
Abstract
Heparin is a well-known glycosaminoglycan extracted from porcine intestines. Increased vigilance for transmissible spongiform encephalopathy in animal-derived pharmaceuticals requires methods to prevent the introduction of heparin from ruminants into the supply chain. The sensitivity, specificity, and precision of the quantitative polymerase chain reaction (PCR) make it a superior analytical platform for screening heparin raw material for bovine-, ovine-, and caprine-derived material. A quantitative PCR probe and primer set homologous to the ruminant Bov-A2 short interspersed nuclear element (SINE) locus (Mendoza-Romero et al. J. Food Prot. 67:550–554, 2004) demonstrated nearly equivalent affinities for bovine, ovine, and caprine DNA targets, while exhibiting no cross-reactivity with porcine DNA in the quantitative PCR method. A second PCR primer and probe set, specific for the porcine PRE1 SINE sequence, was also developed to quantify the background porcine DNA level. DNA extraction and purification was not necessary for analysis of the raw heparin samples, although digestion of the sample with heparinase was employed. The method exhibits a quantitation range of 0.3–3,000 ppm ruminant DNA in heparin. Validation parameters of the method included accuracy, repeatability, precision, specificity, range, quantitation limit, and linearity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Heparin is a sulfated polysaccharide that has been used clinically as an anticoagulant for many years. It is composed of alternating 1 → 4 linked residues of uronic acid and glucosamine with various levels of sulfation. Heparin can be found in a many tissues (e.g., lung, intestines, liver, skin) from a number of animal species (e.g., ox, dog, chicken, sheep, whale) [1]. Porcine intestines are the most significant source of heparin currently used in clinical applications. The concern with disease transmission, most notably prions associated with transmissible spongiform encephalopathy from ruminant tissues [2], makes it necessary to ensure that the supply chain for animal-derived pharmaceuticals is monitored with regard to the source of the material. The possibility of comingling tissue of other animal species such as bovine (cattle), ovine (sheep), and caprine (goat) at multi-product-processing sites creates the need for analytical methods that can distinguish the source of raw heparin.
The detection of ruminant material in animal-derived materials has been extensively described [3–8]. These detection methods include immunological, microscopic, spectroscopic, and nucleic acid detection techniques. Microscopic and spectroscopic analytical methods lack sensitivity, with detection limits as high as 1% and the potential for inaccurate results. Although ELISA offers improved sensitivity, with detection limits as low as 0.01%, this platform can exhibit poor accuracy owing to matrix interferences, false positives from cross-reactivity, and poor robustness due to sample heterogeneity [8]. Additionally, none of these technologies can be used to determine if chemical or biological adulterants have been added to the crude heparin material to mask the source. In contrast, nucleic acid detection, in general, and quantitative polymerase chain reaction (PCR), in particular, offer significant advantages in sensitivity, specificity, accuracy, and robustness [9, 10]. Heparin-mediated inhibition of PCR has been previously described and requires particular precautions to ensure accurate detection and measurement of target sequences [11]. The use of heparinase to degrade the heparin present in the sample is one technique to overcome heparin-mediated quantitative PCR inhibition, and the inclusion of a ruminant spike prior to heparinase digestion ensures accurate quantitation. A quantitative PCR analytical method was developed, validated, and is in current use to assess the animal origin of commercially available crude heparin.
Materials and methods
Materials and equipment
The PCR instrument used in this study was an ABI Prism 7900HT sequence detection system (Applied Biosystems, Foster City, CA, USA). Quantitative PCR requires the use of DNA primers and probes to amplify the target DNA if present. Primers and internal probes specific for short interspersed nuclear elements (SINEs) present in both the ruminant and the porcine genomes were used to detect ruminant DNA in crude heparin as well as to verify the presence of porcine DNA in this material. SINEs were selected as the target DNA owing to their abundant copy number in genomes and the consequent high level of sensitivity and specificity which they offers [12]. The ruminant Bov-A2 SINE consensus sequence has been previously described [9] and exhibits broad specificity across bovine, ovine, and caprine genomes. The absence of detectable levels of porcine DNA suggests either chemical or biological adulteration of the crude heparin. Therefore, primers and an internal probe corresponding to the porcine PRE1 SINE sequence were designed by Applied Biosystems and used in the assay and validation described. All primers and probes used in the analysis are shown in Figs. 1 and 2.

