
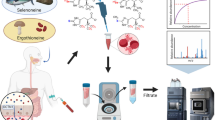
Abstract
Queuosine (Q) is a hypermodified 7-deaza-guanosine nucleoside exclusively synthesized by bacteria. This micronutrient and its respective nucleobase form queuine (q) are salvaged by humans either from gut microflora or digested food. Depletion of Q-tRNA in human or mouse cells causes protein misfolding that triggers endoplasmic reticular stress and the activation of the unfolded protein responses. In vivo, this reduces the neuronal architecture of the mouse brain affecting learning and memory. Herein, a sensitive method for quantifying free q and Q in human blood was developed, optimised and validated. After evaluating q/Q extraction efficiency in several different solid-phase sorbents, Bond Elut PBA (phenylboronic acid) cartridges were found to have the highest extraction recovery for q (82%) and Q (71%) from pooled human plasma. PBS with 4% BSA was used as surrogate matrix for method development and validation. An LC–MS/MS method was validated across the concentration range of 0.0003–1 µM for both q and Q, showing excellent linearity (r2 = 0.997 (q) and r2 = 0.998 (Q)), limit of quantification (0.0003 µM), accuracy (100.39–125.71%) and precision (CV% < 15.68%). In a sampling of healthy volunteers (n = 44), there was no significant difference in q levels between male (n = 14; mean = 0.0068 µM) and female (n = 30; mean = 0.0080 µM) participants (p = 0.50). Q was not detected in human plasma. This validated method can now be used to further substantiate the role of q/Q in nutrition, physiology and pathology.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
It has been reported that over 170 RNA modifications in RNA molecules occur across all domains of life [1]. Amongst all the RNA molecules, tRNA is the most extensively modified RNA. A human nucleus-encoded tRNA molecule contains on average 13 modifications per molecule, of which an average of approximately 17% of the total residues are modified [2, 3]. tRNA modifications can direct mRNA decoding when it occurs in the anticodon loop (position 34). Modifications occurring downstream of the anticodon nucleotides (position 37) affect the stability, folding, localization and translational dynamics of tRNA [2, 4]. Amongst all the tRNA modifications, queuosine (Q) is the only nucleoside that is synthesized in bacteria de novo and salvaged in eukaryotes from their environment [5]. It is a hypermodified 7-deaza-guanosine nucleoside that replaces the unmodified guanine at wobble position 34 of the anticodon GUN for four tRNAs acceptors for the amino acids tyrosine, asparagine, aspartic acid and histidine [6, 7]. The Q biosynthesis pathway has been well studied in eubacteria [8, 9]. However, in eukaryotes, the biosynthesis pathways and cellular roles of Q remain to be fully elucidated. Humans obtain Q as a micronutrient from diet or gut microflora. An enzyme called QNG1 (previously known as DUF2914) hydrolyses Q or a Q-monophosphate to release queuine (q), which in turn can be salvaged and reincorporated into tRNA by the eukaryotic tRNA guanine transglycosylase (eTGT) which is comprised of the QTRT1 and QTRT2 subunits [10].
It has been demonstrated that the depletion of Q-tRNA in both mouse and human cells leads to protein misfolding, resulting in endoplasmic reticular stress and activation of the unfolded protein response [11, 12]. The levels of tetrahydrobiopterin (BH4) are decreased when human HepG2 cells are deprived of q and also in mice with an engineered deficiency of Q-tRNA (through the disruption of the TGT enzyme) [13]. BH4 is a critical cofactor in the production of numerous biogenic amine neurotransmitters, and this has led to speculation that the Q-tRNA status could be an important factor in neurological and neuropsychiatric diseases [14]. Indeed, the administration of an analog of q led to full remission of multiple sclerosis, by reducing effector immune cell proliferation and activation, in a mouse model of this disease [15]. Furthermore, the addition of synthesised q to an in vitro model of Alzheimer’s disease and Parkinson’s disease indicates that it has neuroprotective effects [16]. Recently, Q-tRNA modification was found to occur ubiquitously across all mouse tissues, but tissues with the highest levels of modification were heart, brain and skeletal muscle [17]. Perhaps the most convincing evidence of Q’s physiological importance so far has been observed in the brain. The disruption of the TGT enzyme complex in mice to prevent Q-tRNA modification leads to alterations in the structure of hippocampal pyramidal neurons, reduction in cell density and global imbalance in the speed of codon-biased translation. This appears to impact on learning and memory formation in the mouse, which is particularly pronounced in females [17].
