-called ‘Combinatory SYBR®Green PCR Screening’ (CoSYPS). Though limited in designing multiplex experiments using the SYBR®Green dye, SYBR®Green real-time qPCR method is considered to offer a number of advantages over the other approaches: (1) SYBR®Green qPCR monitors the increase in total fluorescence throughout the amplification, allowing detection of specific amplification and also of non-specific amplification, (2) melting temperature analysis allows post-PCR identification of the amplification not solely of the expected target but also of closely related target(s), (3) the validation of simplex PCR methods is technically as feasible and well documented as other PCR chemistries and (4) the SYBR®Green technology is the least costly qPCR approach since no dye-labelled oligonucleotide probes are required. CoSYPS implies the analysis of a sample for GMO presence using a plant kingdom marker (the chloroplastic rbcl gene [13]), a number of crop-specific markers (e.g. for soybean, maize cotton, oilseed rape), common GMO control element targets (35S and tNOS [14]) and trait-specific targets [CP4-EPSPS, CryIAb, PAT, Bar (in preparation)]. As CoSYPS aims to perform all screening tests within a single analytical PCR run, the choice of the respective markers and the development of the SYBR®Green methods demand uniform PCR conditions and equivalent method performances [6, 12].
When looking at crop-specific markers, special attention needs to be paid to their specificity, stability and copy number. Preferably, crop-specific markers should represent biological taxonomic entities that are used as ingredients in commercial food/feed applications (e.g. soybean, maize,…). A crop marker for such an entity will need careful evaluation when variability in the genome constitution is well established (e.g. for oilseed rape (OSR) and cotton [15, 16]). With respect to copy number of the ingredient marker, a low copy number gene (preferentially a single copy) that is stably inherited and present in the most important cultivars would be the optimal choice. Finally, the target sequences in the marker gene have to be carefully evaluated as in SYBR®Green PCR the specificity of the reaction is solely guaranteed by the primers.
Within the commercial applications for GM Food & Feed, several reference genes for the respective crops/ingredients have already been proposed: the lectin gene for soybean, the adh, hmg, SSIIb and invertase genes for maize, the acc, cruA and fat A genes for oilseed rape, the pld gene for rice, the sad1 and sah7 gene for cotton, and the GS gene for sugar beet [17–28]. Several of these and other newly developed markers have been tested independently for each of these crops both in qualitative screening and/or in GM quantification [29]. However, most of these methods use Taqman chemistry in which the use of a probe as a fluorescent reporter can mask possible aspecific amplification. In this context, the specificity of many methods is not documented adequately enough to rule out such aspecific reactions.
In this study, different sets of primers issued from existing Taqman qPCR were selected as candidate for integration in the COSYPS as crop-specific marker genes. Since this means a switch to SYBR®Green chemistry and application of all methods under the same PCR amplification conditions, each method was first assayed for its specificity in SYBR®Green modus. In case, any aspecific amplification was observed, a new SYBR®Green qPCR method was developed for the respective target. Further, each of the methods was tested for its sensitivity and efficiency by serial dilution analysis.
Materials and methods
Plant materials
Soybean seeds (Monsanto Company), maize seeds (Aveve, Landen, BE) and OSR seeds (Aveve, Landen, BE) were sown and grown in house (plant growth chamber (Snijders Scientific S1084, Tilburg, NL) on a 16/8 h day/night regime at 25 °C and 80% humidity) to collect leave material. Other plant materials were either Certified Reference Materials (CRM) (rice and cotton from AOCS, sugar beet from IRMM) or Reference Material (RM) [potato, wheat, oat, sunflower, winter wheat, barley and German wheat, soybean, maize and oilseed rape from various sources (see Table 1)]. All tested materials, except the sugar beet material, are produced from wild-type (WT) species.