Ruminant primers and probe

Porcine primers and probe
Commercially available bovine DNA (part no. 69231, EMD Chemicals, Gibbstown, NJ, USA) was used to prepare the ruminant DNA reference solutions used for quantitation. Commercially available porcine DNA (part no. 69230, EMD Chemicals, Gibbstown, NJ, USA) was used to prepare the porcine DNA reference solutions. Commercially available ovine DNA (part no. GSHE, Zyagen, San Diego, CA, USA) was solubilized in molecular biology grade water and used to verify the specificity of the ruminant and porcine primers and probes. Caprine DNA was prepared by weighing 485 mg of goat meat in a 15-mL sterile tube, adding 6 mL of nuclei lysis solution (Wizard genomic DNA purification kit, part no. A1120, Promega, Madison, WI, USA), and macerating the sample for 10 min. The sample was incubated for 30 min at 65 °C. The tissue debris was removed by centrifugation and 600 μL of the supernatant was removed. The caprine DNA was purified from the supernatant following the kit instructions and was used to determine the specificity of the ruminant and porcine primers and probes. The concentration and purity of the ovine and caprine DNA solutions were determined from the absorbance at 260, 280, and 320 nm.
Sample, standard, and spike preparation
Crude heparin samples for analysis were prepared by weighing approximately 300 mg crude heparin into a sterile tube. Ten milliliters of 50 mM tris(hydroxymethyl)aminomethane–HCl/5 mM CaCl2 (pH 8.0), which is referred to as heparinase digestion buffer, was added and the sample was mixed vigorously to dissolve the solids. The dissolved sample was diluted 1:99 with heparinase digestion buffer to create a 0.03% (w/v) working crude heparin solution. Standard curves were created by adding appropriate volumes of each reference DNA solution (porcine, bovine, ovine, or caprine) in heparinase digestion buffer to achieve concentrations of 0.1, 1, 10, 100, and 1,000 pg DNA/μL. An additional standard at 2,000 pg DNA/μL was included for the porcine DNA assay to accommodate the high concentration of host DNA in crude heparin.
Control samples included a solution of each 0.03% crude heparin sample spiked with 200 pg DNA/μL, a sample of heparinase digestion buffer spiked with 200 pg DNA/μL, and a sentinel negative control of unspiked heparinase digestion buffer. All controls and standards were heparinase-digested and analyzed in parallel with each test article. Additionally, nontemplate controls (NTCs) were included with the amplification phase of the method.
DNAse inactivation and heparinase digestion
All standard curve dilutions, test articles, and control samples were incubated at 80 ± 5 °C for 5 ± 1 min to inactivate any DNase that may be present and were subsequently placed on ice for a minimum of 10 min. Two units of heparinase I (product no. H2519, Sigma-Aldrich, St. Louis, MO, USA) was added to each standard curve dilution, test article, and control prior to incubation for 18 ± 0.5 h at 28 ± 2 °C.
Quantitative PCR amplification
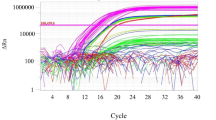
The heparinase-treated materials were subjected to quantitative PCR using the TaqMan universal PCR master mix chemistry and the species-specific DNA primers and probes. All standards, test articles, and controls were analyzed in triplicate. System suitability criteria applied to the analytical method included a standard curve coefficient of determination (R 2) of 0.9 or greater, a slope of -4.0 or greater, recovery of ±0.6 log of the 200 pg DNA/μL spike in the test article, relative standard deviation (RSD) of the threshold cycle (C T) for replicates of 10% or less, and a verification that the mean C T of the sentinel negative control and NTC were both greater than the mean C T of the lowest standard curve sample.
