
Abstract
Hydrophilic interaction liquid chromatography (HILIC) provides an alternative approach to effectively separate small polar compounds on polar stationary phases. The purpose of this work was to review the options for the characterization of HILIC stationary phases and their applications for separations of polar compounds in complex matrices. The characteristics of the hydrophilic stationary phase may affect and in some cases limit the choices of mobile phase composition, ion strength or buffer pH value available, since mechanisms other than hydrophilic partitioning could potentially occur. Enhancing our understanding of retention behavior in HILIC increases the scope of possible applications of liquid chromatography. One interesting option may also be to use HILIC in orthogonal and/or two-dimensional separations. Bioapplications of HILIC systems are also presented.

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A theoretical description of analyte retention in high-performance liquid chromatography (HPLC) has been the subject of various publications. There are several ways to model the separation mechanism: partition, adsorption, ion exchange, and size exclusion. This mechanism is based on specific and nonspecific interactions [1–4]. Nevertheless, there is currently no detailed quantitative retention model that would allow the chromatographic parameters for individual analytes separated under given conditions to be accurately predicted.
Normal or reversed-phase liquid chromatography can be used for analysis. In normal phase liquid chromatography (NP-LC), the stationary phase is more polar than the mobile phase. The retention increases as the polarity of the mobile phase decreases, and thus polar analytes are more strongly retained than nonpolar ones. The opposite situation occurs in reversed-phase liquid chromatography (RP-LC) [5]. NP-LC has been widely used to separate various compounds, from nonpolar to highly polar compounds (note that chromatography was first introduced as a method used in separation science). Although RP-LC systems were previously commonly used by scientists, NP-LC methods are currently undergoing a renaissance.
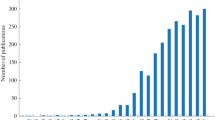
Hydrophilic interaction liquid chromatography (HILIC) is an alternative high-performance liquid chromatography (HPLC) mode for separating polar compounds. For historical reasons, it has been reported that HILIC is a variant of normal phase liquid chromatography, but the separation mechanism used in HILIC is more complicated than that in NP-LC. While the acronym HILIC was first suggested by Alpert in 1990 [6], the number of publications on HILIC has increased substantially since 2003 (Fig. 1), as outlined in the well-constructed review by Hemström and Irgum [7].

SciFinder Scholar search results documenting the continuously growing research area of HILIC, based on the numbers of publications per year
Like NP-LC, HILIC employs traditional polar stationary phases such as silica, amino or cyano [8–12], but the mobile phase used is similar to those employed in the RP-LC mode [7, 11, 12]. HILIC also allows the analysis of charged substances, as in ion chromatography (IC). Figure 2 shows how HILIC complements other areas of chromatography and extends the range of separation options.

HILIC combines the characteristics of the three major methods in liquid chromatography
HILIC has many specific advantages over conventional NP-LC and RP-LC. For example, it is suitable for analyzing compounds in complex systems that always elute near the void in reserved-phase chromatography. Polar samples always show good solubility in the aqueous mobile phase used in HILIC, which overcomes the drawbacks of the poor solubility often encountered in NP-LC. Expensive ion pair reagents are not required in HILIC, and it can be conveniently coupled to mass spectrometry (MS), especially in the electrospray ionization (ESI) mode. In contrast to RP-LC, gradient elution HILIC begins with a low-polarity organic solvent and elutes polar analytes by increasing the polar aqueous content [13]. A desirable mobile phase would contain high organic content for better sensitivity and also show good on-column retention for polar ionic compounds. Hydrophilic interaction liquid chromatography has established itself as the separation mode of choice for uncharged highly hydrophilic and amphiphilic compounds that are too polar to be well retained in RP-LC but have insufficient charge to allow effective electrostatic retention in ion-exchange chromatography. HILIC separation is currently attracting a lot of interest since it solves many previously difficult separation problems, such as the separation of small organic acids, basic drugs, and many other neutral and charged substances. It has been successfully applied to the analysis of carbohydrates [14, 15], peptides [8, 16, 17] and polar pharmaceuticals [11, 18], etc.
This paper covers fundamental developments in hydrophilic interaction liquid chromatography. The objective of the present work is to review options for the characterization of HILIC stationary phases and their applications to separations of polar compounds in complex matrices. Gaining a thorough understanding of retention behavior in HILIC enhances the scope of applications of liquid chromatography. The separation mechanism can depend on many factors, such as the physicochemical properties of the stationary phase and hydroorganic mobile phase, and the structures of the samples investigated. Precisely defining which mechanism prevails is currently a difficult and complicated task. This phenomenon is still waiting for theoretical elucidation.
Stationary phases for the HILIC mode
Any polar chromatographic surface can be used for HILIC separations. Typical HILIC stationary phases consist of classical bare silica or silica gels modified with many polar functional groups. Polymer-based stationary phases can also be used.
The first generation of HILIC mode separations started in 1975. Linden et al. [19] separated carbohydrates by an amino-silica phase, Bondapak (Waters, Milford, MA, USA) in a mixture of acetonotrile and water (75:25 v/v). The next generation of stationary phases for HILIC used DIOL- and amide-silica. The DIOL-silica column has mainly been used for the separation of proteins [20, 21]. According to Tosoh, producer of TSKgel Amide-80, amide-silica columns have been available since at least 1985. This particular phase is described as consisting of nonionic carbamoyl groups that are chemically bonded to the silica gel, but it is commonly known as an amide-bonded silica. After Yoshida [22] applied these phases to the separation of peptides, the amide-silica phase soon found common usage in HILIC. Chemically bonded stationary phases with specific structural properties have been prepared by Buszewski et al. [23–25]. One of them contains aminopropyl ligands bonded to silica (SG–NH2); others are an alkylamide packing phase (SG-AP) and a mixed phase (SG-MIX) containing different types of ligands (–NH2, –CN, –Ph, –C8, –C18) bonded to the support.