The two commonly used techniques to measure Q-tRNA modification are, firstly, acryloylaminophenyl boronic acid (APB) gel electrophoresis and, secondly, LC–MS/MS analysis. In the presence of APB, the cis-diol group on Q slows the migration of Q-tRNA, therefore separating the Q-tRNA from other unmodified tRNA [18]. Then, the Q-tRNA band can be quantified by northern blot [19]. The APB gel technique can directly detect Q-tRNA in individual tRNAs but obviously does not detect free q or Q which are more likely to circulate in blood. LC–MS/MS is another sensitive and accurate technique for measuring the molecular modification in a large variety of biological materials including RNA [20,21,22]. First, the RNA is isolated and purified from biological samples. Then, the nucleosides and nucleotides are enzymatically hydrolysed from the RNA and individually quantified using LC–MS/MS analysis [23]. Although these methods have previously been applied for Q-tRNA modification analysis, no validated LC–MS/MS method for extracting and quantifying free q and Q in complex biological samples has yet been reported.
The aim of this study was therefore to establish an accurate and reliable LC–MS/MS method for quantifying free q and Q in blood samples and to assess the influence of blood collection and processing conditions in its measurement.
Methods
Materials
Q standard was provided by Professor Vincent Kelly (Trinity College Dublin, Ireland) and a Q stock solution (1mM in water) was provided by Professor Valerie de Crecy-Lagard (University of Florida, USA). Indomethacin, butylated hydroxytoluene (BHT), diethylenetriaminepentaacetate (DTPA), sodium carbonate, sodium hydroxide, phosphate buffered saline (PBS), ammonia, ammonium acetate and methanol were purchased from Merck (Missouri, USA). Serum, EDTA and lithium heparin blood collection tubes were purchased from BD Vacutainers (Wokingham, UK). LC–MS grade acetonitrile, methanol and formic acid (FA) were purchased from Honeywell (North Carolina, USA).
Subjects
To optimise the experimental protocol for blood collection, processing and extraction, blood samples were obtained from volunteers at the Centre for Public Health, Queen’s University Belfast. Ethical approval was not required for these method development studies, and the blood samples were anonymised and pooled.
To assess q/Q levels across a range of healthy participants, plasma samples (n = 44) were accessed from the biobank of the German study on Aging, Cognition and Dementia (AgeCoDe) [24]. The study ethical approval and study protocol are detailed in the Supplementary material.
Preparation of stabilization cocktail
A stabilization cocktail was prepared using indomethacin (20 mM in 5%NaHCO3 and then diluted to 0.2 mM with 1xPBS), BHT (5 mM in ethanol), and DTPA (10 mM in PBS, pH 7.4 adjusting with 6M NaOH firstly and then with 1M NaOH). The cocktail was added to the collection tubes before blood collection composed of per 1 mL of blood: 75 µL of indomethacin, 4 µL of BHT and 10 µL of DTPA.
Blood sample collection
For blood collection method optimisation, blood samples from 3 volunteer participants were collected using EDTA, lithium heparin and serum tubes. One set of samples was treated with stabilisation cocktail, kept at 4°C for 30 min, and then centrifuged at 2400 × g for 20 min at 4°C. Untreated EDTA and lithium heparin samples were centrifuged immediately after collection. Untreated serum samples were allowed to clot before centrifugation. The stabilization cocktail was prepared freshly on each day of collection. The three blood samples collected in the same tube were then pooled before q/Q extraction.
Selection of SPE cartridges and sample purification
Six types of SPE cartridges, including Oasis HLB, Oasis MCX, Oasis MAX and Oasis SAX, EBVI CARB and Bond Elut PBA, were selected for purification treatment. Oasis HLB, Oasis MCX, Oasis MAX and Oasis SAX were obtained from Waters (Milford, USA). ENVI CARB was purchased from Merck (Darmstadt, Germany). Bond Elut PBA (phenylboronic acid) cartridge and 96-well plate were purchased from Agilent (Santa Clara, USA). 1 µM of q and Q was spiked in human plasma and extracted with SPE cartridges. The detailed extraction procedures can be found in Supplementary Table 1.
Preparation of calibration standards and quality controls
Queuine standard was dissolved in ultrapure water to prepare 1 mM stock solution. Phosphate-buffered saline (PBS) with 4% bovine serum albumin (BSA) was used as surrogate matrix. Both q and Q stock solutions were aliquoted and stored at − 20 ֯C. Calibration standards (1, 0.3, 0.1, 0.03, 0.01, 0.003, 0.001 and 0.0003 µM) and 3 quality controls (QCs; 0.8, 0.02 and 0.0008 µM) were prepared freshly either in ultrapure water or spiked in matrix surrogate and then extracted using a PBA cartridge on the day of analysis.