Results
Specificity
A standard curve of ovine DNA was prepared in heparinase digestion buffer at concentrations of 0.01, 0.1, 1, 10, 100, and 1,000 pg/μL and analyzed using the ruminant DNA primers and probe. Statistical and visual analysis of the response obtained with the 0.01 pg/μL standard found that this ovine DNA concentration could not be distinguished from the background response of the NTC and was below the quantitation limit of the method. Exclusion of the 0.01 pg/μL standard yielded a standard curve with an R 2 of 0.99 and a slope of -2.79, with an amplification efficiency of 128%. Therefore, it was concluded that the ruminant primers and probe can be used to quantitate also ovine DNA in solution at concentrations greater than 0.1 pg/μL.
A standard curve of caprine DNA was similarly prepared in heparinase digestion buffer at concentrations of 0.1, 1, 10, 100, and 1,000 pg/μL and was analyzed using the ruminant DNA primers and probe set. The 0.1 pg/μL response was indistinguishable from the background response of the NTC and was determined to be below the quantitation limit. When the 0.1 pg/μL standard was removed from the analysis, the standard curve exhibited an R 2 of 0.99 and a slope of -3.2, corresponding to an amplification efficiency of 105%. Therefore, the ruminant primers and probe can be used to quantitate caprine DNA in solution at concentrations greater than 1 pg/μL.
Porcine DNA at a concentration of 1,000 pg/μL in heparinase digestion buffer was indistinguishable from the response of the background (NTC) and sentinel extraction control (SEC) when analyzed with the ruminant primers and probe. An analogous assessment of ruminant DNA at a concentration of 1,000 pg/μL in heparinase digestion buffer was indistinguishable from the response of both the NTC and the SEC when tested with the porcine primers and probe.
Linearity
The ruminant DNA standard curve was prepared as described in “Materials and methods” and analyzed using the ruminant primers and probe set on four separate occasions. The standard curve was created by plotting the mean C T versus the DNA concentration. Semilogarithmic plots of C T versus DNA content exhibited R 2 values of 0.997, 0.985, 0.999, and 0.998 for the separate standard curves.
Linearity of the porcine DNA standard curve was determined in a manner similar to that used for the ruminant DNA linearity determination. The R 2 values for four separate porcine DNA standard curves were 0.998, 0.995, 0.996, and 0.988.
All ruminant DNA standard curves and the porcine standard curves met the target criterion of R 2 ≥ 0.90. No curvature or trends were observed. The linear correlation results combined with the visual inspection of the standard curve residual values demonstrated that the analytical method met the target linearity for both ruminant and porcine DNA analysis.
Accuracy
Accuracy was determined across the range of the analytical method by spiking a crude heparin sample prior to heparinase digestion with known amounts of ruminant DNA at concentrations of 0.1, 1.0, 200, and 1,000 pg/μL . Instrument sensitivity and the logarithmic nature of PCR, which has been previously reported [13], were considered in the design of an acceptance criterion for accuracy. The nonlinear response of PCR and the ability of the instrument to distinguish a twofold to fourfold difference in target DNA content justified a spike recovery acceptance criterion of ±0.6 log of the theoretical response. The acceptance criterion was derived from the logarithm of 4 (i.e., fourfold sensitivity of the target DNA), which is 0.6. The results obtained with ruminant DNA and ruminant primers and probe are shown in Table 1.
Porcine DNA is expected to be present in crude heparin of porcine origin. The quantitation of porcine DNA in crude heparin therefore serves as an internal control demonstrating that the sample has not been adulterated or otherwise compromised as to alter the response of DNA in the analytical method. Additionally, the quantitative recovery of a porcine DNA spike from the crude heparin sample demonstrates that the sample does not exhibit matrix interference effects on the analytical method. To determine the accuracy of the porcine DNA quantitation in crude heparin, three separate crude heparin samples were spiked with 200 pg/μL of porcine DNA prior to heparinase digestion and analyzed. The results obtained with porcine DNA and porcine primers and probe are shown in Table 1.
The analytical method exhibited appropriate accuracy for the quantitation of ruminant DNA and porcine DNA by meeting the target criteria.
Precision
The repeatability of the ruminant quantitative PCR analytical method was assessed by spiking a crude heparin sample prior to heparinase digestion with ruminant DNA at 0.1, 1.0, 200, and 1,000 pg/μL and assaying each sample in triplicate in three repeated analyses. The relative standard deviation (RSD) was 1% for all sample analyses, including the unspiked crude heparin sample.