Over the last 15 years, HILIC has progressed into second and third generation implementations, most of which involve mixed or multiple-interaction solid phases. Many column vendors sell both traditional HILIC and its more sophisticated relatives. Some novel separation materials for HILIC have attracted increasing attention in recent years [7, 18, 26–31]. Hence, the structural variations of HILIC-type stationary phases are wider than those found in reversed-phase systems. HILIC phases can be grouped into neutral polar or ionic surfaces. Table 1 shows the different structures of stationary phases that are applicable to HILIC-mode separation. These special separation materials for HILIC show good selectivity and reproducibility for the separation of polar compounds. Although the number of commercially available columns designed especially for HILIC is growing, there is still no versatile stationary phase like C18 in RP-LC. However, different types of separation materials for HILIC have different retention characteristics and separation selectivities.
Unmodified bare silica gel has some advantages for HILIC, in contrast to chemically bonded stationary phases. Type A silica gels, prepared by precipitation from the solutions of silicates, are acidic because they are polluted with certain metals that activate surface silanol groups and form complexes with some chelating solutes, causing strong retention or asymmetric peaks. Type B silica gels are formed by the aggregation of silica sols in air, contain very low amounts of metals, and are more stable at intermediate and higher pH values (up to at least pH 9) than xerogel-type materials. They generally provide better separations, especially for basic samples, because they are highly purified, less acidic “sol-gel” spherical silica particles [57]. At higher pH values, silanol groups are ionized and cation exchange plays a important role in retention, especially for positively charged basic compounds. Suppressing silanol ionization through the addition of TFA may promote the ion-pairing mechanism. Similar effects have also been observed in HILIC on monolithic silica gel columns, which offer higher permeability than the particle-packed HILIC columns [58]. Silica gel type C with a hydrosilated surface populated with nonpolar silicon hydride Si–H groups instead of silanol groups may have up to 95% of its original silanols removed, making it less polar than silica gels with higher populations of silanol groups [59]. It can be used to separate acids or bases in the HILIC mode in buffered mobile phases containing more than 50–70% organic solvent (acetonitrile).
DIOL, amino, amide and other bonded phases used in HILIC are usually prepared by chemically modifying the silica gel surface, like the C18 phases used for RP-LC [8, 60]. Chemically bonded DIOL phases demonstrate high polarity and hydrogen bonding properties, and do not contain ionizable groups other than unreacted residual silanols, meaning that they are appropriate for the HILIC mode [61]. Bonded amino-silica columns are relatively often used in the HILIC mode. While basic analytes are in general strongly retained on silica gel by hydrogen bonding and ion-exchange interactions with silanol groups, acidic compounds show increased affinities to amino-silica columns, which can sometimes even lead to irreversible adsorption [8]. Chemically bonded phases with other functionalities, such as polyethylene glycol or alkyls with embedded amide or carbamate groups, are generally proposed for RP applications in water-rich mobile phases. On the other hand, when the percentage of organic solvent is high, the retention of many compounds increases with increasing concentration of acetonitrile, showing typical NP behavior [6, 47, 62]. Cyclodextrin-silica stationary phases that possess several linked glucopyranoside units and have chiral recognition properties are useful for HILIC chiral separations [40].
Zwitterionic sulfoalkylbetaine stationary phases have also been introduced for HILIC separations. The active layer, which is grafted onto wide-pore silica gel or a polymer support, contains both strongly acidic sulfonic acid groups and strongly basic quaternary ammonium groups separated by a short alkyl spacer. Ion-exchange interactions of the zwitterionic stationary phase are assumed. The sulfoalkylbetaine bonded phases strongly adsorb water by hydrogen bonding, and the bulk layer of water, which forms part of the stationary phase, then largely controls the retention mechanism. Zwitterionic columns are commercially available under the tradenames ZIC-HILIC (on a silica gel support) and ZIC-pHILIC (on a polymer support) [12].
The separation of neutral compounds on ion exchangers under typical HILIC conditions has been known about for a very long time. On both cation-exchange and anion-exchange styrene-divinylbenzene resins, only the retentions of some polar compounds (e.g., carbohydrates and related substances) increase with increasing ethanol concentration in the mobile phase. For other compounds, the opposite effects have been observed [62–64]. Due to the presence of ion-exchange groups, a mixed-mode HILIC/ion-exchange mechanism controls the retention, which may cause specific selectivity effects. The mixed anion-exchange/cation-exchange/HILIC mechanism that occurs on silica-based, small-pore, weak ion-exchange resins was found to be useful for the analysis and purification of compounds from natural products [18].
Mobile phase selection
A typical mobile phase for HILIC chromatography includes water-miscible polar organic solvents such as acetonitrile with a small amount of water [6]. However, any aprotic solvent that is miscible with water (e.g., tetrahydrofuran, THF, and/or dioxane) can be used. Alcohols can also be adopted, although a higher concentration is needed to achieve the same degree of retention of the analyte relative to an aprotic solvent–water combination [7].
An eluotropic row is useful for selecting a suitable organic modifier for the mobile phase. This lists solvents according to increasing elution strength. Relative solvent strengths in HILIC can be approximately summarized as follows:
HILIC separations are performed either in isocratic mode with a high percentage of organic solvent or with gradients starting with a high percentage of organic solvent and ending with a high proportion of aqueous solvent [6].
It is commonly believed that in HILIC, the mobile phase forms a water-rich layer on the surface of the polar stationary phase vs. the water-deficient mobile phase, creating a liquid/liquid extraction system. The analyte is distributed between these two layers [6, 33].
Mobile phase additives
Ionic additives, such as ammonium acetate and ammonium formate, are typically used to control the mobile phase pH and ion strength. In HILIC, they can also contribute to the polarity of the analyte, resulting in differential changes in retention. For ionizable analytes, such as aminoglycoside antibiotics, the pH must be adjusted to ensure that the analyte will be in a single ionic form. Increasing the buffer concentration decreases the retention if ion exchange controls the retention, while the opposite effect may occur, affecting the solvation, in the absence of ion exchange under HILIC conditions. If this is not done, an asymmetric peak shape, chromatographic peak “tailing,” and/or poor recovery from the stationary phase will be observed. No buffer is needed to separate neutral polar analytes (e.g., carbohydrates). The use of other salts (such as 100–300 mM sodium perchlorate) that are soluble in high organic solvent mixtures (ca. 70% acetonitrile) can be used to increase the polarity of the mobile phase in order to achieve elution. These salts are not volatile, so this technique is less useful with a mass spectrometer as a detector [6, 65].