Chemicals, PCR reagents and primers
All applied chemical products were analytical grade (NaCl, EDTA, Tris, boric acid, HCl, CTAB, chloroform, isopropanol and ethanol). The applied enzyme products were Ribonuclease A and Proteinase K from Sigma–Aldrich (Bornem, BE), EcoRI and T4 DNA polymerase from Invitrogen (Merelbeke, BE). DNase- and RNase-free water was purchased from ACROS organics (Geel, BE).
All kit-based protocols were carried out according to the manufacturer recommendation: Quant-iT™ PicoGreen® dsDNA Assay Kit, TOPO TA Cloning® Kit, pCR® 2.1-TOPO® vector, TOP10F’ competent cells (Invitrogen, Merelbeke, BE); Genome lab, Dye Terminator Cycle Sequencing (DTCS) kit (Beckman Coulter/Analis, Suarlée, BE), QIAGEN Plasmid Midi kit (QIAGEN, Venlo, NL).
Pre-casted “Ready Agarose™ 96 Plus Gel (3%)” (BioRad, Nazareth, BE) and “EZ Load HT molecular weight markers (100 bp–2 kb)” (Biorad, Nazareth, BE) were used for agarose gel analysis.
In the PCRs, Oligold® oligonucleotides (Eurogentec, Ougrée, BE) and SYBR®Green PCR Mastermix (Diagenode, Liège, BE) were used.
Development of primer pairs
The primer sets for the respective crop-specific markers were at first retrieved from the literature (Table 1). When not suitable as such for the SYBR®Green approach, new primers were developed in the context of this study (ADH short, CruA and PLD-alt, Table 1), based on bioinformatic analysis of the respective DNA sequences according to Barbau-Piednoir et al. [14].
Extraction of genomic DNA
Genomic DNA (gDNA) was extracted using a CTAB-based method [14]. The extracted gDNA was quantified using a VersaFluorTM Fluorometer (Biorad, Nazareth, BE) and the Quant-iT™ PicoGreen® dsDNA Assay Kit. The DNA was stored at −20 °C.
Real-Time PCR
All qPCR assays were performed on an ABI 7300 PCR System (Applied Biosystems, Lennik, BE) in 25-μl reaction volume containing 1× SYBR®Green PCR Mastermix and 250 nM of each primer, except for the RBCL primers for which a concentration of 1 μM was used. The following thermal programme was applied: a single cycle of DNA polymerase activation for 10 min at 95 °C followed by 40 amplification cycles of 15-s denaturing step (95 °C) and 1-min annealing–extension step (60 °C). Afterwards, melting temperature analysis of the obtained amplification products was performed using standard machine settings. Note that the fluorescent reporter signal was normalized against the internal reference dye (ROX) signal and that threshold limit setting was performed in automatic mode, according to the ABI Sequence Detection Software version 1.4, unless manual adjustment was considered necessary.
Amplicon cloning, sequencing and plasmid deposit
PCR fragments obtained using the different qPCR methods were cloned and characterized by dideoxy sequence analysis according to Barbau-Piednoir et al. [14]. The so-called “SYBR®Green amplicons” (abbreviated as “Sybricons”) and “pENGL plasmids” (= plasmid deposited in the context of the European Network of GMO Laboratories) were registered under “Safe Deposit” or “Patent deposit” at the “Belgian Culture Collection for Micro-organisms” (BCCM/LMBP) (Gent, BE).
SYBR®Green qPCR specificity assessment
Primer pair specificity was tested by amplification of the target in 10-ng reference material gDNA. A sample is considered as positive if a specific signal is generated with a SYBR®Green qPCR method according to the following criteria [14]: (1) (exponential) amplification (increase of fluorescence) should be obtained using template DNA containing the target sequence(s), while negative controls (No Template Controls (NTC) and/or gDNA from non-target-plants) do not yield amplification. (2) For all positive reactions, the obtained PCR product should represents a single peak in melting analysis with a unique Tm value corresponding to the nominal Tm value obtained for the corresponding Sybricon or pENGL template (with an acceptable difference of ±1 °C), while no specific peaks should be detectable in the negative controls. Also (3), a single band should be obtained in agarose gel analysis with a molecular weight corresponding to the predicted amplicon size (SD ± 10 bp).
