
Abstract
Lipids are biomolecules with a broad variety of chemical structures, which renders them essential not only for various biological functions but also interestingly for biotechnological applications. Rhamnolipids are microbial glycolipids with surface-active properties and are widely used biosurfactants. They are composed of one or two l-rhamnoses and up to three hydroxy fatty acids. Their biosynthetic precursors are 3-hydroxy(alkanoyloxy)alkanoic acids (HAAs). The latter are also present in cell supernatants as complex mixtures and are extensively studied for their potential to replace synthetically derived surfactants. The carbon chain lengths of HAAs determine their physical properties, such as their abilities to foam and emulsify, and their critical micelle concentration. Despite growing biotechnological interest, methods for structural elucidation are limited and often rely on hydrolysis and analysis of free hydroxy fatty acids losing the connectivity information. Therefore, a high-performance liquid chromatography-mass spectrometry method was developed for comprehensive structural characterization of intact HAAs. Information is provided on chain length and number of double bonds in each hydroxy fatty acid and their linkage by tandem mass spectrometry (MS/MS). Post-column photochemical derivatization by online Paternὸ–Büchi reaction and MS/MS fragmentation experiments generated diagnostic fragments allowing structural characterization down to the double bond position level. Furthermore, the presented experiments demonstrate a powerful approach for structure elucidation of complex lipids by tailored fragmentation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
3-(3-Hydroxyalkanoyloxy)alkanoic acids (HAAs) are known as the hydrophobic building blocks of the surface-active rhamnolipids (RLs), which are glycolipids with a hydrophilic part composed of one to two glycosidic-linked rhamnose moieties. Both the free HAAs and the glycolipids are promising alternatives to the petroleum-derived surfactants, with the latter being reported to suffer from a low biodegradation and high aquatic toxicity [1,2,3]. With the striving for higher environmental awareness, the microbial-derived biosurfactants are increasingly gaining attention, since they originate from renewable resources and are biodegradable [1, 4]. The glycolipid product derived from the pathogenic Pseudomonas aeruginosa was first described by Jarvis and Johnson in 1949 [5]. Although many other bacterial species were claimed as RL producers in recent years, P. aeruginosa is still regarded as the primary RL-producing microorganism [6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21].
Besides as esterified residue to the rhamnose moiety, HAAs also appear in their free form in RL extracts. Furthermore, studies explain the presence by their role as precursor for the synthesis of RLs rather than as a result of a degradation process [22]. The HAA moiety is composed of two esterified β-hydroxy fatty acids [6, 23, 24]. Moreover, with respect to variations in the chain length and the additional presence of double bonds as well as the combinatorial variety of the two β-hydroxy fatty acid chains, they present the predominate factor for the congeners’ diversity. In general, the length of hydrocarbon chains is ranging from 8 to 14 carbon atoms, with different predominant lengths depending on the RL-producing organism [6, 10, 24, 25]. Besides that, the chain can contain one to two double bonds [26, 27]. In order to vary the corresponding congeners’ properties, novel studies focus on the production of new structures divergent from the naturally synthesized, intending RLs as well the mere HAA moiety as biosurfactants [28, 29].
Nevertheless, the elucidation on double-bond level remains challenging, and corresponding reports on the pinpointing of double-bond positions in HAAs are rather limited. Furthermore, the rare studies are predominantly performed by means of nuclear magnetic resonance spectroscopy (NMR) or are GC–MS-based and thereby require preceding hydrolysis and derivatization steps. For instance, Řezanka et al. demonstrated a GC–MS method by utilizing the reaction agent dimethyl disulfide after hydrolysis of the ester bond and derivatization to the corresponding fatty acid methyl ester [26, 27]. However, these alternatives for the NMR-based approach suffer from complex preparation steps. Moreover, necessary hydrolysis steps are accompanied by the loss of structural information, especially concerning the linkage of corresponding HAA moieties.
Nevertheless, in the field of lipidomics, the overall awareness for the necessity of the structural elucidation of lipids down to the double-bond positional level is rapidly growing. This holds true especially for complex lipids like phospholipids and glycolipids. Conventional tandem mass spectrometry (MS/MS) using low-energy collision-induced dissociation (CID) is mostly insufficient for double-bond localization. Nevertheless, respective fragmentation patterns are often based on charge-directed fragmentation, yielding information on lipid class, number of carbon atoms, and degree of unsaturation of the bound fatty acid residues. As a result, a growing number of mass spectrometric approaches is reported in the literature for the challenging task of pinpointing double-bond positions in complex lipids. Reported methods are based on most varied approaches, such as charge-remote fragmentation (CRF) [30,31,32,33,34,35,36,37] and radical-induced fragmentation mechanisms [38,39,40,41,42,43,44,45,46] as well as ultraviolet photodissociation (UVPD) [47, 48]. One of the most straightforward approaches, based on an ozone-induced fragmentation (OzID), was implemented by Blanksby and co-workers [49, 50]. Nonetheless, OzID-based methods require a constant supply of high ozone concentrations to the mass spectrometer, which demands its modification.
A further promising and increasingly applied mass spectrometric approach was implemented by Ma et al. in 2014, focusing on photochemical in-source functionalization [51]. The underlying name reaction, the so-called Paternὸ–Büchi (PB) reaction, is based on the UV light activation of a carbonyl group, such as acetone. As a result, the molecule with the excited carbonyl group reacts with an olefin, forming an oxetane ring, and a mixture of two site isomers per double bond is generated [52]. Furthermore, successive mass spectrometric low-energy collision-induced dissociation (CID) experiments result for each derivatized double bond in a pair of diagnostic fragments with a characteristic mass difference of 26 Da (cf. Fig. 1). Thus, these diagnostic fragments enable a precise localization of double-bond positions in lipids.

PB reaction and fragmentation scheme for the reaction of acetone with a carbon–carbon double bond (exemplified by an indicated lipid structure). Diagnostic fragments are generated with a mass difference of 26 Da
Since 2014, a number of related approaches was presented and successfully applied to a variety of biological samples [53,54,55,56,57,58]. Moreover, modifications of the initially introduced setup were reported, tackling particular drawbacks of this approach. For example, Wäldchen et al. combined the PB reaction by ultraviolet photodissociation (UVPD), striving for an even higher selectivity in terms of the fragmentation via altered dissociation process [59]. Based on a different approach, Esch et al. demonstrated the effect of charge switching, via derivatization with acetylpyridine, on the conversion to the targeted diagnostic fragments. Thus, by means of the directed cleavage in the positive ionization mode, improvements for the fragmentation pattern were reported compared to the negative ionization mode [60]. Besides that, further investigations concerning the variation of the carbonyl compound were reported with respect to targeted higher quantum yields or the suitability for tissue-based studies via matrix-assisted laser dissociation/ionization (MALDI) [61, 62].