Separation mechanism in HILIC mode
The mechanism and theoretical description of analyte retention in HPLC has been the subject of many articles. Different research groups and scientific schools still disagree about the most realistic retention mechanism and the best theory to describe and predict it [66, 67].
There are essentially three possible ways to model the separation mechanism. The first is analyte partitioning between the mobile and stationary phases [68, 69]; the second is the adsorption of the analyte onto the surface of the adsorbent [70, 71]; the third assumes the preferential adsorption of the organic mobile phase modifier onto the adsorbent surface, followed by the partitioning of this analyte into the adsorbed layer [72]. The retention phenomenon in HPLC simultaneously depends on various types of intermolecular interactions between the solute and the stationary phase, the solute and the mobile phase, and the stationary and mobile phases. Known intermolecular interaction types are given in Table 2.
Present theory proposes that HILIC retention is caused by partitioning. This phenomenon still lacks a thorough theoretical explanation. In this mode, the separation mechanism is based on the differential distribution of the injected analyte solute molecules between the acetonitrile-rich mobile phase and a water-enriched layer adsorbed onto the hydrophilic stationary phase [6, 7] (see Fig. 3). The more hydrophilic the analyte, the more the partitioning equilibrium is shifted towards the immobilized water layer on the stationary phase, and thus, the more the analyte is retained. In other words, a separation based on the polarities of the compounds and the degree of solvation takes place. As a result of the preferential solvation of the stationary phase surface, it is very likely that the gradient of a given solvent concentration from the adsorbent surface into a bulk mobile phase is formed.

Schema of the separation mechanism in a HILIC system
When the concentration of acetonitrile (or some other organic solvent) increases, water interacts more strongly with the surface of the polar stationary phase (e.g., bare silica). In this case, acetonitrile cannot interact with residual silanols on the stationary phases, so they are uncovered and water molecules can be adsorbed onto them [74–78]. Figure 4a shows the largest excess adsorption of acetonitrile on the surface area of an HILIC adsorbent. It was found for water-rich mobile phases containing about 80% water by volume. This mirrors the preferential adsorption of acetonitrile onto siloxane bridges. The largest deficit of acetonitrile is found for a water concentration of close to 20% (v/v). This corresponds to the maximum excess adsorption of water onto the silica, and illustrates the preferential adsorption of water molecules onto free, accessible silanol groups (≡Si–OH) [79]. The water adsorption is higher when the proportion of water molecules in the mobile phase decreases [78]. Figure 4b shows the plots obtained when following the convention that total adsorption isotherms have a horizontal plateau or rather inflection point. This convention represents the minimum adsorbed amount of eluent, because the total amount adsorbed is a strictly monotonously increasing function of its bulk concentration [79].

Surface excesses of acetonitrile and water on the HILIC adsorbent (a). Total adsorbed amounts of acetonitrile and water per unit surface area of HILIC adsorbent as a function of the volume fraction of acetonitrile in the binary eluent (b). Based on data from [79]
When the acetonitrile volume fraction is between 30 and 70%, the adsorbed phase consists of at least three adsorbed layers of acetonitrile and water mixture. This agrees with previous studies that demonstrated the formation of an adsorbed multilayer of water on bare silica. The number of adsorbed monolayers of pure acetonitrile is around 2, while that of pure water is close to 3 [79].
To sum up, when the concentration of water in the mobile phase is lower than 20%, water adsorption can be multilayer in nature, and it can create an excess of adsorbed water in comparison with the concentration of water in the eluent. McCalley and Neue [78] concluded that about 4–13% of the pore volume of the silica phase is occupied by a water-enriched layer when there is 75–90% acetonitrile in the eluent. The solvent adsorption governed by these specific interactions may have a large influence on the selectivity of the separation in the discussed HILIC systems [76].
Although it is well established that a hydrophilic surface holds water when exposed to mixtures of organic solvent and water, the HILIC partitioning theory is based on only circumstantial evidence. HILIC is more than just simple partitioning, and includes hydrogen donor interactions between neutral polar species, as well as weak electrostatic mechanisms under the high organic solvent conditions used for retention. This indicates that the mechanism of HILIC is distinct from that of ion-exchange chromatography. There are studies that point towards a more multimodal separation mechanism [6, 7]. Alpert [6] considered dipole–dipole interactions, and hydrogen bonds may also contribute to partitioning into the stationary phase layer. He noted charged that basic groups in a solute lead to pronounced hydrophilicity and retention, so these interactions make important contributions to the mechanism of separation. Yoshida [16] similarly considered that HILIC retention encompassed both hydrogen bonding (which depends on Lewis acidity/basicity) and dipole–dipole interactions (dependent on dipole moments and the polarizability of molecules).
It was shown the elution pattern was similar to that in (nonaqueous) normal phase liquid chromatography, so it was proposed that the mechanism must be similar too. Hydrogen bonding, especially when using low-water mobile phases, probably also contributes to the retention mechanism in HILIC [80].
On the other hand, electrostatic interactions can play an important role in HILIC, due to either ionic groups that have been intentionally incorporated onto the stationary phase or less intentional residual charges, such as those of dissociated silanol groups [65]. These residual silanol groups on the silica surface, which cannot be removed or blocked due to steric effects of bonded ligands, are partially ionized. Residual silanols may influence the separation of polar analytes, especially basic compounds and biopolymers, because of the strong polar (hydrogen-bonding and dipole–dipole) interactions between their basic groups and the ionized residual silanols of the support. Adsorbed water molecules suppress the ionization of surface silanols, which leads to negative charging of the silica gel surface. This layer of negative charges on the silica gel surface creates a negative electric field in the contacting mobile phase. Positive charges attracted by the field create the second layer of the electric double layer (Fig. 5) [81].