Extraction of q and Q
For spiked samples, 500 µL of surrogate matrix was mixed with 50 µL of spiking standards and 450 µL of ammonia solution (1M, pH 11.35). In case of study samples, 500 µL of blood was mixed with 500 µL of ammonia solution (1M, pH 11.35). The Bond Elut PBA 1cc column was conditioned with 1 mL of acetonitrile–water (3:7, v/v) with 1% formic acid and equilibrated with 1 mL of ammonia solution (1M, pH 11.35). The diluted samples were loaded onto the column, followed by washing with 1 mL of 5% acetonitrile in ammonia solution (1M, pH 11.35). The analytes were eluted using acetonitrile–water (3:7,v/v) with 5% methanol and 1% formic acid. The elute was then vacuum dried and reconstituted in 100 µL of water with 5% methanol. Similarly, for validation studies, 1ml PBA 96-well plates were used, and the volume of serum samples used was 200 µL and diluted using 200 µL of ammonia solution (1M, pH 11.35) before processing through the plate.
Chromatographic and mass spectrometric conditions
LC–MS/MS analysis was performed on a system consisting of an AB SCIEX ExionLC system (Foster, USA) coupled to an AB SCIEX Triple Quad 5500 + mass spectrometer (Foster, USA). The mass spectrometer was operated in ESI positive mode using multiple reaction monitoring (MRM). Chromatographic separation was achieved with a Waters XSelect HSS T3 column (100 × 4.6 mm, 3.5 μm, Milford, USA) at 45°C using a gradient of 0.1% formic acid in water (mobile phase A) and 0.1% formic acid in acetonitrile (mobile phase B) at a flow rate of 1.0 mL/min. The mobile gradient and mass spectrometric parameters were set as previously described [25]. A chromatogram for q and Q is in the supplementary (Supplementary Fig. 1).
Method validation
The established UPLC-MS/MS method was evaluated and validated for linearity, sensitivity, matrix effect, recovery and precision. Recovery and precision were evaluated by spiking q or Q into 4% BSA PBS at three different concentrations.
-
a) Linearity and sensitivity
Linearity was evaluated by the 8-point calibration curve of q and Q in the range of 0.3 nM to 1.0 µM. A linear regression model with was applied by plotting the peak areas against their corresponding concentrations. The linearity was evaluated using R2 values and further confirmed by residual analysis. The sensitivity of method was assessed by the limit of detection (LOD) and limit of quantitation (LOQ) for q and Q at signal-to-noise (S/N) ratio of 3 and 10, respectively [26].
-
b) Accuracy and precision
Five replicated sets of QCs (0.0008, 0.02 and 0.8 µM) were prepared in a surrogate matrix to determine the accuracy and precision of the assay. These experiments were carried out intraday (same day) and inter-day (after 24 h) during 5 days. The accuracy was calculated by [(the measured concentration) / (the spiked concentration)] × 100%, while the precision of the method was expressed by the coefficient of variation [(standard deviation) / mean] × 100%.
-
c) Extraction recovery, matrix effects and parallelism
Extraction recovery was measured by comparing the response of analytes added to extracted surrogate matrix (post-extraction spiked samples) and responses of analytes added to surrogate matrix before extraction (pre-extraction spiked samples).
$$\mathrm{Extraction}\;\mathrm{recovery}\%=\frac{\mathrm{peak}\;\mathrm{area}\;\mathrm{of}\;\mathrm{pre}-\mathrm{spiked}\;\mathrm{samples}}{\mathrm{peak}\;\mathrm{area}\;\mathrm{of}\;\mathrm{post}-\mathrm{spiked}\;\mathrm{samples}}\times100\%$$The assessment of on the ionization suppression/enhancement were carried out by comparing the response of analytes added to extracted surrogate matrix (post-extraction spiked samples) and the response of analytes in solvent.
$$\mathrm{Matrix}\;\mathrm{effects}=\frac{\mathrm{peak}\;\mathrm{area}\;\mathrm{of}\;\mathrm{post}-\mathrm{spiked}\;\mathrm{samples}}{\mathrm{Peak}\;\mathrm{area}\;\mathrm{in}\;\mathrm{solvent}}\times100\%$$Evaluation of parallelism is of critical importance for endogenous analytes when surrogate matrix is applied for quantification. Batter of parallelism indicates that there is no significant difference between the surrogate and the authentic matrix. Two sets of curves were prepared either in surrogate matrix (4% BSA in PBS) or in human plasma with q and Q. Both sets of curves were prepared following the same preparation procedure. After LC–MS/MS analysis, linear regression equations were generated employing a linear regression model in each matrix, plotted using Microsoft Excel®, and the relative percentage difference (RPD) was calculated.
d) Carryover
Carryover was evaluated by assaying solvent blank samples injected after the analysis of a surrogate matrix spiked with the highest QC concentration.