SYBR®Green qPCR sensitivity assessment
In this study, the sensitivity of the assays was estimated according to the former AFNOR Norm XP V03-020-2 [30], as adapted by Barbau-Piednoir et al. [14]: each crop-specific marker was tested in a dilution series of corresponding gDNA ranging from 20,000 haploid genome copies (HGE) to a single haploid genome copy (crop genome size according to Arumugunathan & Earle [31]). Each dilution analysis was performed in hexaplicate (n = 6). The detection limit is defined as the lowest level haploid genome copies in the serial dilution for which each of the 6 repeats provides a specific positive signal (n = 6; 6/6 specific signals), hence the name, LOD6.
Agarose gel analysis
Agarose gel electrophoresis (3% precast gels, Biorad) was performed using 1× TBE (45 mM TRISborate- 1 mM EDTA) at 100 volts for 15 min, including a 100-bp–2-kb Molecular Marker (BioRad, Nazareth, BE).
Results and discussion
Tm value determination for the crop SYBR®Green qPCR amplicons with reference plasmids as DNA template
SYBR®Green PCR offers the possibility to apply post-run the melting temperature (Tm) analysis of the PCR products as an additional decision criterion for identifying positive signals [14]. Since the Tm is primarily dependent on the base pair composition of the amplified products, all specific PCR products obtained with a particular primer pair should have a similar melting temperature. To minimize bias due to the genetic background in determining the nominal reference value of the Tm for each target in test samples, the respective qPCR products were cloned into a uniform plasmid background. Such plasmids are considered the material of choice to define the nominal Tm value for the respective amplicons and may serve as the target reference. The Tm values for the different cloned targets are listed in Table 2. To take into account the potential variability in measured Tm, a prediction interval of ±1 °C around the nominal Tm value was applied as acceptance range [14].
Assessment of the primer set specificity
Primer sets for crop-specific markers that were already validated under Taqman chemistry or by conventional PCR (see Table 1) were used as candidates for development of SYBR®Green assays. Their specificity was tested against the major commodity crops commercialized worldwide for which GM cultivars exist, namely soybean, maize, OSR, rice, cotton, potato and sugar beet [32]. In addition, the specificity was also evaluated against wheat, barley, sunflower and oat that represent important crops commonly present in food and/or feed.
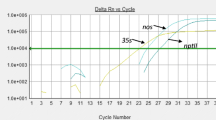
The presence of amplifiable gDNA in all extracts was controlled using the generic plant qPCR method targeting the rbcl chloroplastic gene (Figs. 1l, 2) [13]. Based on the specificity criteria defined in the materials and methods, all commodity crops tested with the Rbcl method are positive, indicating that the RBCL SYBR®Green qPCR method is suitable to detect the presence of said materials. Next, the respective gDNAs were further assayed with the following primers: the LECTIN long and short primers (soybean), the ADH long primers (maize), the ACC primers (OSR), the PLD primers (rice), the SAH7 and the SAD1 primers (cotton) and the GluA3 primers (sugar beet). Results of the specificity test are presented in Figs. 1, 2 and Table 2. The LECTIN short & long, the ADH long, the ACC, the GluA3 and the Sad1 primer sets meet the criteria for the amplification, Tm specificity assessment and evaluation by agarose gel electrophoresis (Fig. 2).