Model of the electric double layer on the stationary bonded-phase surface. Based on data from [81]
The interactions of basic and acidic analytes with the stationary phase are expected to be based on both hydrophilic interactions and electrostatic forces. However, the final separation mechanism of the elution process is most probably a superposition of partitioning and electrostatic interactions or hydrogen bonding to the stationary phase [6, 7].
The extent to which each mechanism dominates is dependent on the actual type of stationary phase used and the buffer conditions, including the level and type of organic solvent, the type and concentration of salt, and the pH [47].
Increasing the concentration of water in the eluent decreases retention when—as is typical in HILIC—high acetonitrile concentrations are used (Fig. 6) [10, 57]. Increasing the admixture of an organic component in a partially aqueous mobile phase generally leads to a decrease in the relative static permittivity of the mixed mobile phase. This is indeed the case with acetonitrile, the solvent typically used in HILIC. Lower relative static permittivity is accompanied by decreased ion stabilization, and this is manifested in increased pK a values for solutes [65, 82]. Above 60% v/v acetonitrile, strong deviations start to take place due to homo- and heteroconjugation when the amount of water available to form hydrogen bonds with the buffer compounds and solutes decreases [65].

Effect of the mobile-phase aqueous buffer concentration on the HILIC retentions of the test compounds obtained using HILIC monolithic columns. Adapted from the data in [83]
The presence of buffering salts in the mobile phase can decrease electrostatic interactions through disruption [42, 65]. Additionally, the same salt-based disruption can decrease the retentions of analytes, and can be useful during elution [6]. However, in some instances, a higher salt concentration might drive the more solvated salt ions into the water-enriched layer formed on the particles, yielding an increase in the volume of the water layer and therefore an increase in retention [42, 65, 84]. A thickening of the water layer on the stationary phase through hydration takes place at the same time [57]. Another possible cause has been hypothesized: electrostatic repulsion between the stationary phase and the analytes is weakened by the higher salt concentration [85].
Another factor that influences the retention characteristics in HILIC is the pH of the buffer. Whether the buffer pH is above or below the pK a of the analyte determines its charge state, which in turn affects the hydrophilicity of the analyte and likewise the interaction with the stationary phase [7, 33]. For example, acidic solutes had low retention or showed exclusion in ammonium formate buffers, but were strongly retained when trifluoroacetic acid (TFA) buffers were used. This is possibly due to suppression of the repulsion of the solute anions from ionized silanol groups at the low pH values of TFA solutions of aqueous acetonitrile. At high buffer pH, the ionization of weak bases was suppressed, reducing ionic (and possibly hydrophilic) retention, leading to further opportunities to manipulate selectivity [57].
Charged stationary phases, such as the above mentioned anion or cation exchangers and deprotonated silanol groups, are most likely to display some degree of electrostatic interaction [7].
The effect of column temperature on HILIC separations is often rather small (considerably less than in RP-LC), but ultimately this depends on the nature of the retained molecule [86]. The retention in the RP mode decreases at increasing temperature and is principally controlled by the enthalpic contribution. However, the retention factors in the HILIC range of the mobile phase are almost independent of the temperature. In HILIC mode, the retention is probably controlled by entropic contributions, possibly originating in different levels of sample solvation in the stationary and bulk mobile phases [87].
Polar analytes show very different selectivities under HILIC and RP-LC conditions [88]. Sometimes polar compounds are well retained, and the elution order in HILIC is one of increasing hydrophilicity, which is roughly the reverse of that seen in RP-LC. For example, Olsen [10] has compared the properties of various stationary phases used for the separation of pyrimidines and purines (polar compounds of significant biological and pharmaceutical interest). Figure 7 shows changes in the selectivity of the separation of these types of polar compound on the HILIC and RP-LC columns.

Comparison of the selectivities obtained for polar pharmaceuticals using a reversed-phase ion pairing (LiChrospher RP-Select B column) and b HILIC (Zorbax NH2) separation modes. Compound numbering is the same for both chromatograms. Adapted from the data in [10]
It was demonstrated recently that the mechanism of HILIC separation involves various combinations of hydrophilic interactions, ion exchange, and reversed-phase retention by the siloxane on the silica surface, which contribute to various degrees depending on the particular conditions employed [33, 34].
Characterization and selection of HILIC separation systems
The selectivity depends not only on the stationary phase but also strongly on the mobile phase [89].
In reversed-phase systems, the effects of the volume fraction of a strong organic solvent (φ) in a binary mobile phase on the retention factor k over a limited concentration range of acetonitrile–water can be described by a widely used semi-empirical equation [90–93]:
The constant a in Eq. 1 should theoretically be equal to the logarithm of the retention factor of the solute in pure water; the constant m increases with decreasing polarity of the organic solvent and with increasing size of the sample molecule. Equation 1 was assumed to apply generally to the systems where partition or nonlocalized adsorption controls the retention, except for mobile phases with low concentrations of organic solvents. Because the retention in HILIC is proportional to the polarity of the solute and inversely proportional to the polarity of the mobile phase (opposite to the retention in RP systems), Eq. 1 with φ taken to mean the volume fraction of water rather than that of the organic solvent can be used to describe the effects of the mobile phase on the retention in HILIC systems, where the partition between the adsorbed water and the bulk mobile phase is assumed to control retention [94].
For conventional nonaqueous normal phase systems, where retention is based on surface adsorption, the following equation was found to adequately describe the retention model [95, 96]:
where φ is the volume fraction of the polar solvent in the binary organic mobile phase, k ′ 0 is the retention factor in pure polar solvent, and m is the stoichiometric coefficient characterizing the number of molecules of the strong solvent that are needed to displace one adsorbed molecule of the analyte.
The relative polarity of the mobile phase with respect to the stationary phase is a distinguishing feature that can be used to classify NP and RP systems. Hence, a dual retention mechanism, where the NP and RP effects contribute simultaneously to the retention, is a rather common phenomenon in hydrophilic interaction liquid chromatography [89].