Application of the method to clinical human plasma samples
The validated method was applied to quantify q and Q in bio-banked human EDTA plasma from healthy volunteers. Mann–Whitney test was applied to determine if the levels of q and Q are significantly different between male and female participants.
Results
Selection of SPE cartridge
Six SPE cartridges, Oasis HLB, Oasis MCX, Oasis WCX and Oasis SAX, EBVI CARB and Bond Elut PBA, were selected to investigate the extract efficiency of spiked q and Q from human plasma (Fig. 1). The results showed that the Bond Elut PBA cartridge had the best extraction ability with the recovery of 82% for q and 71% for Q, followed by the Oasis MCX cartridge with the recovery of 44.5% for q and 54% for Q.

Recovery of q and Q extracted by different SPE cartridges
Selection of blood collection tubes and inclusion of stabilization cocktail
Three blood collection tubes with or without stabilization cocktail were applied to evaluate the interference of anticoagulant and stabilization cocktail in the LC–MS/MS measurement. It was found that the EDTA plasma collected without stabilization cocktail obtained highest average q levels, while the serum with/without stabilization cocktail showed lowest experimental variability during the q/Q extraction procedure (Fig. 2). Adding stabilization cocktail did not improve the quantifiable levels of q. Thus, it is unnecessary to add stabilization cocktail during blood collection. Serum samples are the first preference for measurement for q in future studies, and the next preference would be EDTA plasma without any stabilization cocktail.

Measured concentration of q in human blood collected using different tubes with/without stabilization cocktail
Validation of LC–MS/MS methods
-
a)
Linearity, LOD and LOQ.
The calibration curves were generated at 8 concentration levels. It was shown that the linearity was good in the range from 0.3 nM to 1 µM. The mean regression coefficients (r2) for q is 0.997 ± 0.002 (n = 5) and for Q is 0.998 ± 0.003 (n = 5). The residual plot for all calibration curves are in the Supplementary Fig. 2. The residues randomly distributed in a horizontal band centred on the concentration axis, which indicated a good fitting of the linear model. The LOD and LOQ of the developed method were 0.1 nM and 0.3 nM, respectively, for both q and Q.
-
b)
Accuracy and precision.
The intra- and inter-assay accuracies and precisions for surrogate matrix are shown in Table 1. For q, the intra-day assay accuracy of the QCs in surrogate matrix ranged from 100.39 to 119.48% with %CV less than 12.24% across all levels. The inter-assay accuracy of QCs in surrogate matrix ranged from 104.28 to 113.41% with %CV less than 9.38% across all levels. For Q, the inter-assay accuracy of surrogate QCs ranged from 103.91 to 125.71% with %CV less than 15.68% across all levels. The inter-assay accuracy of surrogate QCs ranged from 104.89 to 124.67% with %CV less than 14.69%.
-
c)
Matrix effects, extraction recovery and parallelism in surrogate matrix.
For both q and Q, the matrix effect at the low QCs exceeded 200%, which indicated the ionization enhancement by the coeluting undetected matrix components for the low QC (Table 2). The recovery was assessed to evaluate the extraction efficiency of analytes. For q, the extraction recoveries are all higher than 70%. However, for Q, the recoveries are lower than 50%, which indicates loss of Q in the solid phase extraction (Table 2).
Parallelism was performed to evaluate the equivalence of the surrogate matrix (4%BSA in PBS) to the authentic matrix (human plasma) for q and Q (Fig. 3). Both sets of curves were prepared following the same preparation procedure. The RPD of the slopes between human plasma and surrogate matrix curves for q and Q was 6.61% and 4.89%, respectively. Currently, there is no regulatory guidance on acceptance criteria for parallelism between the authentic and matrix calibration curves, most published guidance suggests that if the slopes from the two sets of curves with the RPD are below 15%, the curves are considered to be adequately parallel [27, 28].
-
d)
Carry-over
No deviation from noise was observed following a highest QC injection. Carryover was thus considered negligible in the optimised method. QC levels during the method validation were not affected by injection order.