Specificity analysis, qPCR runs: Primers listed in Table 1 were evaluated for their specificity for the plant species described in Table 2. The relative fluorescence (Delta Rn) registered during the qPCR is plotted against the number of qPCR cycles (a). For each amplification curve, the amplicon is analysed for its melting profile (b). a LECTIN long primers, b LECTIN short primers, c ADH long primers, d ADH short primers, e ACC primers, f CruA primers, g PLD primers, h PLD-alt primers, i SAH-7 primers, j SAD1 primers, k GluA3 primers, l RBCL primers

Specificity analysis, gel electrophoresis. qPCR products are loaded on an ethidium bromide stained gel to approximate the molecular weight of amplicon. The expected molecular size of each PCR product is indicated in regard to the SYBR®Green qPCR method name. Lanes 1 Soybean, Lanes 2 Maize, Lanes 3 OSR, Lanes 4 Rice, Lanes 5 Cotton, Lanes 6 Potato, Lanes 7 Sugarbeet, Lanes 8 Wheat, Lanes 9 NTC, Lanes 10 Oat, Lanes 11 Sunflower, Lanes 12 Barley, Lanes 13 Winter Wheat, Lanes 14 German Wheat, MW Molecular weight marker EZ load HT
The PLD primers (rice) and SAH7 primers (cotton) show cross-reactivity, respectively, with maize and with potato. Both primer sets were thus excluded, and new primers were designed for rice using the pld gene sequence as a template (accnr AB001919). After in silico testing, a single set of PLD primers was retained and the results of the specificity testing demonstrated that this novel primer set, indicated as PLD-alt, meet the specificity criteria (see Figs. 1, 2; Table 2).
Sensitivity of the SYBR®Green qPCR methods
The primers fulfilling the above specificity criteria were then evaluated at the level sensitivity as described in the materials and methods (see Table 3). The LOD6 of each qPCR method was equal or below 25 HGE, except for the ADH long method (50 HGE). Thus, a new primer set targeting a shorter ADH sequence was developed (designated as ADH short). This new set of ADH primers was shown to be specific (see Figs. 1, 2; Table 2) with a LOD6 set at 25 HGE (see Table 3).
In order to determine the dynamic range of each of the crop-specific SYBR®Green qPCR methods, a regression analysis on the different dilutions was performed, wherein the obtained Ct values were plotted against the logarithm of their target copy number. In this way, an estimated PCR efficiency of the respective methods was determined [33] and checked against the ENGL acceptance criteria [34]. For most of the developed qPCR methods, both the PCR efficiency (between 90 and 110%) (Table 3) and the linear correlation of regression (≥0.98) fell in the ENGL acceptance range. The efficiency of the ACC qPCR is highly variable (not shown) and often fell out of the ENGL acceptance range (PCR efficiency = 81% in Table 3). Therefore, a novel qPCR method for oilseed rape was developed targeting the cruciferin A gene (accession number: X14555.1). The results concerning the CruA SYBR®Green qPCR are also listed in Figs. 1, 2 and Table 2. When tested on the crops listed in Table 1, these new oligonucleotides clearly demonstrated their specificity for OSR. At the level of sensitivity, the CruA primers performed adequately with an LOD6 at 6 HGE and a PCR efficiency of 102.42% (Table 3).
In conclusion, under similar qPCR conditions, 6 crop-specific SYBR®Green methods for detecting commodity crops were successfully developed at the level of specificity and showed to present similar and satisfactory PCR efficiencies.
Genome-specificity assessment of the SAD1 and CruA SYBR®Green qPCR in, respectively, cotton and oilseed rape
As mentioned previously, any crop-specific method to be used in GMO screening should target a stable unique marker representative of its biological taxonomic entity. Cotton and oilseed rape genomes are the result of particular evolutionary processes; as a consequence, they exist as tetraploïd individuals comprising two coexisting genomes [15, 16]. It was, therefore, meaningful to investigate (1) the copy numbers of the candidate reference genes developed in this study in the major commercialized species (Gossypium hirsutum for cotton and Brassica napus for oilseed rape) and (2) whether these targets may be found in other genus species.
The SAD1 SYBR®Green qPCR method was tested among four different cotton species, namely Gossypium hirsutum (which represents the majority of the cotton production worldwide), Gossypium barbadense, Gossypium herbaceum and Gossypium arboreum. Equivalent gDNA amounts of each Gossypium species were analysed. The respective amplicons have a similar electrophoretic mobility (approximately 110 bp) (not shown) and a similar Tm of 78.2 °C ± 0.4 °C, corresponding with the plasmid DNA reference value (78.5 °C) (Table 4A). In addition, as all Gossypium species yield a similar Ct (average Ct = 24.1 ± 0.5, Table 4A) with the SAD1 primers, it is therefore most likely that the sad1 gene is represented at equivalent copy numbers in each of the different genomes.