The retention factor (k) includes the solvophobic and the polar contributions, which depend on the polarity of the solute, the chemistry of the stationary phase, and the composition of the mobile phase [94]. Plots of retention factor versus binary aqueous–organic mobile phase composition show a “U shape” [97, 98]. Minimum retention at the “U turn” composition of the mobile phase corresponds to the transition from the RP to the NP (HILIC in an aqueous–organic mobile phase) mechanism, and the retention at this point is usually very low [94, 99]. The U shape of k versus the volume fraction of an aqueous buffer φ (BUF) can be described by the following equation:
The parameters a 1, m 1 and m 2 of Eq. 3 can be determined by nonlinear regression of the experimental retention factors measured for varying volume fractions of water in the mobile phase. The parameter m 1 characterizes the effect of increasing the water concentration in the mobile phase on the contribution of the RP mechanism to the retention, whereas the parameter m 2 is a measure of the opposite effect, characteristic of the HILIC contribution to the retention in highly organic mobile phases. a 1 is an empirical constant [100].
To characterize the retention in liquid chromatography, quantitative structure–retention relationships (QSRRs) are widely used. QSRRs are the most extensively studied manifestations of linear free-energy relationships (LFERs). These are the statistically derived relationships that model the analyte retention as a function of properties (descriptors) related to the molecular structure of the analyte and the physicochemical properties of both the stationary and mobile phase [73]. Using the QSRR approach, the chromatographic column can be regarded as a “free-energy transducer,” translating differences in the chemical potentials of solutes resulting from differences in their structures into the chromatographic retention parameter [101]. The QSRR method can assist in elucidating the molecular mechanism for separation that occurs in a given chromatographic system, and can aid in the optimization of this process [102]. QSRR models can be used to characterize and compare the suitability of columns for both RP [103] and HILIC separations [104, 105]. Buszewski et al. [106] grouped analytical columns based on the retention data for peptides. They showed that different columns for RP-LC and HILIC can be clearly distinguished using the QSRR method (Fig. 8). Jandera et al. [99] investigated the retention of the antioxidants phenolic acid and flavone under HILIC and RP conditions, and characterized the selectivities of various HILIC systems using a linear solvation energy relationship model with molecular structure descriptors.

Comparison of columns through the cluster analysis of retention data for peptide separation. Adapted from data in [106]
The main problem with QSRR prediction models is that the results are affected by the composition of the mobile phase, which may become more or less adsorbed in the stationary phase and thus change its true nature. Therefore, comparing the properties of HILIC stationary phases in order to predict the retention of polar compounds would be probably more complicated than comparing RP columns [73]. Additional study in this direction is necessary to show the real merits of the QSRR approach in HILIC. Possibly, comparing the retention data, either isocratic or obtained in gradient experiments with increasing salt and/or water concentration(s), may prove useful for the selection of appropriate stationary–mobile phase combinations in HILIC of different sample types [94].
Orthogonal and two-dimensional separations of HILIC
In 2D (LC × LC) (Fig. 9), the orthogonality between the two dimensions is of paramount importance. It is generally understood that a 2D separation is orthogonal if the two separation mechanisms used are independent of each other and show distinct retention profiles; or, in other words, they provide different selectivities [107–110].

Scheme of the LC × LC sample transfer interface with two sampling loops or two X-Terra trapping columns. Adapted from the data in [111]
The concept and meaning of orthogonality is presented schematically in Fig. 10 using practical examples of NP-LC × RP-LC and RP-LC × RP-LC separations of lemon oil and steroids, respectively. Low correlation (high orthogonality) is observed in Fig. 10a, in which the compounds are randomly distributed across the entire separation space, whereas there is a high linear correlation for the separation shown in Fig. 10b. In this case, the addition of an extra separation dimension leads to an unnecessary increase in the complexity of the system [112].

Theoretical representation of orthogonality in comprehensive LC: a NP-LC × RP-LC separation of lemon oil, b RP-LC × RP-LC separation of steroids. Adapted from the data in [113]
True orthogonality is technically difficult to achieve, as orthogonality depends not only on the separation mechanisms but also on the properties of the solutes and the separation conditions. Successful orthogonal combinations can be achieved when the appropriate stationary and mobile phases are carefully chosen with respect to the physicochemical properties of the sample constituents, including size, charge, polarity, hydrophobicity, etc. A variety of stationary phases are presently available, with differences in surface chemistries, support material, carbon load, pore size, etc., whereas the characteristics of the mobile phase can be altered by changing the modifier, pH, temperature, or by adding ion pair agents. These parameters play important roles in the mixed-mode HILIC retention mechanism and can be flexibly tuned to suit specific separation problems.
Because of its selectivity is highly complementary to RP-LC, HILIC is ideally suited to 2D LC separations. Therefore, an attractive feature of HILIC is its 2D separation capabilities when used together with RP-LC [94, 99]. However, the high-organic mobile phases used in HILIC systems have very high elution strengths in RP systems, and vice versa: mobile phases with high concentrations of water (common in RP systems) are very strong eluents in HILIC systems. This causes serious problems when attempting to use on-line 2D HILIC × RP-LC setups with direct fraction transfer via a switching valve interface. The first-dimension mobile phase in which the fractions are transferred to the second dimension is an excessively strong eluent in the second dimension, which often causes low resolution, peak asymmetry, and even split peaks in the second dimension [99].
For this reason, off-line setups are generally used in HILIC × RP-LC systems, with trapping columns and/or make-up flow post-column adjustment of the mobile-phase elution strength employed [114], which is often time-consuming and not very convenient. Another possibility for orthogonal HILIC × RP separations on a single polar column is to use repeated sample injection on a single column with alternating gradients of increasing (RP) and decreasing (HILIC) organic solvent concentration in the mobile phase [99]. Figure 11 illustrates the concentration profile of ACN in a sequence of 10 min alternating gradient runs over the full range of mobile phase composition (HILIC with 100–0% ACN and RP with 0–100% ACN), with 10 min equilibration periods inserted between the alternating gradient runs.