Nonetheless, despite the diversity of the methods presented, the PB approach has primarily been applied to direct-infusion experiments. However, complex lipid mixtures lead to poor detection sensitivities, especially originating from the increased sample’s complexity after UV light exposure resulting in ion suppression effects [51, 57]. Furthermore, the shotgun approach suffers from limited structural differentiation of isomers. To tackle the latter issue, occurring from isomeric/isobaric compounds, Franklin et al. [63] demonstrated a method introduced as charge inversion, which was combined by the PB approach. As a result, the differentiation of phosphatidylcholines and phosphatidylethanolamines was attained, attributed to the differences in the reaction ability between a primary amine in the last-mentioned phospholipid class and a quaternary amine in the former phospholipid class. Nevertheless, for addressing the number of mentioned drawbacks, especially with respect to the known challenges of complex biological samples, hyphenation with LC is advantageous. Accordingly, only recently, we implemented an online approach utilizing a post-column derivatization (LC-PB-MS) [64]. Furthermore, subsequently, a second online approach was presented by Zhang et al. [65], focused on a comparable hyphenation by means of preceding HILIC separation, confirming the necessity of the chromatographic aspect. Hence, the combined approaches are increasingly recognized as essential for the improved elucidation of lipids’ double-bond positions as well as additional structural features and have to be explored further.
Subsequent to the successful hyphenation, in this work, the succeeding conversion to the essential targeted diagnostic fragments was examined more closely, since it displays a further decisive key role for the unambiguous allocation of double-bond positions, especially with respect to structurally more complex lipids. Aiming at spectra of high informative value, including abundant diagnostic fragments, the influence of the precursor ion was investigated. Hence, the effect of competing fragmentation mechanisms is discussed with respect to the two major reaction processes known as charge-driven fragmentation (CDF) and charge-remote fragmentation. Corresponding experiments are exemplified by means of the introduced structurally challenging and biotechnologically relevant HAAs.
Materials and methods
Chemicals and nomenclature
The HAA sample production strain Escherichia coli C43(DE3) carrying the rhlA gene from Pseudomonas fluorescens LMG05825 was constructed previously [29]. Furthermore, a fraction of the main congener HAA 22:1 was obtained by means of preparative reversed-phase-HPLC.
Water was purified by a Milli-Q® Academic-System (18.2 MΩ cm; 0.22 μm filter; Millipore, Molsheim, France). Lithium acetate (anhydrous, ≥ 99% for analysis) and hydrochloric acid as well as the solvents isopropanol, methanol, ACN (HPLC gradient grade), and diethyl ether (≥ 99.7%) were purchased from VWR International (Darmstadt, Germany). Acetone (HPLC grade), methyl tert-butyl ether (MtBE), boron trifluoride (BF3, 14% in methanol), sodium thiosulfate (≥ 99.5%), and dimethyl disulfide (≥ 99.0%) were obtained from Sigma-Aldrich (Steinheim, Germany). Ammonium acetate (≥ 99.99%) and acetic acid (≥ 99.99%) were delivered by Fluka GmbH (Darmstadt, Germany). Formic acid (99–100% p.a.) was purchased from Th. Greyer GmbH & Co. KG (Renningen, Germany). Chloroform (≥ 99.8%) and n-hexane (GC–MS grade) were obtained from Merck KGaA (Darmstadt, Germany). Sodium hydroxide (≥ 98%) and iodine were purchased from Honeywell International Inc. (Charlotte, NC, USA).
The lipids’ nomenclature was performed according to the standardized shorthand notation for lipid structures implemented by Liebisch et al. [66]. Furthermore, localized double-bond positions are indicated according to Δ nomenclature.
Sample preparation
Purification of the aqueous supernatant from E. coli was carried out as described by Behrens et al. [67] to reveal the examined HAA sample. The purified HAA 22:1 sample was diluted hundredfold in ACN:H2O (50:50). Moreover, the photochemical direct-infusion experiments, based on the latter, were conducted by a sample diluted in acetone:H2O (70:30).
Fractionation of HAAs by preparative HPLC
For the chromatographic separation, a preparative HPLC system consisting of the AZURA pump P6.1L and the AZURA autosampler 3950 (both Knauer GmbH, Berlin, Germany) connected to the SEDEX 85 LT-ELSD detector (SEDERE Olivet, France) and the fraction collector Foxy R1 (Teledyne ISCO Lincoln, USA) using a VP250/21 NUCLEODUR C18 HTec column (Macherey-Nagel GmbH & Co. KG, Düren, Germany) was employed. The flow rate was set to 10 mL/min and 3 mL sample were injected for every run. As eluent, ACN and water supplied with 0.2% (v/v) formic acid were used. The ACN concentration was linearly increased from 70% to 80% between 5 min and 35 min, and to 100% until 37 min. It was decreased back to 70% from 42 min to 52 min. The measurement was terminated after 60 min. The HAA 22:1 congener was collected between 45 min and 48 min. The ELSD detector was operated with compressed air at 3.5 bar and 40 °C. Further settings were the following: signal offset (mV), − 005; gain, 1; output signal value after AZ, + 000 mV; filter, 1S; and stray light, 100%.
Hydrolysis and derivatization for GC–MS
For the confirmation of the HPLC–MS/MS results via gas chromatography–mass spectrometry (GC–MS), an aliquot of the preparative HPLC fraction was blown to dryness under a gentle stream of nitrogen and hydrolyzed with a 0.5 M NaOH in MeOH-H2O solution (9:1 v/v, 2 mL, 70 °C, 1 h). The solution was then acidified to pH 3 with 1 M HCl, and fatty acids were extracted with chloroform (3 × 3 mL). Afterwards, the solvent was removed and fatty acid methyl esters (FAMEs) were prepared by adding 100 μL of BF3 (14% in MeOH) and heating (75 °C, 1 h). Then, 2 mL H2O was added and the FAMEs extracted with chloroform (3 × 2 mL). After removal of the chloroform with a gentle stream of nitrogen, the alkylthiolation of the double bond was carried out according to the method of Shibahara et al. [68]. Therefore, 200 μL of an iodine/dimethyl disulfide solution (13 mg/mL) was added and heated (35 °C, 30 min). Then, the iodine was reduced with an aqueous Na2S2O4 solution (5% w/v) and 0.4 mL n-hexane/diethyl ether (1:1 v/v) was added. The organic phase was extracted, blown to dryness under a gentle stream of nitrogen, re-dissolved in 200 μL n-hexane, and used for GC–MS analysis.