A similar analysis was performed for the cruA gene, where the SYBR®Green qPCR was also tested against other Brassica species listed in Table 4B. All the tested Brassica species could be specifically amplified with an average Tm of 79.4 °C ± 0.2 °C. This clearly demonstrated that the cruA gene is to be considered as genus-specific marker (Brassicacae), rather than a Brassica napus species marker. The CruA qPCR Ct values obtained with the respective Brassica species were again very similar (average Ct = 20.1 ± 0.5), if not identical; it was thus most likely that the cruA gene was represented at similar copy number in each of the different genomes.
From these results, it appears that the SAD1 and CruA primers are suitable for the respective identification of cotton and oilseed rape. Attention should be paid when using these markers for quantitative purposes as they are not only present in GM crops but also in different subspecies of non-transgenic material. As such, the final quantification will be biased if these non-transgenic species are present in high amounts [35].
Conclusion
In this study, species-specific SYBR®Green qPCR methods that performed at similar efficiencies were successfully developed to allow the identification of main commodity crops. These species methods are associated to GMO screening methods (p35S, tNos …) in the recently described CoSYPS [12]. Such association is a powerful tool to find out the GM events in food/feed products.
References
CERA’s database of safety information website: http://cera-gmc.org/index.php?action=gm_crop_database&mode=Synopsis
GMO compass web site: http://www.gmo-compass.org/eng/grocery_shopping/crops/
Commission Regulation (EC) 1829/2003 of the European Parliament and of the Council of 22 September 2003 on genetically modified food and feed. Official journal of the European communities (2003) L 268:1–23. http://ec.europa.eu/food/food/biotechnology/gmfood/legisl_en.htm
Commission Regulation (EC) 1830/2003 of the European Parliament and of the Council of 22 September 2003 concerning the traceability and labeling of genetically modified organisms and the traceability of food and feed products produced from genetically modified organisms and amending directive 2001/18/EC. Official Journal of the European Communities (2003) L 268:24–28. http://ec.europa.eu/food/food/biotechnology/gmfood/legisl_en.htm
Holst-Jensen A, Rønning SB, Løvseth A, Berdal KG (2003) PCR technology for screening and quantification of genetically modified organisms (GMOs). Anal Bioanal Chem 375(8):985–993
Querci M, Van den Bulcke M, Žel J, Van den Eede G, Broll H (2010) New approaches in GMO detection. Anal Bioanal Chem 396:1991–2002
Marmiroli N, Maestri E, Gullì M, Malcevschi A, Peano C, Bordoni R, De Bellis G (2008) Methods for detection of GMOs in food and feed. Anal Bioanal Chem 392:369–384
Leimanis S, Hamels H, Naze F, Mbongolo Mbella G, Sneyers M, Hochegger R, Broll H, Roth L, Dallmann K, Micsinai A, Luis La Paz J, Pla M, Brünen-Nieweler C, Papazova N, Taverniers T, Hess N, Kirschneit B, Bertheau Y, Audeon C, Laval V, Busch U, Pecoraro S, Neumann K, Rösel S, van Dijk J, Kok E, Bellocchi G, Foti N, Mazzara M, Moens W, Remacle J, Van Den Eede G (2008) Validation of the performance of a GMO multiplex screening assay based on microarray detection. Eur Food Res Technol 227:1621–1632
Morisset D, Dobnik D, Hamels S, Žel J, Gruden K (2008) NAIMA: target amplification strategy allowing quantitative on-chip detection of GMOs. Nucleic Acids Res 36:e118. doi:10.1093/nar/gkn524
Compton J (1991) Nucleic acid sequence-based amplification. Nature 350:91–92