Schematic diagram of the ACN concentration profile in a sequence of alternating HILIC and RP gradients on a single column. Adapted from the data in [99]
It has been concluded on several occasions that compounds which display large k values on HILIC are not well retained by RP-LC and vice versa [112]. HILIC × RP-LC is the most orthogonal combination of liquid separation techniques when orthogonality is defined as the fraction of coverage of a 2D separation space [115]. The benefits of the orthogonality of HILIC × RP-LC can also be utilized by running the same sample in both HILIC and RP-LC modes and combining the data. This provides more information on the total sample and can, for example, be used for enhanced protein identification via database searches or in approaches with sample fingerprint analysis. For example, selected LC modes including RP-LC and HILIC were combined in different 2D LC setups for the separation of the natural antioxidants phenolic acid and flavone (Fig. 12) by Jandera et al. [116].

Three-dimensional chromatogram for the comprehensive LC × LC separation of phenolic acid and flavone test standards with an optimized two-dimensional parallel gradient. Adapted from the data in [116]
Other examples are the separation of peptides in 2005 by Gilar et al. [115], and of flavonoid glycosides in both RP-LC and HILIC modes on a β-cyclodextrin column by Feng et al. in 2010 [46]. 2D techniques combining RP and HILIC chromatography represent powerful tools in the analysis of pharmaceuticals and their degradation products, and in the metabolic profiling of physiological fluids [61]. Two-dimensional liquid chromatography is often used to reduce the complexity of a proteomic sample prior to tandem mass spectrometry analysis.
An elegant way to utilize the advantageous orthogonality of HILIC and RP-LC is to use solid-phase extraction (SPE) to fractionate the sample before separation. The two fractions are then separated in either the HILIC or the RP-LC mode, depending on the polarity [34]. Other techniques, such as IC and size exclusion chromatography (SEC), result in more peak grouping or poorer resolution in comparison to HILIC [115].
Applications of HILIC
There are many examples of the application of HILIC to the analysis of small polar molecules, including biomarkers, nucleosides, nucleotides/oligonucleotides, amino acids, peptides and proteins, saccharides, glycosides, oligosaccharides, hydrophilic drugs, alkaloids, carbohydrates, and other small polar or ionizable compounds, which contribute to the fields of pharmaceutical chemistry, proteomics, metabolomics, glycomics, medical science, agricultural and food chemistry. The HILIC mode is used intensively to separate some biomolecules through differences in polarity, as well as organic and some inorganic molecules. Its popularity has increased due to the simple biological sample preparation employed when looking for metabolites, since the metabolic process generally results in the addition of polar groups to enhance elimination from the cellular tissue. Table 3 shows several examples of applications of HILIC systems.
HILIC separations are very easy to combine with several detection techniques, such as ultraviolet light absorbance (UV), fluorescence (FL), refractive index (RI), evaporative light scattering (ELSD), charged aerosol (CAD), and mass spectrometry (MS) [127–129]. In addition it is ideally suited to the sensitive LC-MS analysis of water-soluble polar compounds, because the high organic content in the mobile phase leads to rapid evaporation of the solvent during electrospray ionization. HILIC can offer a tenfold increase in sensitivity over reversed-phase chromatography because the organic solvent is much more volatile.
Concluding remarks
Recent years have witnessed an increased interest in HILIC. More versatile and diverse stationary phases have become available, leading to reports of an exciting and broad range of applications. The unique separation ability of HILIC and its orthogonality towards RP make it an ideal method for multidimensional chromatography, which can extend separation power. As far as selectivity is concerned, HILIC can complete well with RP, which is one of the main chromatographic techniques applied today.
Its applications now encompass most categories of polar compounds, charged as well as uncharged, although HILIC is particularly well suited to solutes that lack charge (meaning that Coulombic interactions cannot be used to mediate retention). This approach has recently gained a great deal of attention because of the increased need to analyze polar compounds in complicated mixtures. Another reason for its popularity is the widespread use of MS coupled to HPLC, as HILIC mobile phases are very compatible and give high sensitivity. Recently, HILIC has found great popularity in bioanalytical applications because drugs and their metabolites are often polar structures.