Instrumentation
Preliminary HPLC–MS/MS investigation
Preliminary investigations, regarding the analysis of HAA samples, were performed by means of an Agilent 1200 series HPLC system (Agilent Technologies, Santa Clara, CA, USA) coupled with a Thermo Finnigan LTQ ion trap mass spectrometer (Thermo Scientific, Bremen, Germany). The instrument was operated in the negative ionization mode using a heated electrospray source (HESI II) for ionization. Data acquisition was performed in full scan MS and MSn scan mode using an isolation width of 2 Da. For a detailed description of experimental parameters of the mass spectrometric detection and the liquid chromatographic separation using an Accucore C18 column, please refer to the Electronic Supplementary Material (ESM) chapter “Preliminary HPLC-MS/MS investigation.”
Furthermore, direct-infusion experiments in the negative ionization mode, with respect to the HAA 22:1 sample, were recorded by means of the Q Exactive plus Orbitrap mass spectrometer (Thermo Fischer Scientific, Waltham, MA, USA) equipped with an electrospray ionization (ESI) probe. A syringe provided a constant flow rate of 10 μL/min. Data acquisition was performed in full-scan MS and MS/MS scan mode using an isolation width of 0.5 Da. The selected precursor ions were fragmented using higher-energy C-trap dissociation (HCD). For a detailed description of experimental parameters, please refer to the ESM chapter “Preliminary HPLC-MS/MS investigation.”
Photochemical activation
The online PB reaction was performed according to Jeck et al. [64] (cf. ESM Fig. S1) by means of post-column derivatization and a utilized micro-flow reactor, consisting of a deactivated fused-silica capillary and a low-pressure mercury lamp (primary emission at 254 nm, model 80-1057-01; BHK, Ontario, Canada). Prior to the reaction, the eluent flow was combined with a constant acetone flow. The latter was provided by an external syringe and adjusted to the same flow rate as the former, achieving a one-to-one dilution. The lithium-based experiments were conducted by means of an additional supply of diluted lithium acetate (0.7 mM, water-based; flow rate, 4 μL/min). With respect to a minimized system contamination, the addition was performed directly before the injection into the ionization device.
Supplementary investigations via direct-infusion were carried out at optimized reaction conditions and flow rates set to 10 μL/min. The LTQ was operated in the negative ionization mode using ESI. Data acquisition was performed in full-scan MS and MSn scan mode using an isolation width of 2 Da. A detailed description of MS conditions can be found in the ESM chapter “Photochemical activation.”
Hyphenation with capillary HPLC
Measurements based on capillary HPLC were performed by an Agilent 1200 series capillary pump (Agilent Technologies, Waldbronn, Germany). The chromatographic separations were conducted using an Ascentis® Express C18 column (150 × 0.5 mm, 2.7 μm, Supelco®, Bellefonte, USA), and the column oven was operated at 40 °C. For experiments conducted in the positive ionization mode, by means of lithiated compounds, the eluent flow was combined by a constant flow (2 μL/min, for analysis without acetone addition) of an aqueous lithium acetate solution (0.7 mM) directly prior to the introduction into the ionization device.
Analysis of the E. coli extract was performed by reversed-phase capillary chromatography coupled to high-resolution MS. For a detailed description of LC–MS conditions, please refer to the ESM chapter “Hyphenation with capillary HPLC.”
Precursors for higher-energy C-trap dissociation (HCD) by the Q Exactive plus mass spectrometer were dynamically selected by means of an inclusion list, which was generated by MZmine 2.34 [69, 70] and contained a database of potential HAA species and their corresponding PB products, in a range of 16 to 32 carbon atoms and 0 to 2 double bonds in both acyl chains combined.
Confirmation of double-bond position by GC–MS
For GC–MS analysis, a GCMS-QP-2020 (Shimadzu, Kyoto, Japan) equipped with a Nexis GC-2030 gas chromatograph (Shimadzu, Kyoto, Japan) was used. Samples (1 μL injection volume by AOC-20i Plus autosampler) were separated on a 30-m, 0.25-mm-i.d., 0.25-μm film thickness DB-5MS column (J&W Scientific, Folsom, CA, USA). Mass spectra were obtained by electron ionization (EI, 70 eV) in the mass range m/z 50–500. Further experimental details are compiled in the ESM in chapter “GC-MS method.”
Results and discussion
First investigations were based on the examination of chromatographic as well as fragmentation behavior of HAAs. These examinations were carried out based on a cell supernatant derived from E. coli, as no commercial authentic standard of HAAs is available. The initial investigations were conducted in the negative ionization mode, with regard to the HAAs’ proton donor properties, attributed to their free carboxyl group. However, the MS/MS experiments suffered from disadvantageous fragmentation processes driven by their structural characteristic β-hydroxy group. Therefore, further attempts were focused rather on complementary fragmentation processes by means of the so-called CRF. A successful approach by use of alkaline metal precursor ions, in this case lithiated HAAs, and an online hyphenation with the PB reaction is presented in the following.
Chromatographic behavior and fragmentation patterns
Since HAAs mainly differ in the length of their hydrocarbon chain, their chromatography was conducted with regard to their differing hydrophobicity. Accordingly, a RP chromatography on a C18 phase was performed. In order to achieve an optimal separation, a gradient elution was applied by a mobile phase composed of ACN as organic phase and an acidified aqueous phase (5% ACN and 0.1% formic acid). As a result, a baseline separation of congeners with respect to their hydrocarbon chain length as well as their degree of unsaturation was attained. While longer chains resulted in stronger interaction with the stationary phase and longer retention times, the presence of double bonds leads to the opposite effect (cf. ESM Fig. S2). The determined sample composition was a mixture ranging from HAA 22:X to HAA 26:X, varying between no and one double bond (i.e., X = 0 or X = 1). Moreover, the chromatography revealed the presence of isomeric compounds by displaying additional baseline separated peaks with the same m/z or prominent peak shoulders.
For gaining information on the HFA level, fragmentation experiments were conducted. The characteristic fragmentation pattern obtained in the negative ionization mode is exemplified by HAA 22:1 and displayed in Fig. 2 and ESM Fig. S3 for the proposed two isomers, eluting after 8.3 and 9.1 min, respectively.

LC–MS/MS spectrum in the negative electrospray ionization mode of HAA 22:1 with precursor m/z 383 ([M−H]−) and retention time 8.83–9.49 min. Mass spectra of HAA 22:1 (corresponding to HAA 12:1/10:0). Displayed is the MS/MS (a) and subsequent MS3 spectrum of HFA 12:1 (b), with indicated diagnostic fragment for the double bond in position Δ 5 (cf. Fig. 3b)
The obtained MS/MS spectra displayed in Fig. 2 and in ESM Fig. S3 show different fragments and can thus be allocated to different HFA combinations. For example, the later eluting and abundant peak indicates fragments that can be allocated to HFA 10:0 and HFA 12:1. According to Lépine et al. [22], the fragments solely derive from the cleaved terminal HFAs at the hydroxyl end. Hence, the fragments can be attributed to two different isomers HAA C10:0/C12:1 vs. HAA C12:1/C10:0 with the terminal HFA interchanged (cf. Fig. 3a, indicating the fragmentation).