Parallelism of q (left) and Q (right) quantitation in surrogate matrix (4% BSA in PBS) and authentic matrix (human plasma) at their respective curve ranges
Application of the method to clinical human plasma samples
No significant difference in q levels between female (mean = 8.0 nM, min = 0.10 nM, max = 26.45 nM and IQR = 9.17 nM) and male (mean = 6.8 nM, min = 0.01 nM, max = 15.89 nM and IQR = 8.18 nM) participants was detected (p = 0.50) (Fig. 4). Q was not detected in human plasma.

Plasma q levels in healthy male and female subjects
Discussion
LC–MS/MS-based methods have previously been used to quantify Q-tRNA modification [20]. LC–MS/MS has also be developed for measuring as many as 65 other free nucleosides and nucleotides [29], but unfortunately not yet for q nor Q. To our knowledge, there is one study that has analysed q in human plasma with HPLC–MS/MS [16]. However, this report did not describe any validation for the method, something that is considered essential for reliable and accurate quantification of clinical samples. Thus, given this limitation and q/Q’s emerging importance in nutrition, physiology and pathology, we present an optimised and validated method which has considered various blood processing approaches, analyte extraction efficiency and quantification of q and Q. Firstly, the influence of applying stabilization cocktail in the blood collection were evaluated. The stabilization cocktail can prevent the formation of ex-vivo prostaglandins which may interfere with the measurement of q and Q using LC–MS/MS. This study found that the addition of a stabilization cocktail does not improve the detected levels of q in either plasma or serum, and therefore, stabilization cocktail is not necessarily required for sample collection. Either serum or EDTA plasma samples (collected without stabilization cocktail) could be used for measurement of free q in human blood. Both serum and plasma were used for measurement of nucleosides and nucleotides in previous research [29,30,31,32]. Serum, however, would be preferential for measuring q/Q because the serum samples showed lower variability in the SPE extraction and LC–MS/MS quantification. For extraction, the use of a PBA cartridge is a necessity because it is capable of extracting q at recovery rates greater than 80% in biological samples. The PBA sorbent retains the diol-containing analytes via reversible covalent bonds [33], which can ensure that the nucleic acid successfully binds to the PBA SPE cartridge under alkaline conditions (ammonia solution, pH 11.35), and subsequently, this can be eluted by an acidic mobile phase (acetonitrile–water (3:7,v/v) with 5% methanol and 1% formic acid). It should be noted that the extraction recovery for q and Q using the 96-well PBA plate is 10% and 30% lower, respectively, than that using individual PBA cartridges. This is likely due to the smaller amount of sorbent, but clearly, there are throughput vs recovery trade-offs to be considered.
The described LC–MS/MS method employed here has been successfully applied by our research group to quantify preQ1, Q-5’MP, Q-3’MP, q and Q in the intracellular extracts and culture medium [10]. However, human blood is a more complex matrix, containing high levels of phospholipids, with implications for ion suppression or enhancement in LC–MS/MS analyses [34]. Furthermore, q is an endogenous and ubiquitous analyte, for which, and the lack of analyte-free (blank) matrix is a challenge for quantification. The approach using PBS with an additive of 4% BSA to mimic the blank matrix is commonly used to address this challenge [35]. However, the surrogate matrix has a potential limitation in that does not contain any anticoagulant (as would be present in actual samples), which could potentially alter the responses of analytes. Another potential limitation is the lack of commercially available isotope-labelled q or Q which could be applied as an internal standard to compensate for the matrix effect, since the matrix effect should affect the relative ionization efficiency similarly for both the analyte and its isotope-labelled internal standard. The synthesis of q and Q is reportedly challenging [36, 37] but the development of commercial isotope-labelled internal standards for these micronutrients would be recommended to progress this field, particularly given the recent guidance of the FDA on this matter [38]. In such circumstances the surrogate matrix and biological matrix could be compared. Nonetheless, the validation results here demonstrate this method is accurate and reliable using a surrogate matrix without isotope-labelled standards. The accuracy for Q exceeds the FDA acceptance criteria which should be within ± 20% for LLOQ.
To demonstrate the application of this validated method samples from 44 healthy participants were analysed. It was found that q levels in plasma tended to be higher in female subjects than male, but the difference was not statistically significant. The one previously published study found mean plasma q levels to be significantly higher in female volunteers than in males [16]. A key observation of the present study was that Q was not detectable in the human plasma samples. It could be the case that q is the major circulating isoform of this micronutrient. It is also possible that all Q metabolites present in food are digested and absorbed as q and/or that these are processed by the liver, which might only facilitate the entry of q into the blood circulation. It could also be the case that both q and Q enter the blood circulation, but that cells and organs have a stronger absorbative preference for Q. Rapid Q absorption could make it less detectable in blood. Further studies are required to understand the metabolism and pharmacokinetics of q/Q, and the method outlined here will be a key enabler for this. Furthermore, with this validated method, either free q/Q levels or q/Q levels in blood samples it is now possible to evaluate the various influences of diet, microbiome and disease. It would be interesting to assess the levels of Q in cell-free RNA and tRNA fragments in blood. An obvious next step will be to assess circulating levels of the micronutrient in cases of neurodegenerative disease, such as AD, and to assess whether they differ from healthy control subjects.