References
Jaroniec M (1993) J Chromatogr A 656:37–50
Gritti F, Félix G, Achard MF, Hardouin F (2001) J Chromatogr A 922:51–61
Wolcott RG, Dolan JW, Snyder LR, Bakalyar SR, Arnold MA, Nichols JA (2000) J Chromatogr A 869:211–230
Krupczyńska K, Buszewski B, Jandera P (2004) Anal Chem 76:226–234
Jandera P (2005) Liquid chromatography—normal phase. In: Encyclopedia of analytical science, 2nd edn. Elsevier, Oxford, pp 142–152
Alpert AJ (1990) J Chromatogr A 499:177–196
Hemström P, Irgum K (2006) J Sep Sci 29:1784–1821
Oyler AR, Armstrong BL, Cha JY, Zhou MX, Yang Q, Robinson RI, Dunphy R, Burinsky DJ (1996) J Chromatogr A 724:378–383
Garbis SD, Melse-Boonstra A, West CE, van Breemen RB (2001) Anal Chem 73:5358–5364
Olsen BA (2001) J Chromatogr A 913:113–122
Li R, Huang J (2004) J Chromatogr A 1041:163–169
Guo Y, Gaiki S (2005) J Chromatogr A 1074:71–80
Cubbon S, Bradbury T, Wilson J, Thomas-Oates J (2007) Anal Chem 79:8911–8918
Alpert AJ, Shukla M, Shukla AK, Zieske LR, Yuen SW, Ferguson MAJ, Mehlert A, Pauly M, Orlando R (1994) J Chromatogr A 676:191–202
Churms SC (1996) J Chromatogr A 720:151–166
Yoshida T (2004) J Biochem Biophys Meth 60:265–280
Hao ZG, Lu C, Xiao BM, Wenig ND, Parker B, Knapp M, Ho C (2007) J Chromatogr A 1147:165–171
Strege MA, Stevenson S, Lawrence SM (2000) Anal Chem 72:4629–4633
Linden JC, Lawhead CL (1975) J Chromatogr A 105:125–133
Regnier FE, Noel R (1976) J Chromatogr Sci 14:316–320
Rubinstein M (1979) Anal Biochem 98:1–7
Yoshida T (1998) J Chromatogr A 811:61–67
Buszewski B, Jezierska M, Wełniak M, Kaliszan R (1999) J Chromatogr A 845:433–445
Buszewski B, Jezierska-Świtała M, Kowalska S (2003) J Chromatogr B 792:279–286
Kowalska S, Krupczyńska K, Buszewski B (2005) J Sep Sci 28:1502–1511
Strege MA (1998) Anal Chem 70:2439–2445
Iwasaki Y, Hoshi M, Ito R, Saito K, Nakazawa H (2006) J Chromatogr B 839:74–79
Oertel R, Neumeister V, Kirch W (2004) J Chromatogr A 1058:197–201
Jiang ZJ, Smith NW, Ferguson PD, Taylor MR (2007) Anal Chem 79:1243–1250
Horie K, Ikegami T, Hosoya K, Saad N, Fiehn O, Tanaka N (2007) J Chromatogr A 1164:198–205
Jiang Z, Reilly J, Everatt B, Smith NW (2009) J Chromatogr A 1216:2439–2448
Naidong W, Shou W, Chen YL, Jiang X (2001) J Chromatogr B 754:387–399
Naidong W (2003) J Chromatogr B 796:209–224
Grumbach ES, Wagrowski-Diehl DM, Mazzeo JR, Alden B, Iraneta PC (2004) LC-GC N Am 10:1010–1023
Tanaka H, Zhou X, Masayoshi O (2003) J Chromatogr A 987:119–125
Kaczmarski K, Prus W, Kowalska T (2000) J Chromatogr A 869:57–64
Tolstikov VV, Fiehn O (2002) Anal Biochem 301:298–307
Ytterberg JA, Ogorzalek-Loo RR, Boontheung P, Loo JA (2007) In: Proc 55th ASMS Conf Mass Spectrometry, Indianapolis, IN, USA, 3–7 June 2007
Alpert AJ (2008) Anal Chem 80:62–76
Yoshida T (1997) Anal Chem 69:3038–3043
Risley DS, Strege MA (2000) Anal Chem 72:1736–1739
Koh HL, Lau AJ, Chan ECY (2005) Rapid Commun Mass Spectrom 19:1237–1244
Dallet Ph, Labat L, Kummer E, Dubost JP (2000) J Chromatogr B 742:447–452
Rong L, Takeuchi T (2004) J Chromatogr A 1042:131–135
Guo Z, Jin Y, Liang T, Liu Y, Xu Q, Liang X, Lei A (2009) J Chromatogr A 1216:257–263
Feng J, Guo Z, Shi H, Gu J, Jin Y, Liang X (2010) Talanta 81:1870–1876
Fu Q, Liang T, Zhang X, Du Y, Guo Z, Liang X (2010) Carbohydr Res 345:2690–2697
Xue M, Huang H, Ke Y, Chu Ch, Jin Y, Liang X (2009) J Chromatogr A 1216:8623–8629
Tobias J, Patrik A (2004) LC-GC Eur 17:40–41
Núñez O, Nakanishi K, Tanaka N (2008) J Chromatogr A 1191:231–252
Hägglund P, Bunkenborg J, Elortza F, Jensen ON, Roepstorff P (2004) J Proteome Res 3:556–566
Guo Z, Lei A, Zhang Y, Xu Q, Xue X, Zhang F, Liang X (2007) Chem Commun (Camb) 24:2491–2493
Fourdinier M, Bostyn S, Delépée R, Fauduet H (2010) Talanta 81:1281–1287
Thermo Electron Corp. (2003) BioBasic SCX and AX columns (TG02-01). Thermo Electron Corp., Waltham, http://www.md-scientific.com/thermo/biobasic_scx_ax.pdf
Lindner H, Sarg B, Meraner C, Helliger W (1996) J Chromatogr A 743:137–144
Lömmerhofer M, Richter M, Wu J, Nogueira R, Bicker W, Lindner W (2008) J Sep Sci 31:2572–2588
McCalley DV (2007) J Chromatogr A 1171:46–55
Pack BW, Risley DS (2005) J Chromatogr A 1073:269–275
Sandoval JE, Pesek JJ (1989) Anal Chem 61:2067–2075
Yoshida T, Okada T (1999) J Chromatogr A 840:1–9
Wang X, Li W, Rasmussen HT (2005) J Chromatogr A 1083:58–62
Karlsson G, Winge S, Sandberg H (2005) J Chromatogr A 1092:246–249
Samuelson O, Swenson B (1962) Acta Chem Scand 16:2056
Samuelson O, Swenson B (1962) Anal Chim Acta 28:426–432
Hong Bui NT, Verhage JJ, Irgum K (2010) J Sep Sci 33:2965–2976
Vailaya A, Horváth Cs (1998) J Chromatogr A 829:1–27
Gritti F, Guiochon G (2005) J Chromatogr A 1099:1–42
Dill KA (1987) J Phys Chem 91:1980–1988
Ying PT, Dorsey JG, Dill KA (1989) Anal Chem 61:2540–2546
Wang HL, Duda U, Radke CJ (1978) J Colloid Interface Sci 66:153–165
Riedo F, Kováts ES (1982) J Chromatogr A 239:1–26