Fragmentation pattern of examined HAA 22:1 (corresponding to HAA 12:1/10:0), conducted in the negative ionization mode ([M−H]−) (a). Subsequent obtained fragmentation pattern of HFA 12:1, with indicated diagnostic fragment m/z 87.0 (b). Proposed charge-remote allylic fragmentation (R1 and R2 representing the carboxyl group and the terminal end of the hydrocarbon chain, respectively) (c)
The earlier eluting low abundant peak, therefore, can be allocated to two isomeric compounds composed of HFA 8:0 and HFA 14:1 (HAA C8:0/C14:1 vs. HAA C14:1/C8:0). Accordingly, the HAAs with the shorter saturated HFA chain (HFA 8:0) have shorter retention times than those with the larger saturated HFA chain (HFA 10:0) due to reduced hydrophobic interaction with the stationary phase.
Interestingly, a subsequent fragmentation of the generated HFA product ions revealed further distinctive structural information. The fragmentation of [HFA 12:1-H]− resulted, on the one hand, in diagnostic cleavages at the polar carboxylic side that point to the hydroxylation in the β position (m/z 167.1 and 153.0; cf. Fig. 2b). Corresponding proposed product ion structures are consistent with previous CID fragmentation experiments on HFAs investigated by Marshall et al. [71]. On the other hand, an additional low abundant fragment at m/z 86.9 was obtained. The fragment is assumed to result from a doubly activated cleavage, which is attributed to a double bond located in position Δ 5 (see Fig. 3b). More precisely, the carbon–carbon bond cleavage is facilitated by the bond’s beneficial position adjacent to the hydroxy group and allylic to the double bond. The particular dissociation is assumed to involve a charge-remote allylic fragmentation as suggested for comparable experiments demonstrated in the literature [72, 73]. In comparison, the equivalent spectra generated from [HFA 12:0-H]− (cf. ESM Fig. S4) merely display mentioned cleavages attributed to the β-hydroxy group. Moreover, fragmentation experiments that involved different unsaturated HFAs with divergent hydrocarbon chain length suffered from the same lack of diagnostic fragments. Both experiments, however, confirm the distinctive diagnostic fragment originating from the double-bond position Δ 5. Moreover, the fragmentation patterns of the further examined HFAs, reversely, indicate a divergent double-bond position. For verification, subsequent investigations by means of HRMS and direct-infusion setup were conducted, which confirmed the assumption via accurate masses (e.g., m/z 87.0088, C3H3O3−, Δ m/z = 0.4 ppm).
Capillary flow rate and PB-LC-MS
PB direct- infusion
In order to establish a method that pinpoints double bonds, regardless of their particular position in HAAs, further investigations were based on the application of a LC–PB–MS hyphenation, introduced by Jeck et al. [64]. However, prior to the hyphenation experiments, pretests were conducted via direct-infusion experiments and micro-flow reaction. Therefore, a purified and enriched fraction of HAA 22:1 was used which has been obtained by preparative HPLC (see section “Materials and methods”). The resulting mass spectra of HAA 22:1, recorded in the negative ionization mode, are displayed in Fig. 4.

Spectra received from derivatized HAA 22:1, obtained via direct-infusion and micro-flow reaction, conducted in the negative ionization mode ([M−H]−). Displayed is the MS/MS (a) and subsequent MS3 spectrum of the derivatized HFA 12:1 (b) as well as the derivatized HFA 10:0 (c). Indicated are the acetone losses (58 Da) and fragments received for a dominant side reaction (SR fragm.; cf. Fig. 5)
According to previous experiments, acetone addition to the targeted HAA was observed as a response to the applied UV light irradiation. However, the MS/MS spectrum indicates an acetone addition to both HFA moieties (m/z 245.2 and 271.2), regardless of the existence of a double bond. Furthermore, no diagnostic fragments resulting from the derivatized HFA 12:1 were obtained (cf. Fig. 4c). Especially the former aspect suggests a competing addition reaction, which is independent of the presence of a double bond. One of the main known side reactions, occurring in the course of the PB reaction, is the hydrogen abstraction at the allylic position of the double bond. The reaction is a result of the labile hydrogen atom and the relatively stable radical intermediate [52, 74]. HAAs, however, possess a further labile hydrogen atom (α position), which is attributed to its hydroxylation in the β position. Furthermore, a hydrogen abstraction by the excited acetone produces the well-known 2-hydroxy-2-propyl radical [75, 76]. A subsequent radical recombination, lastly, forms the suggested side product (see Fig. 5a). Interestingly, further evidence for the proposed conversion is given by the obtained MS3 spectra. Alongside the fragments indicating an acetone loss, two further characteristic fragments at m/z 98.9 and 116.9 were obtained, which can be explained by a cleavage between the α and β position (see Fig. 4b, c). Furthermore, the fragmentation of the derivatized double bond appears to be disadvantageous compared to further obtained unspecific fragmentation processes. Hence, no diagnostic fragments were observed for pinpointing the position. Based on this, further attempts were focused on a complementary fragmentation approach directed by lithiated precursor ions. Since alkaline metals are known to promote CRFs, an improvement of the fragmentation pattern was expected. In accordance with that, the conducted experiments resulted in a significant reduction of unspecific fragments originating from a CDF mechanism. Moreover, abundant signals for the essential diagnostic fragments were obtained, which will be discussed in detail below.

Postulated side reaction, attributed to a hydrogen abstraction in the α position by an excited acetone, the latter yielding a 2-hydroxy-2-propyl radical [75, 76], and a subsequent radical recombination. Displayed is the suggested reaction scheme, with R corresponding to the attached HFA 10:0 residue (a) as well as the obtained characteristic fragments (b)
LC–PB–MS
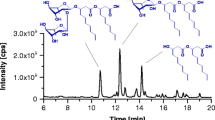
With regard to the online hyphenation by means of the LC–PB–MS approach, the initial chromatographic separation was transferred to capillary flow rates. The HPLC flow rate was set to 13 μL/min and combined, directly prior to the introduction to the ESI probe, by an additional flow of an aqueous lithium acetate solution (2 μL/min, for analysis without acetone addition). The applied gradient was, compared to the previous one, adjusted to a steeper increase of the organic content over time. Thus, the large dwell volume of the utilized capillary pump was compensated to a certain degree although it was accompanied by a loss of separation efficiency. Moreover, the hyphenation by the micro-flow reactor resulted in further moderate peak broadening. Nevertheless, a sufficient separation was attained, which presents a baseline separation for HAA congeners of different carbon numbers and degrees of unsaturation, which prevents interferences by 13C isotopes. In addition, isomeric structures could be differentiated by partly chromatographically separated peaks. Based on attained separation, the LC–PB–MS measurements were performed with a reaction time of 7 s. The particular correlation of peaks corresponding to unsaturated HAAs and their respective derivatization products is demonstrated in Fig. 6. The recorded characteristic MS/MS spectra, obtained for the HAAs and their PB products, are displayed in Fig. 7 and Fig. 9, exemplified by HAA 22:0 and HAA 22:1, respectively.