The repeatability of the porcine quantitative PCR analytical method was assessed in a similar manner, except a crude heparin sample was spiked with porcine DNA at only one concentration (i.e., 200 pg/μL). The samples were analyzed in triplicate in four repeated analyses, and the RSD was 1% or less for each analysis.
The target criterion for precision (repeatability) was 10% or less RSD. The analytical method exceeded the target criteria for both ruminant and porcine DNA and was determined to exhibit acceptable repeatability.
The intermediate precision of the ruminant DNA analytical method was assessed by spiking a crude heparin sample and heparinase digestion buffer with ruminant DNA at 0.1, 1.0, 200, and 1,000 pg/μL and assaying each sample in triplicate on three separate days by two different analysts. The pooled RSD (n = 9) for the spiked heparin sample and spiked heparinase buffer was 4% or less at all concentrations and for both sample types.
The intermediate precision of the porcine DNA analytical method was assessed in a manner similar to that for the ruminant DNA analysis, except the crude heparin sample and heparin digestion buffer were spiked with porcine DNA at only one concentration (i.e., 200 pg/μL) before the samples were analyzed in triplicate on four separate days by two analysts. The pooled RSD (n = 12) for the porcine-DNA-spiked crude heparin sample and for the spiked digestion heparinase digestion buffer was 3% or less for both sample types.
These results meet the target criterion of a pooled RSD of 10% or less and it was determined that the analytical method exhibited acceptable intermediate precision.
Quantitation limit
The quantitation limit of the analytical method was defined as the lowest DNA spike concentration in crude heparin that achieved the acceptance criterion for accuracy of ±0.6 log spike recovery, precision of 10% or less RSD, and fell within the linear response of the analytical method. As shown by the previous data, the quantitation limit was determined to be 0.1 pg DNA/μL for both ruminant and porcine DNA in crude heparin. When the sample preparation procedure is taken into consideration, this limit corresponds to 0.3 ppm ruminant or porcine DNA in crude heparin powder.
Detection limit
Bovine serum albumin (BSA) may be included as a stabilizer for both DNA polymerase and heparinase. Both enzymes are used in the analytical procedure; therefore, a background response in the NTC and the SEC is expected owing to trace levels of ruminant DNA associated with BSA. SEC replicates contained all reagents used during the analysis, including heparinase and DNA polymerase, whereas the NTC contained only heparinase digestion buffer and DNA polymerase. During these experiments the C T from the SEC was always lower than that from the NTC, suggesting the presence of minute, yet detectable quantities of ruminant DNA in the heparinase. The presence of ruminant DNA in the reagents could not be controlled, and has the potential of changing between reagent lots. This observation represents a limitation of this method. On the basis of statistical analysis and visual examination of the data, it was difficult to discern the 0.01 pg/μL (i.e., 0.03 ppm) ruminant DNA response from the background routinely. Consequently, in practice the quantitation limit 0.1 pg/μL (i.e., 0.3 ppm ruminant DNA in crude heparin) was also assigned as the detection limit. A detection limit of 0.3 ppm ruminant DNA is comparable to other reported detection limits for similar analytical methods [6].
Discussion
The identity of the animal origin of crude heparin must be determined to ensure the safety of the supply chain when the introduction of transmissible spongiform encephalopathy is a possibility. Quantitative PCR is an ideal tool for this purpose.