Waiblinger H-U, Grohmann L, Mankertz J, Engelbert D, Pietsch K (2010) A practical approach to screen for authorized and unauthorized genetically modified plants. Anal Bioanal Chem 396:2065–2072
Van den Bulcke M, Lievens A, Barbau-Piednoir E, Mbongolo Mbella EG, Roosens N, Sneyers M, Leunda Casi A (2010) A theoretical introduction to “Combinatory SYBR®Green qPCR Screening”, a matrix-based approach for the detection of materials derived from genetically modified plants. Anal Bioanal Chem 396:2113–2123
Debode F, Marien A, Dardenne P, Berben G (2004) Développement d’une méthode qualitative et quantitative pour la détection des transgènes applicable en routine aux productions agricoles et aux denrées alimentaires : la technique PCR real-time. Convention de recherche S-6062 financée par le SPF Santé Publique
Barbau-Piednoir E, Lievens A, Mbongolo Mbella EG, Roosens N, Sneyers M, Leunda-Casi A, Van den Bulcke M (2010) SYBR®Green qPCR screening methods for the presence of ‘‘35S promoter’’ and ‘‘NOS terminator’’ elements in food and feed products. Eur Food Res Technol 230:383–393
Organisation for economic co-operation and development. Series on harmonisation of regulatory oversight in biotechnology N°7, Consensus document on the biology of cotton of Brassica napus L. (OILSEED RAPE). OCDE/GD (1997) 63.(http://www.olis.oecd.org/olis/1997doc.nsf/LinkTo/NT00000A16/$FILE/05E79505.PDF)
Organisation for economic co-operation and development. Series on harmonisation of regulatory oversight in biotechnology N°45. Consensus document on the biology of cotton (Gossypium spp.). ENV/JM/MONO (2008) 33 (http://www.olis.oecd.org/olis/2008doc.nsf/LinkTo/NT0000794A/$FILE/JT03257047.PDF)
Berben G, Dardenne P (2001) Traçage et authentification des produits à base d’organismes génétiquement modifiés, Recherches financées par les SSTC dans le cadre du programme de ‘Recherche pré-normative dans le secteur alimentaire’, CRAW, rapport final contract NP/42/026
Vaïtilingom M, Pijnenburg H, Gendre F, Brignon P (1999) Real-time quantitative PCR detection of genetically modified maximizer maize and roundup ready soybean in some representative foods. J Agric Food Chem 47:5261–5266
Hernandez M, Duplan M-N, Berthier G, Vaïtilingom M, Hauser W, Freyer R, Pla M, Bertheau Y (2004) Development and comparison of four Real-Time Polymerase Chain Reaction systems for specific detection and quantification of Zea mays L. J Agric Food Chem 52:4632–4637
Paterno A, Marchesi U, Gatto F, Verginelli D, Quarchioni C, Fusco C, Zepparoni A, Amaddeo D, Ciabatti I (2009) Finding the joker among the maize endogenous reference genes for genetically modified organism (GMO) detection. J Agric Food Chem 57:11086–11091
Kuribara H, Shindo Y, Matsuoka T, Takubo K, Futo S, Aoki N, Hirao T, Akiyama H, Goda Y, Toyoda M, Hino A (2002) Novel reference molecules for quantitation of genetically modified maize and soybean. J AOAC int 85(5):1077–1089
Hernandez M, Rıo A, Esteve T, Prat S, Pla M (2001) A rapeseed-specific gene, Acetyl-CoA Carboxylase, can be used as a reference for qualitative and Real-Time quantitative PCR detection of transgenes from mixed food samples. J Agric Food Chem 49:3622–3627
Community Reference Laboratory for GM Food and Feed. Event-specific method for the quantification of the Oilseed Rape Line T45 using Real-time PCR. (http://gmocrl.jrc.ec.europa.eu/summaries/T45_validated_RTPCR_method.pdf)
Monsanto Biotechnology Regulatory Sciences (2004) A recommended procedure for Real-Time Quantitative TaqMan PCR for Roundup Ready Canola RT73. (http://gmo-crl.jrc.ec.europa.eu/detectionmethods/MON-Art47-pcrGT73rapeseed.pdf)