Obtained chromatographic separation for HAA congeners of the cell supernatant derived from E. coli, conducted by means of LC–PB–MS and recorded in the positive ionization mode ([M+Li]+). Illustrated is the correlation of peaks corresponding to unsaturated HAAs (a) and their corresponding derivatized counterparts (b)

MS/MS spectra received from HAA 22:1 (7.35–8.21 min) (a) and HAA 22:0 (9.18–10.03 min) (b), obtained via LC–PB–MS, conducted in the positive ionization mode ([M+Li]+). Displayed is the characteristic fragmentation pattern, with the indicated diagnostic fragment for the former (m/z 265.1613; cf. Fig. 8)
With respect to the educts, similarities can be observed by comparing the MS/MS spectra with the spectra conducted in the negative ionization mode. Both approaches resulted in abundant product ions that can be assigned to the terminal HFA at the hydroxyl end. However, in the case of the lithiated precursor ion, additional low-abundant fragments can be observed, e.g., attributed to a subsequent water loss. More interestingly, the fragmentation of [HAA 22:1 + Li]+ leads to the generation of an additional signal at m/z 265.1613. The product ion can be explained by a cleavage induced by the double bond in the position Δ 5. Furthermore, the fragment confirms the suggested origin of the previously discussed m/z 86.9, which was obtained in the negative ionization mode after a subsequent fragmentation of HFA 12:1. By means of the Li+ coordinated precursor ion, however, the underlying CRF is assumed to be promoted. Hence, the cleavage occurs without an additional fragmentation step and, furthermore, the preceding scission of the HFA moieties (cf. fragmentation scheme in Fig. 8).

Fragmentation pattern of examined HAA 22:1 (corresponding to HAA 12:1/10:0), conducted in the positive ionization mode ([M+Li]+), with indicated diagnostic fragment m/z 265.1622
Besides that, a significant improvement was observed regarding the fragmentation pattern of the PB products. By means of the Li+ coordination, abundant signals were obtained which can be allocated to the cleavage of the derivatized double bonds. Two sets of diagnostic fragments were generated enabling the unambiguous pinpointing of double-bond positions. The generated fragments resulted either from the sole cleavage of the oxetane ring or an additional subsequent loss of the esterified second HFA moiety. Consequently, a further confirmation of the positional information is given, as well as a distinct assignment to a particular HFA moiety. The derivatized HAA 22:1, for example, resulted in the two diagnostic pairs of fragments at m/z 309.1874 and 335.2392 as well as m/z 139.0573 and 165.1092 (cf. Fig. 9). The first pair indicated neutral losses that can be assigned to a ω 7 double bond. However, due to the second pair, a loss of a HFA 10:0 moiety is pointed out, which enabled the distinct allocation to a Δ 5 position of a HFA 12:1 moiety. Moreover, further confirmation about the structural arrangement of the two HFA moieties is provided by additional signals, which are comparable to the pattern received form the educt.

MS/MS spectrum received from derivatized HAA 22:1 (corresponding to HAA 12:1/10:0; 7.69 min) obtained via LC–PB–MS, conducted in the positive ionization mode ([M+Li]+). Displayed is the characteristic fragmentation pattern, with indicated acetone loss (58 Da) and pairs of diagnostic fragments (diagn. frag. I/II)
Noteworthily, the results for HAA 22:1 are in accordance with the previous suggested double-bond position by means of the fragments m/z 86.9 (LTQ) and 265.1613 (Q Exactive plus), received from the non-derivatized precursor ion in the negative and positive ionization mode, respectively. Interestingly, the latter fragment was also obtained by means of the lithiated derivatized precursor ion (m/z 265.1612).
According to the same pattern, the further congeners of the mixture were examined concerning their HFA composition and double-bond position. The results are listed in Table 1. With respect to the Δ nomenclature, double-bond positions ranging from Δ 5 to 9 were unambiguously discovered. However, applying the ω nomenclature, the variance of the HAAs’ double-bond position showed to be limited to the ω 7 position, which represents a known double-bond position for HAAs derived from strains such as P. aeruginosa. Only the spectrum obtained for HAA 10:1/10:0 suffered from limited informative value, preventing a localization, which is attributed to its very low abundance and, furthermore, the coelution with HAA 12:1/8:0. Nevertheless, obtained data is consistent with previous reports for HAAs derived from a related species, Pseudomonas aeruginosa [27, 77].
The approach by means of lithiated precursor ions proved to be valuable for the structural elucidation of HAAs, especially with respect to the pinpointing of double bonds. Although the structure rather suggests an ionization conducted in the negative ionization mode, for yielding the highest ionization efficiencies, the success of the LC–PB–MS approach appeared to rather depend on the precursor ion.
GC–MS results
The assignment of double-bond positions in HAAs by LC–PB–MS was confirmed by an independent method, i.e., GC–MS. Similar to Kendel et al. and Řezanka et al., we performed GC–MS after hydrolysis of the ester bond and a double derivatization strategy [26, 27, 78]. First, corresponding fatty acid methyl esters (FAMEs) were formed, and second, double bonds were reacted with dimethyl disulfide. Electron ionization (EI) of the derivatives yielded diagnostic fragments to pinpoint the double-bond position. For the derivative of HFA 12:1, the ions at m/z 145 and at m/z 177 indicate the position of the double bond as ω7/Δ5 (cf. ESM Fig. S5). Further characteristic ions are m/z 74 (McLafferty rearrangement) and m/z 103 (α cleavage next to the hydroxyl group) and confirm the position of the hydroxyl group as 3-OH.
In comparison to the presented LC–PB–MS method, pinpointing of double-bond positions by GC–MS required a hydrolysis step which is accompanied by the loss of structural information, especially concerning the linkage of corresponding HAA moieties. Therefore, the here presented method based on online PB reaction provides a more comprehensive picture of HAAs’ structure.