In conclusion, this method has fulfilled the requirements for a fully validated quantification method for q and Q in biological samples using a surrogate matrix. It was sufficiently validated in terms of accuracy, precision, linearity, recovery, matrix effect and parallelism following the recent FDA guidance. This assay was successfully applied to human plasma samples to support further studies for understanding the physiological and pathophysiological roles of q and Q.
References
Cappannini A, Ray A, Purta E, Mukherjee S, Boccaletto P, Moafinejad SN, Lechner A, Barchet C, Klaholz BP, Stefaniak F, Bujnicki JM (2023) MODOMICS: a database of RNA modifications and related information. 2023 update. Nucleic Acids Research. https://doi.org/10.1093/nar/gkad1083.
Pan T. Modifications and functional genomics of human transfer RNA. Cell Res. 2018;28(4):395–404. https://doi.org/10.1038/s41422-018-0013-y.
Jackman JE, Alfonzo JD. Transfer RNA modifications: nature’s combinatorial chemistry playground. Wiley Interdisciplinary Reviews-Rna. 2013;4(1):35–48. https://doi.org/10.1002/wrna.1144.
Suzuki T. The expanding world of tRNA modifications and their disease relevance. Nat Rev Mol Cell Biol. 2021;22(6):375–92. https://doi.org/10.1038/s41580-021-00342-0.
Fergus C, Barnes D, Alqasem MA, Kelly VP. The queuine micronutrient: charting a course from microbe to man. Nutrients. 2015;7(4):2897–929. https://doi.org/10.3390/nu7042897.
Katze JR, Basile B, McCloskey JA. Queuine, a modified base incorporated post-transcriptionally into eukaryotic transfer RNA: wide distribution in nature. Science. 1982;216(4541):55–6.
Boland C, Hayes P, Santa-Maria I, Nishimura S, Kelly VP. Queuosine formation in eukaryotic tRNA occurs via a mitochondria-localized heteromeric transglycosylase. J Biol Chem. 2009;284(27):18218–27. https://doi.org/10.1074/jbc.M109.002477.
de Crécy-Lagard V, Jaroch M. Functions of bacterial tRNA modifications: from ubiquity to diversity. Trends Microbiol. 2021;29(1):41–53. https://doi.org/10.1016/j.tim.2020.06.010.
Quaiyum S, Yuan Y, Kuipers PJ, Martinelli M, Jaroch M, de Crécy-Lagard V. Deciphering the diversity in bacterial transporters that salvage queuosine precursors. Epigenomes. 2024;8(2):16.
Hung SH, Elliott GI, Ramkumar TR, Burtnyak L, McGrenaghan CJ, Alkuzweny S, Quaiyum S, Iwata-Reuyl D, Pan XB, Green BD, Kelly VP, De Crécy-Lagard V, Swairjo MA. Structural basis of Qng1-mediated salvage of the micronutrient queuine from queuosine-5′-monophosphate as the biological substrate. Nucleic Acids Res. 2023;51(2):935–51. https://doi.org/10.1093/nar/gkac1231.
Suzuki T, Yashiro Y, Kikuchi I, Ishigami Y, Saito H, Matsuzawa I, Okada S, Mito M, Iwasaki S, Ma D, Zhao XW, Asano K, Lin H, Kirino Y, Sakaguchi Y, Suzuki T (2020) Complete chemical structures of human mitochondrial tRNAs. Nature Communications 11 (1). https://doi.org/10.1038/s41467-020-18068-6.
Tuorto F, Legrand C, Cirzi C, Federico G, Liebers R, Müller M, Ehrenhofer-Murray AE, Dittmar G, Gröne HJ, Lyko F (2018) Queuosine-modified tRNAs confer nutritional control of protein translation. Embo Journal 37 (18). https://doi.org/10.15252/embj.201899777.
Rakovich T, Boland C, Bernstein I, Chikwana VM, Iwata-Reuyl D, Kelly VP. Queuosine deficiency in eukaryotes compromises tyrosine production through increased tetrahydrobiopterin oxidation. J Biol Chem. 2011;286(22):19354–63. https://doi.org/10.1074/jbc.M111.219576.