Knox JH, Pryde A (1975) J Chromatogr A 112:171–188
Kaliszan R (2007) Chem Rev 107:3212–3246
Kazakevich YV, McNair HM (1995) J Chromatogr Sci 33:321–327
Buszewski B, Krupczyńska K, Gadzała-Kopciuch RM, Rychlicki G, Kaliszan R (2003) J Sep Sci 26:313–321
Gritti F, Kazakevich YV, Guiochon G (2007) J Chromatogr A 1169:111–124
Bocian S, Vajda P, Felinger A, Buszewski B (2008) J Chromatogr A 1204:35–41
McCalley DV, Neue UD (2008) J Chromatogr A 1192:225–229
Gritti F, dos Santos Pereira A, Sandra P, Guiochon G (2009) J Chromatogr A 1216:8496–8504
Berthod A, Chang SSC, Kullman JPS, Armstrong DW (1998) Talanta 47:1001–1012
Buszewski B, Bocian S, Dziubakiewicz E (2010) J Sep Sci 33:1529–1537
Kazakevich YV, LoBrutto R (2007) HPLC for pharmaceutical scientists. Wiley, Hoboken
Jandera P, Urban J, Škeřiková V, Langmaier P, Kubíčková R, Planeta J (2010) J Chromatogr A 1217:22–33
Jandera P, Churáček J, Taraba B (1983) J Chromatogr 262:121–140
Zhou T, Lucy CA (2010) J Chromatogr A 1217:82–88
Kobayashi J, Kikuchi A, Sakai K, Okano T (2001) Anal Chem 73:2027–2033
Škeřiková V, Jandera P (2010) J Chromatogr A 1217:7981–7989
Liu Y, Xue X, Guo Z, Xu Q, Zhang F, Liang X (2008) J Chromatogr A 1208:133–140
Jandera P (2006) J Sep Sci 29:1763–1783
Soczewiński E, Wachtmeister CA (1962) J Chromatogr A 7:311–320
Snyder LR, Dolan JW, Gant JR (1979) J Chromatogr A 165:3–30
Valko K, Snyder LR, Glajch JL (1993) J Chromatogr A 656:501–520
Snyder LR, Dolan JW (1998) Adv Chromatogr 38:115–185
Jandera P (2008) J Sep Sci 31:1421–1437
Jandera P, Churáček J (1974) J Chromatogr 91:207–221
Jin G, Guo Z, Zhang F, Xue X, Jin Y, Liang X (2008) Talanta 76:522–527
Pesek JJ, Matyska MT, Larrabee S (2007) J Sep Sci 30:637–647
Jandera P, Hájek T (2009) J Sep Sci 32:3603–3619
Jandera P, Hájek T, Škeřiková V, Soukop J (2010) J Sep Sci 33:841–852
Urban J, Škeřiková V, Jandera P, Kubičková R, Pospišolová M (2009) J Sep Sci 32:2530–2543
Romanelli GP, Autino JC, Castro EA (2002) Turk J Chem 26:335–343
Szepesy L (2002) J Chromatogr A 960:69–83
Noga S, Michel M, Buszewski B (2011) Chromatographia 73:857–864
Michel M, Bączek T, Studzińska S, Badzioch K, Jonsson T, Kaliszan R, Buszewski B (2007) J Chromatogr A 1175:49–54
Jinno K, Quiming NS, Denola NL, Saito Y (2009) Anal Bioanal Chem 393:137–153
Buszewski B, Kowalska S, Kowalkowski T, Rozpędowska K, Michel M, Jonsson T (2007) J Chromatogr B 845:253–260
Giddings JC (1995) J Chromatogr A 703:3–15
Liu Z, Patterson DG Jr, Lee ML (1995) Anal Chem 67:3840–3845
Slonecker PJ, Li X, Ridgway TH, Dorsey JG (1996) Anal Chem 68:682–689
Jandera P (2007) LC-GC Eur 20:510
Cacciola P, Jandera P, Hajdú Z, Česla P, Mondello L (2007) J Chromatogr A 1149:73–87
François I, Sandra K, Sandra P (2009) Anal Chim Acta 641:14–31
François I, de Villiers A, Tienpont B, David F, Sandra P (2008) J Chromatogr A 1178:33–42
Wilson SR, Jankowski M, Pepaj M, Mihailova A, Boix F, Truyols GV, Lundanes E, Greibrokk T (2007) Chromatographia 66:469–474
Gilar M, Olivova P, Daly AE, Gebler JC (2005) Anal Chem 77:6426–6434
Jandera P, Česla P, Hájek T, Vohralík G, Vyňuchalová K, Fischer J (2008) J Chromatogr A 1189:207–220
Kind T, Tolstikov V, Fiehn O, Weiss RH (2007) Anal Biochem 363:185–195
Bonanno LM, Denizot BA, Tchoreloff PC, Puisieux F, Cardot PJ (1992) Anal Chem 64:371–379
Schlichtherle-Cerny H, Affolter M, Cerny C (2003) Anal Chem 75:2349–2354
Doi T, Kajimura K, Takatori S, Fukui N, Taguchi S, Iwagami S (2009) J Chromatogr B 877:1005–1010
Quiming NS, Denola NL, Ueta I, Saito Y, Tatematsu S, Jinno K (2007) Anal Chim Acta 598:41–50
Zheng M, Zhang M, Peng G, Feng Y (2008) Anal Chim Acta 625:160–172
Qin F, Zhao YY, Sawyer MB, Li X (2008) Anal Chim Acta 627:91–98
Dugo P, Škeříková V, Kumm T, Trozzi A, Jandera P, Mondello L (2006) Anal Chem 78:7743–7750
Antonio C, Larson T, Gilday A, Graham I, Bergström E, Thomas-Oates J (2008) Rapid Commun Mass Spectrom 22:1399–1407
van der Werf MJ, Overkamp KM, Muilwijk B, Coulier L, Hankemeier T (2007) Anal Biochem 370:17–25
Petritis K, Elfakir C, Dreux M (2002) J Chromatogr A 961:9–21
Mitchell CR, Bao Y, Benz NJ, Zhang S (2009) J Chromatogr B 877:4133–4139
Douša M, Břicháč J, Gibala P, Lehnert P (2011) J Pharmaceut Biomed 54:972–978
Acknowledgements
The work was supported by European Social Fund, the Polish National Budget, Kujawsko-pomorskie Vovidship Budget—“Stypendia dla doktorantów 2008/2009—ZPORR.” Financial support from the Foundation for Polish Science (Professor’s Subsidy “Mistrz”) is gratefully acknowledged.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in the 10th Anniversary Issue.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Buszewski, B., Noga, S. Hydrophilic interaction liquid chromatography (HILIC)—a powerful separation technique. Anal Bioanal Chem 402, 231–247 (2012). https://doi.org/10.1007/s00216-011-5308-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-011-5308-5