Conclusions
Subsequent to the successful hyphenation of high-performance liquid chromatography (HPLC) with the online Paternò–Büchi (PB) reaction, as presented previously [64], this work focused on the just as important and challenging successive conversion to targeted diagnostic fragments. This conversion was explored by a comparison of different precursor ions with respect to the known major reaction processes, classified as charge-driven fragmentation (CDF) and charge-remote fragmentation (CRF) reaction [79, 80, 81]. As expected, a gain of control concerning the spectra’s information value, and thus the abundance of diagnostic fragments, was achieved. However, the investigations carried out on the structurally challenging lipid class of HAAs demonstrated the limitations of the approach performed by deprotonated precursor ions. Furthermore, it presented the possibilities of lithium-coordinated precursor ions. Initial investigations were examined in the negative ionization mode, with respect to the HAAs’ acidic and, thus, proton donor properties. However, the fragmentation experiments suffered from disadvantageous cleavages originating from the structural characteristic β-hydroxy group. Consequently, diagnostic fragments were neither generated by means of single nor multiple fragmentation experiments. Interestingly, fragmentation experiments based on the mere underivatized HAA 22:1 resulted, in comparison, in a distinct product ion induced by the double bond in the Δ 5 position. However, this particular fragmentation and thus the elucidation on the double-bond positional level was solely observed for the specific position. The specific carbon–carbon bond cleavage is assumed to be doubly activated by the bond’s beneficial position adjacent to the hydroxy group and allylic to the double bond. Nevertheless, subsequent experiments were conducted by means of lithiated precursor ions, leading to spectra of higher informative value, including abundant diagnostic fragments. Based on this, the analysis of the cell supernatant of a recombinant Escherichia coli strain carrying the gene for the acyltransferase RhlA from P. fluorescens LMG05825 was successfully conducted by means of LC–PB–MS, with respect to the HAAs’ congener composition and its detailed structural elucidation. Furthermore, the proven gain of control concerning the spectra’s informational value, demonstrated by HAAs as model compounds, presents a powerful tool and can equally be applied to the elucidation of further complex lipid classes.
References
Van Bogaert INA, Zhang J, Soetaert W. Microbial synthesis of sophorolipids. Process Biochem. 2011;46:821–33.
Mann RM, Bidwell JR. The acute toxicity of agricultural surfactants to the tadpoles of four Australian and two exotic frogs. Environ Pollut. 2001;114:195–205.
Mann RM, Boddy MR. Biodegradation of a nonylphenol ethoxylate by the autochthonous microflora in lake water with observations on the influence of light. Chemosphere. 2000;41:1361–9.
Claus S, Van Bogaert INA. Sophorolipid production by yeasts: a critical review of the literature and suggestions for future research. Appl Microbiol Biotechnol. 2017;101:7811–21.
Jarvis FG, Johnson MJ. A glyco-lipide produced by Pseudomonas aeruginosa. J Am Chem Soc. 1949;71:4124–6.
Abdel-Mawgoud AM, Lepine F, Deziel E. Rhamnolipids: diversity of structures, microbial origins and roles. Appl Microbiol Biotechnol. 2010;86:1323–36.
Chong H, Li Q. Microbial production of rhamnolipids: opportunities, challenges and strategies. Microb Cell Factories. 2017;16:137.
Priji P, Sajith S, Unni KN, Anderson RC, Benjamin S. Pseudomonas sp. BUP6, a novel isolate from Malabari goat produces an efficient rhamnolipid type biosurfactant. J Basic Microbiol. 2017;57:21–33.
Rooney AP, Price NPJ, Ray KJ, Kuo TM. Isolation and characterization of rhamnolipid-producing bacterial strains from a biodiesel facility. FEMS Microbiol Lett. 2009;295:82–7.
Dubeau D, Déziel E, Woods DE, Lépine F. Burkholderia thailandensis harbors two identical rhl gene clusters responsible for the biosynthesis of rhamnolipids. BMC Microbiol. 2009;9:263.
Christova N, Tuleva B, Lalchev Z, Jordanova A, Jordanov B. Rhamnolipid biosurfactants produced by Renibacterium salmoninarum 27BN during growth on n-hexadecane. Z Naturforsch C J Biosci. 2004;59:70–4.
Tavares LFD, Silva PM, Junqueira M, Mariano DCO, Nogueira FCS, Domont GB, et al. Characterization of rhamnolipids produced by wild-type and engineered Burkholderia kururiensis. Appl Microbiol Biotechnol. 2013;97:1909–21.
Häußler S, Nimtz M, Domke T, Wray V, Steinmetz I. Purification and characterization of a cytotoxic exolipid of Burkholderia pseudomallei. Infect Immun. 1998;66:1588–93.
Wittgens A, Tiso T, Arndt TT, Wenk P, Hemmerich J, Müller C, et al. Growth independent rhamnolipid production from glucose using the non-pathogenic Pseudomonas putida KT2440. Microb Cell Factories. 2011;10:80.
El-Amine Bendaha M, Mebrek S, Naimi M, Tifrit A, Belaouni H, Abbouni B. Isolation and comparison of rhamnolipids production in Pseudomonas aeruginosa P.B:2 and Pseudomonas fluorescens P.V:10. Sci Rep. 2012;1:544.
Nalini S, Parthasarathi R. Production and characterization of rhamnolipids produced by Serratia rubidaea SNAU02 under solid-state fermentation and its application as biocontrol agent. Bioresour Technol. 2014;173:231–8.
Nordin N, Zakaria MR, Halmi MIE, Ariff A, Zawawi RM, Wasoh H. Isolation and screening of high efficiency biosurfactant-producing Pseudomonas sp. J Biochem Microbiol Biotechnol. 2013;1:25–31.
Gunther NW IV, Nuñez A, Fett W, Solaiman DKY. Production of rhamnolipids by Pseudomonas chlororaphis, a nonpathogenic bacterium. Appl Environ Microbiol. 2005;71:2288–93.
Vasileva-Tonkova E, Galabova D, Stoimenova E, Lalchev Z. Production and properties of biosurfactants from a newly isolated Pseudomonas fluorescens HW-6 growing on hexadecane. Z Naturforsch C J Biosci. 2006;61:553–9.
Tuleva BK, Ivanov GR, Christova NE. Biosurfactant production by a new Pseudomonas putida strain. Z Naturforsch C J Biosci. 2002;57:356–60.
Martínez-Toledo A, Ríos-Leal R, Vázquez-Duhalt R, M del C G-C, Esparza-García JF, Rodríguez-Vázquez R. Role of phenanthrene in rhamnolipid production by P. putida in different media. Environ Technol. 2006;27:137–42.
Lépine F, Déziel E, Milot S, Villemur R. Liquid chromatographic/mass spectrometric detection of the 3-(3-hydroxyalkanoyloxy)alkanoic acid precursors of rhamnolipids in Pseudomonas aeruginosa cultures. J Mass Spectrom. 2002;37:41–6.
Hauser G, Karnovsky ML. Rhamnose and rhamnolipide biosynthesis by Pseudomonas aeruginosa. J Biol Chem. 1957;224:91–105.
Déziel E, Lépine F, Dennie D, Boismenu D, Mamer OA, Villemur R. Liquid chromatography/mass spectrometry analysis of mixtures of rhamnolipids produced by Pseudomonas aeruginosa strain 57RP grown on mannitol or naphthalene. Biochim Biophys Acta Mol Cell Biol Lipids. 1999;1440:244–52.