Skolnick SD, Greig NH. Microbes and monoamines: potential neuropsychiatric consequences of dysbiosis. Trends Neurosci. 2019;42(3):151–63. https://doi.org/10.1016/j.tins.2018.12.005.
Varghese S, Cotter M, Chevot F, Fergus C, Cunningham C, Mills KH, Connon SJ, Southern JM, Kelly VP. In vivo modification of tRNA with an artificial nucleobase leads to full disease remission in an animal model of multiple sclerosis. Nucleic Acids Res. 2017;45(4):2029–39. https://doi.org/10.1093/nar/gkw847.
Richard P, Kozlowski L, Guillorit H, Garnier P, McKnight NC, Danchin A, Manière X (2021) Queuine, a bacterial-derived hypermodified nucleobase, shows protection in in vitro models of neurodegeneration. Plos One 16 (8). https://doi.org/10.1371/journal.pone.0253216.
Cirzi C, Dyckow J, Legrand C, Schott J, Guo W, Hernandez DP, Hisaoka M, Parlato R, Pitzer C, van der Hoeven F, Dittmar G, Helm M, Stoecklin G, Schirmer L, Lyko F, Tuorto F (2023) Queuosine-tRNA promotes sex-dependent learning and memory formation by maintaining codon-biased translation elongation speed. Embo J. https://doi.org/10.15252/embj.2022112507.
Cirzi C, Tuorto F (2021) Analysis of queuosine tRNA modification using APB northern blot assay. In: McMahon M (ed) RNA Modifications: Methods and Protocols. Springer US, New York, NY, pp 217–230. https://doi.org/10.1007/978-1-0716-1374-0_14.
Zhang W, Xu RY, Matuszek Z, Cai Z, Pan T. Detection and quantification of glycosylated queuosine modified tRNAs by acid denaturing and APB gels. RNA. 2020;26(9):1291–8. https://doi.org/10.1261/rna.075556.120.
Costa A, de Barros JPP, Keith G, Baranowski WB, Desgrès J. Determination of queuosine derivatives by reverse-phase liquid chromatography for the hypomodification study of Q-bearing tRNAs from various mammal liver cells. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;801(2):237–47. https://doi.org/10.1016/j.jchromb.2003.11.022.
Su D, Chan CTY, Gu C, Lim KS, Chionh YH, McBee ME, Russell BS, Babu IR, Begley TJ, Dedon PC. Quantitative analysis of ribonucleoside modifications in tRNA by HPLC-coupled mass spectrometry. Nat Protoc. 2014;9(4):828–41. https://doi.org/10.1038/nprot.2014.047.
Thüring K, Schmid K, Keller P, Helm M. Analysis of RNA modifications by liquid chromatography-tandem mass spectrometry. Methods. 2016;107:48–56. https://doi.org/10.1016/j.ymeth.2016.03.019.
Ammann G, Berg M, Dalwigk JF, Kaiser SM. Pitfalls in RNA modification quantification using nucleoside mass spectrometry. Acc Chem Res. 2023;56(22):3121–31. https://doi.org/10.1021/acs.accounts.3c00402.
Ramirez A, Wolfsgruber S, Lange C, Kaduszkiewicz H, Weyerer S, Werle J, Pentzek M, Fuchs A, Riedel-Heller SG, Luck T, Mösch E, Bickel H, Wiese B, Prokein J, König HH, Brettschneider C, Breteler MM, Maier W, Jessen F, Scherer M, AgeCoDe Study G (2015) Elevated HbA1c is associated with increased risk of incident dementia in primary care patients. J Alzheimers Dis 44 (4):1203-1212. https://doi.org/10.3233/jad-141521.
Dixit S, Kessler AC, Henderson J, Pan XB, Zhao RX, D’Almeida GS, Kulkarni S, Rubio MAT, Hegedusova E, Ross RL, Limbach PA, Green BD, Paris Z, Alfonzo JD. Dynamic queuosine changes in tRNA couple nutrient levels to codon choice in Trypanosoma brucei. Nucleic Acids Res. 2021;49(22):12986–99. https://doi.org/10.1093/nar/gkab1204.
Konieczka P (2012) 2.31 - Validation and regulatory issues for sample preparation. In: Pawliszyn J (ed) Comprehensive sampling and sample preparation. Academic Press, Oxford, pp 699–711. https://doi.org/10.1016/B978-0-12-381373-2.00064-8.
Agrawal K, Shi YF, Del Rosario AM, Jian WY. Method development workflow for quantifying protein biomarkers by hybrid LC-MS/MS. Bioanalysis. 2022;14(14):985–1004. https://doi.org/10.4155/bio-2022-0058.