Community Reference Laboratory for GM Food and Feed. Event-specific method for the quantification of rice line LLRICE62 using Real-time PCR. (http://gmo-crl.jrc.ec.europa.eu/summaries/LLRICE62_validated_Protocol.pdf)
Community Reference Laboratory for GM Food and Feed. Event-specific Method for the quantification of cotton 281-24-236 using Real-time PCR. (http://gmo-crl.jrc.ec.europa.eu/summaries/281-24-36_cotton_Protocol.pdf)
Yang L, Chen J, Huang C, Liu Y, Jia S, Pan L, Zhang D (2005) Validation of a cotton-specific gene, Sad1, used as an endogenous reference gene in qualitative and real-time quantitative PCR detection of transgenic cottons. Plant Cell Rep 24:237–245
Community Reference Laboratory for GM Food and Feed. Event-specific method for the quantification of sugar beet line H7-1 using Real-time PCR. (http://gmo-crl.jrc.ec.europa.eu/summaries/H7-1-Protocol.pdf)
Dong W, Yang L, Shen K, Kim B, Kleter G, Marvin H, Guo R, Liang W, Zhang D (2008) GMDD: a database of GMO detection methods. BMC Bioinformatics 9:260 (http://gmdd.shgmo.org/index/search)
Norme AFNOR XP V03-020-2 (04/2005) Produits alimentaires–Détection et quantification des organismes végétaux génétiquement modifiés et produits dérivés–Partie 2: méthodes basées sur la réaction de polymérisation en chaîne
Arumuganathan K, Earle ED (1991) Nuclear content of some important plant species. Plant Mol Biol Rep 9(3):208–218
James C (2009) ISAAA brief 41-2009: global status of commercialized biotech/GM Crops: 2009. Available at http://www.isaaa.org/resources/publications/briefs/41/executivesummary/default.asp
Rutledge RG, Côte C (2003) Mathematics of quantitative kinetic PCR and the application of standard curves. Nucleic Acids Res 31(16):e93 10.1093/nar/gng093
European Network of GMO Laboratories (ENGL) (2008) Definition of minimum performance requirements for analytical methods of GMO testing. http://gmo-crl.jrc.ec.europa.eu/doc/Min_Perf_Requirements_Analytical_methods.pdf
Wu G, Zhang L, Wu Y, Cao Y, Lu C (2010) Comparison of five endogenous reference genes for specific PCR detection and quantification of Brassica napus. J Agric Food Chem 58:2812–2817
Acknowledgments
The authors would like to greatly thank Els Vandermassen and Dirk Van Geel for their technical support. Gilbert Berben (CRA-W, Belgium) and his team are acknowledged for providing the Rbcl primer sequences prior to publication. CRA-W kindly provided seeds from WT wheat, WT oat (tornade variety), WT sunflower, WT winter wheat (centenaire variety), WT barley (seychelles variety) and WT German wheat (cosmos variety). Seeds from the different Brassicacae and Gossypium species were obtained thanks to the Germplasm Resources Information Network (GRIN) of the U.S. Department of Agriculture’s Agricultural Research Service. This research was financially supported by the GMODETEC project (RT-06/6) of the Belgian Federal Ministry of ‘‘Health, Food Chain Safety and Environment’ and by the European Commission through the Integrated Project Co-Extra, Contract No. 007158, under the 6th Framework Program. All applied analyses were acceptable for accreditation under ISO-17025 by the official Belgian ISO accreditation organisation “Belac” (date: 2006).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Mbongolo Mbella, E.G., Lievens, A., Barbau-Piednoir, E. et al. SYBR®Green qPCR methods for detection of endogenous reference genes in commodity crops: a step ahead in combinatory screening of genetically modified crops in food and feed products. Eur Food Res Technol 232, 485–496 (2011). https://doi.org/10.1007/s00217-010-1408-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-010-1408-2