Manso Pajarron A, de Koster CG, Heerma W, Schmidt M, Haverkamp J. Structure identification of natural rhamnolipid mixtures by fast atom bombardment tandem mass spectrometry. Glycoconj J. 1993;10:219–26.
Řezanka T, Siristova L, Sigler K. Rhamnolipid-producing thermophilic bacteria of species Thermus and Meiothermus. Extremophiles. 2011;15:697–709.
Rendell NB, Taylor GW, Somerville M, Todd H, Wilson R, Cole PJ. Characterisation of Pseudomonas rhamnolipids. Biochim Biophys Acta. 1990;1045:189–93.
Tiso T, Zauter R, Tulke H, Leuchtle B, Li WJ, Behrens B, et al. Designer rhamnolipids by reduction of congener diversity: production and characterization. Microb Cell Factories. 2017;16:225.
Germer A, Tiso T, Müller C, Behrens B, Vosse C, Scholz K, et al. Exploiting the natural diversity of RhlA acyltransferases for the synthesis of the rhamnolipid precursor 3-(3-hydroxyalkanoyloxy)alkanoic acid. Appl Environ Microbiol. 2020;86.
Jensen NJ, Tomer KB, Gross ML. Collisional activation decomposition mass spectra for locating double bonds in polyunsaturated fatty acids. Anal Chem. 1985;57:2018–21.
Tomer KB, Crow FW, Gross ML. Location of double bond position in unsaturated fatty acids by negative ion MS/MS. J Am Chem Soc. 1983;105:5487–8.
Cheng C, Gross ML, Pittenauer E. Complete structural elucidation of triacylglycerols by tandem sector mass spectrometry. Anal Chem. 1998;70:4417–26.
Pittenauer E, Allmaier G. High-energy collision induced dissociation of biomolecules: MALDI-TOF/ RTOF mass spectrometry in comparison to tandem sector mass spectrometry. Comb Chem High Throughput Screen. 2009;12:137–55.
Pittenauer E, Allmaier G. The renaissance of high-energy CID for structural elucidation of complex lipids: MALDI-TOF/RTOF-MS of alkali cationized triacylglycerols. J Am Soc Mass Spectrom. 2009;20:1037–47.
Hsu FF, Turk J. Structural characterization of triacylglycerols as lithiated adducts by electrospray ionization mass spectrometry using low-energy collisionally activated dissociation on a triple stage quadrupole instrument. J Am Soc Mass Spectrom. 1999;10:587–99.
Hsu FF, Turk J. Structural characterization of unsaturated glycerophospholipids by multiple-stage linear ion-trap mass spectrometry with electrospray ionization. J Am Soc Mass Spectrom. 2008;19:1681–91.
Hsu FF, Turk J. Electrospray ionization multiple-stage linear ion-trap mass spectrometry for structural elucidation of triacylglycerols: assignment of fatty acyl groups on the glycerol backbone and location of double bonds. J Am Soc Mass Spectrom. 2010;21:657–69.
Deimler RE, Sander M, Jackson GP. Radical-induced fragmentation of phospholipid cations using metastable atom-activated dissociation mass spectrometry (MAD-MS). Int J Mass Spectrom. 2015;390:178–86.
Li P, Hoffmann WD, Jackson GP. Multistage mass spectrometry of phospholipids using collision-induced dissociation (CID) and metastable atom-activated dissociation (MAD). Int J Mass Spectrom. 2016;403:1–7.
Campbell JL, Baba T. Near-complete structural characterization of phosphatidylcholines using electron impact excitation of ions from organics. Anal Chem. 2015;87:5837–45.
Baba T, Campbell JL, Le Blanc JCY, Baker PRS. In-depth sphingomyelin characterization using electron impact excitation of ions from organics and mass spectrometry. J Lipid Res. 2016;57:858–67.
Baba T, Campbell JL, Le Blanc JCY, Baker PRS. Structural identification of triacylglycerol isomers using electron impact excitation of ions from organics (EIEIO). J Lipid Res. 2016;57:2015–27.
Baba T, Campbell JL, Le Blanc JCY, Baker PRS. Distinguishing cis and trans isomers in intact complex lipids using electron impact excitation of ions from organics mass spectrometry. Anal Chem. 2017;89:7307–15.
Jones JW, Thompson CJ, Carter CL, Kane MA. Electron-induced dissociation (EID) for structure characterization of glycerophosphatidylcholine: determination of double-bond positions and localization of acyl chains. J Mass Spectrom. 2015;50:1327–39.
Pham HT, Ly T, Trevitt AJ, Mitchell TW, Blanksby SJ. Differentiation of complex lipid isomers by radical-directed dissociation mass spectrometry. Anal Chem. 2012;84:7525–32.
Ellis SR, Pham HT, In het Panhuis M, Trevitt AJ, Mitchell TW, Blanksby SJ. Radical generation from the gas-phase activation of ionized lipid ozonides. J Am Soc Mass Spectrom. 2017;28:1345–58.
Klein DR, Brodbelt JS. Structural characterization of phosphatidylcholines using 193 nm ultraviolet photodissociation mass spectrometry. Anal Chem. 2017;89:1516–22.
Ryan E, Nguyen CQN, Shiea C, Reid GE. Detailed structural characterization of sphingolipids via 193 nm ultraviolet photodissociation and ultra high resolution tandem mass spectrometry. J Am Soc Mass Spectrom. 2017;28:1406–19.
Thomas MC, Mitchell TW, Blanksby SJ. Ozonolysis of phospholipid double bonds during electrospray ionization: a new tool for structure determination. J Am Chem Soc. 2006;128:58–9.
Thomas MC, Mitchell TW, Harman DG, Deeley JM, Murphy RC, Blanksby SJ. Elucidation of double bond position in unsaturated lipids by ozone electrospray ionization mass spectrometry (OzESI-MS). Anal Chem. 2007;79:5013–22.
Ma X, Xia Y. Pinpointing double bonds in lipids by Paternò-Büchi reactions and mass spectrometry. Angew Chem. 2014;53:2592–6.
Fréneau M, Hoffmann N. The Paternò-Büchi reaction—mechanisms and application to organic synthesis. J Photochem Photobiol C: Photochem Rev. 2017;33:83–108.
Ma X, Zhao X, Li J, Zhang W, Cheng JX, Ouyang Z, et al. Photochemical tagging for quantitation of unsaturated fatty acids by mass spectrometry. Anal Chem. 2016;88:8931–5.
Zhao X, Zhao Y, Zhang L, Ma X, Zhang S, Zhang X. Rapid analysis of unsaturated fatty acids on paper-based analytical devices via online epoxidation and ambient mass spectrometry. Anal Chem. 2018;90:2070–8.