Yin F, Ling YH, Martin J, Narayanaswamy R, McIntosh L, Li FM, Liu GW (2021) Quantitation of uridine and L-dihydroorotic acid in human plasma by LC-MS/MS using a surrogate matrix approach. J Pharm Biomed Anal 192. https://doi.org/10.1016/j.jpba.2020.113669.
He LQ, Wei XL, Ma XP, Yin XM, Song M, Donninger H, Yaddanapud K, McClain CJ, Zhang X. Simultaneous quantification of nucleosides and nucleotides from biological samples. J Am Soc Mass Spectrom. 2019;30(6):987–1000. https://doi.org/10.1007/s13361-019-02140-7.
Djukovic D, Baniasadi HR, Kc R, Hammoud Z, Raftery D. Targeted serum metabolite profiling of nucleosides in esophageal adenocarcinoma. Rapid Commun Mass Spectrom. 2010;24(20):3057–62. https://doi.org/10.1002/rcm.4739.
Cichna M, Daxecker H, Raab M. Determination of 18 nucleobases, nucleosides and nucleotides in human peripheral blood mononuclear cells by isocratic solvent-generated ion-pair chromatography. Anal Chim Acta. 2003;481(2):245–53. https://doi.org/10.1016/s0003-2670(03)00081-3.
Amalric A, Bastide A, Attina A, Choquet A, Vialaret J, Lehmann S, David A, Hirtz C. Quantifying RNA modifications by mass spectrometry: a novel source of biomarkers in oncology. Crit Rev Clin Lab Sci. 2022;59(1):1–18. https://doi.org/10.1080/10408363.2021.1958743.
Struck W, Siluk D, Yumba-Mpanga A, Markuszewski M, Kaliszan R, Markuszewski MJ. Liquid chromatography tandem mass spectrometry study of urinary nucleosides as potential cancer markers. J Chromatogr A. 2013;1283:122–31. https://doi.org/10.1016/j.chroma.2013.01.111.
King R, Bonfiglio R, Fernandez-Metzler C, Miller-Stein C, Olah T. Mechanistic investigation of ionization suppression in electrospray ionization. J Am Soc Mass Spectrom. 2000;11(11):942–50. https://doi.org/10.1016/s1044-0305(00)00163-x.
Jones BR, Schultz GA, Eckstein JA, Ackermann BL. Surrogate matrix and surrogate analyte approaches for definitive quantitation of endogenous biomolecules. Bioanalysis. 2012;4(19):2343–56. https://doi.org/10.4155/bio.12.200.
Ohgi T, Kondo T, Goto T. Total synthesis of optically pure nucleoside Q -determination of absolute configuration of natural nucleoside Q. J Am Chem Soc. 1979;101(13):3629–33. https://doi.org/10.1021/ja00507a032.
Akimoto H, Imamiya E, Hitaka T, Nomura H, Nishimura S. Synthesis of queuine, the base of naturally-occurring hypermodified nucleoside (queuosine), and its analogs. Journal of the Chemical Society-Perkin Transactions. 1988;1(7):1637–44. https://doi.org/10.1039/p19880001637.
US Food and Drug Administration. M10 bioanalytical method validation and study sample analysis–guidance for industry. 2022.
Funding
This work was funded by Health and Social Care in Northern Ireland (HSCNI) [STL/5460/18] in support of an international research consortium through the US-Ireland programme. SC was supported by the CITI-GENS doctoral training programme, which was funded by the European Union’s Horizon 2020 research and innovation programme under a Marie Skłodowska-Curie Action [No: 945231].
Author information
Authors and Affiliations
Contributions
XP, SC, BG and JW conceived and designed the studies. XP and SC performed laboratory studies and collected and analysed the data. The initial manuscript draft was written by XP. SR, MS, WM and AR obtained, collected and supplied clinical samples. BG and JW obtained research funding and managed and supervised the project. All authors commented on manuscript drafts and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
The original study protocol was approved by the local ethics committees at the following German institutions: University of Bonn, University of Hamburg, University of Düsseldorf, University of Heidelberg/Mannheim and University of Leipzig and the Technical University of Munich. Written informed consent was obtained from all participants.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pan, X., Chandrasekaran, S., Woodside, J.V. et al. Development, validation and application of an LC–MS/MS method quantifying free forms of the micronutrients queuine and queuosine in human plasma using a surrogate matrix approach. Anal Bioanal Chem (2024). https://doi.org/10.1007/s00216-024-05489-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00216-024-05489-1