Ren J, Franklin ET, Xia Y. Uncovering structural diversity of unsaturated fatty acyls in cholesteryl esters via photochemical reaction and tandem mass spectrometry. J Am Soc Mass Spectrom. 2017;765:1–4.
Ma X, Chong L, Tian R, Shi R, Hu TY, Ouyang Z, et al. Identification and quantitation of lipid C=C location isomers: a shotgun lipidomics approach enabled by photochemical reaction. Proc Natl Acad Sci. 2016;113:2573–8.
Stinson CA, Xia Y. A method of coupling the Paternò–Büchi reaction with direct infusion ESI-MS/MS for locating the C=C bond in glycerophospholipids. Analyst. 2016;141:3696–704.
Murphy RC, Okuno T, Johnson CA, Barkley RM. Determination of double bond positions in polyunsaturated fatty acids using the photochemical Paternò-Büchi reaction with acetone and tandem mass spectrometry. Anal Chem. 2017;89:8545–53.
Wäldchen F, Becher S, Esch P, Kompauer M, Heiles S. Selective phosphatidylcholine double bond fragmentation and localisation using Paternò–Büchi reactions and ultraviolet photodissociation. Analyst. 2017;142:4744–55.
Esch P, Heiles S. Charging and charge switching of unsaturated lipids and apolar compounds using Paternò-Büchi reactions. J Am Soc Mass Spectrom. 2018;29:1971–80.
Xu T, Pi Z, Song F, Liu S, Liu Z. Benzophenone used as the photochemical reagent for pinpointing C=C locations in unsaturated lipids through shotgun and liquid chromatography-mass spectrometry approaches. Anal Chim Acta. 2018;1028:32–44.
Bednařík A, Bölsker S, Soltwisch J, Dreisewerd K. An on-tissue Paternò–Büchi reaction for localization of carbon–carbon double bonds in phospholipids and glycolipids by matrix-assisted laser-desorption–ionization mass-spectrometry imaging. Angew Chem Int Ed. 2018;57:12092–6.
Franklin ET, Betancourt SK, Randolph CE, McLuckey SA, Xia Y. In-depth structural characterization of phospholipids by pairing solution photochemical reaction with charge inversion ion/ion chemistry. Anal Bioanal Chem. 2019;411:4739–49.
Jeck V, Korf A, Vosse C, Hayen H. Localization of double-bond positions in lipids by tandem mass spectrometry succeeding high-performance liquid chromatography with post-column derivatization. Rapid Commun Mass Spectrom. 2019;33:86–94.
Zhang W, Zhang D, Chen Q, Wu J, Ouyang Z, Xia Y. Online photochemical derivatization enables comprehensive mass spectrometric analysis of unsaturated phospholipid isomers. Nat Commun. 2019;10.
Liebisch G, Vizcaíno JA, Köfeler H, Trötzmüller M, Griffiths WJ, Schmitz G, et al. Shorthand notation for lipid structures derived from mass spectrometry. J Lipid Res. 2013;54:1523–30.
Engelen J, Behrens B, Hayen H, Tiso T, Blank LM. Characterization of rhamnolipids by liquid chromatography/mass spectrometry after solid-phase extraction. Anal Bioanal Chem. 2016;408:2505–14.
Shibahara A, Yamamoto K, Kinoshita A, Anderson BL. An improved method for preparing dimethyl disulfide adducts for GC/MS analysis. J Am Oil Chem Soc. 2008;85:93–4.
Pluskal T, Castillo S, Villar-Briones A, Orešič M. MZmine 2: modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinf. 2010;11:395.
Korf A, Jeck V, Schmid R, Helmer PO, Hayen H. Lipid species annotation at double bond position level with custom databases by extension of the MZmine 2 open-source software package. Anal Chem. 2019;91:5098–105.
Marshall DL, Saville JT, Maccarone AT, Ailuri R, Kelso MJ, Mitchell TW, et al. Determination of ester position in isomeric (O-acyl)-hydroxy fatty acids by ion trap mass spectrometry. Rapid Commun Mass Spectrom. 2016;30:2351–9.
Murphy RC. Chapter 1. Fatty acids. In: Tandem mass spectrometry of lipids: molecular analysis of complex lipids, vol. 9. Cambridge: Royal Society of Chemistry; 2014. p. 1–39.
Wheelan P, Zirrolli JA, Murphy RC. Electrospray ionization and low energy tandem mass spectrometry of polyhydroxy unsaturated fatty acids. J Am Soc Mass Spectrom. 1996;7:140–9.
Bradshaw JS. Ultraviolet irradiation of carbonyl compounds in cyclohexene and 1-hexene; 1966; Vol. 31.
Zeldes H, Livingston R. Paramagnetic resonance study of liquids during photolysis. II. Acetone and solutions containing acetone. J Chem Phys. 1966;45:1946–54.
Shiraishi Y, Tsukamoto D, Hirai T. Highly efficient methyl ketone synthesis by water-assisted C-C coupling between olefins and photoactivated acetone. Org Lett. 2008;10:3117–20.
Lebrón-Paler A Solution and Interfacial Characterization of Rhamnolipid Biosurfactatnt from P.Aeruginosa ATCC 9027, University of Arizona, 2008;
Kendel M, Barnathan G, Fleurence J, Rabesaotra V, Wielgosz-Collin G. Non-methylene interrupted and hydroxy fatty acids in polar lipids of the alga Grateloupia turuturu over the four seasons. Lipids. 2013;48:535–45.
Adams J. Charge-remote fragmentations: analytical applications and fundamental studies. Mass Spectrom Rev. 1990;9:141–86.
Gross ML. Charge-remote fragmentation: an account of research on mechanisms and applications. Int J Mass Spectrom. 2000;200:611–24.
Demarque DP, Crotti AEM, Vessecchi R, Lopes JLC, Lopes NP. Fragmentation reactions using electrospray ionization mass spectrometry: an important tool for the structural elucidation and characterization of synthetic and natural products. Nat Prod Rep. 2016;33:432–55.
Acknowledgments
The RWTH Aachen University researcher would like to acknowledge the funding from the European Union’s Horizon 2020 research and innovation program under grant agreement 863922 for the project MIX-UP and the German Research Foundation (DFG) under Germany’s excellence strategy within the cluster of excellence 2186 “FSC – The Fuel Science Center.”
Funding
Open Access funding provided by Projekt DEAL. Financial support by the Deutsche Forschungsgemeinschaft (DFG) (Grant No. HA 4319/9-1 and INST 211/802-1 FUGG) is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 522 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jeck, V., Froning, M., Tiso, T. et al. Double bond localization in unsaturated rhamnolipid precursors 3-(3-hydroxyalkanoyloxy)alkanoic acids by liquid chromatography–mass spectrometry applying online Paternò–Büchi reaction. Anal Bioanal Chem 412, 5601–5613 (2020). https://doi.org/10.1007/s00216-020-02776-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-020-02776-5