The analytical method described has been developed to ensure that precautions are taken to avoid a false-negative response. Heparin, a highly charged glycosaminoglycan, is well known to inhibit the PCR analysis. The concentration of crude heparin in the test sample (0.03%) was shown during development to not inhibit the PCR analysis. A heparinase digestion step was added to the sample preparation scheme to degrade the heparin in the sample and ensure that the heparin in the sample did not inhibit the quantitative PCR amplification step. Although exogenous template control DNA may be used as an internal positive control to assess sample inhibition [10, 14], an authentic ruminant DNA spike was chosen to evaluate sample inhibition in the assay described in this evaluation. The ruminant spike was added to each test sample prior to heparinase digestion and a recovery criterion was applied to the spike to verify that accurate results were obtained for all samples during routine analysis. The inclusion of an authentic spike of the analyte mitigates the potential effect of differences in amplification efficiency and experimental bias that may occur between a commercially available internal positive control and the target amplicon. Additionally, this strategy eliminates the possibility of a false negative due to DNase digestion that may occur during the heparin digestion step. The technique of inhibitor-controlled PCR previously described [14] incorporates a semiauthentic internal control which is coamplified in an end-point PCR analytical method and followed by agarose electrophoresis. Although this approach eliminates potential concerns of amplification efficiency between the control and the target analyte, the construction of the inhibitor-controlled PCR control is relatively laborious and the reliance on electrophoresis for detection requires semiquantitive and subjective analysis. Authentic bovine DNA and porcine DNA were chosen as the spike for the positive sample controls in the analytical method to ensure the specificity and sensitivity of the PCR were adequately controlled using the specified analyte. Incorporation of the ruminant and porcine spikes prior to heparinase digestion further reduces the possibility of a false-negative result caused by the presence of DNases in the crude heparin sample. It is also noteworthy that the detection and quantitation limits of this method for ruminant DNA are the lowest reported limits to date in crude heparin [10, 14]
The analytical method was validated for specificity, linearity, accuracy, precision (repeatability and intermediate precision), and quantitation limit. The ruminant DNA primers and probe were found to amplify bovine, ovine, and caprine DNA in a quantitative manner and did not detect porcine DNA. The porcine DNA primers and probe did not amplify ruminant (bovine) DNA. The range of the analytical method, defined as acceptable accuracy, precision, and linearity at the extremes of the analyte concentration and points in between, was found to be 0.3–6,667 ppm DNA in crude heparin for both ruminant and porcine DNA. The method described herein has been used to successfully assess commercial crude heparin since 2005, indicating the stability of the analytical method and its value to the industry.
References
Nader HB, Dietrich CP (1989) In: Lane DA, Lindahl U (eds) Heparin: chemical and biological properties, clinical applications. London, Arnold
Will RG (199) ACTA Paediatr Suppl 433:28-32
Levieux D, Levieux A (2001) J Immunoassay 22:127–145
Levieux A, Rivera V, Levieux D (2001) J Immunoassay Immunochem 22:323–336
Levieux A, Rivera V, Levieux D (2001J) Pharm Biomed Anal 27:305–313
Momcilovic D, Rasooly A (2000) J Food Prot 63(11):1602–1609
Krcmar P, Rencova E (2000) J Food Prot 64(1):117–119
Gizzi G, van Raamsdonk LWD, Baeten V, Murray I, Beben G, Brambilla G, von Holst C (2003) Rev Sci Tech Off Int Epizoot 22(1):311–331
Mendoza-Romero L, Verkaar ELC, Savelkoul PH, Catsburg A, Aarts HJM, Buntjer JB, Lenstra JA (2004) J Food Prot 67:550–554
Houiste C, Auguste C, Macrez C, Dereux S, Derouet A, Anger P (2009) Clin Appl Thromb Hemost 15:50–58
Satsangi J, Jewell DP, Welsh K, Bunce M, Bell JI (1994) Lancet 343(8911):1509–1510
Walker JA, Hughes DA, Anders JA, Shewale J, Sinha SK, Batzer MA (2003) Anal Biochem 316:259–269
Lovatt A (2002) Rev Mol Biotechnol 82:279–300
Huang Q, Yao C-Y, Chen B, Wang F, Huang J-F, Zhang X, Fu W-L (2006) Mol Cell Probes 20:250–258
Acknowledgements
The authors wish to acknowledge Kevin Rust for his work on development of the porcine primers and probe and initial development of the analytical method and Jerry Davis for analysis of crude heparin samples in support of this work. The authors also wish to acknowledge Monika Lorenz and Vladimir Golynskiy for their review, guidance, and support of this work.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in the special issue Heparin Characterization with Guest Editor Cynthia K. Larive.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Concannon, S.P., Wimberley, P.B. & Workman, W.E. A quantitative PCR method to quantify ruminant DNA in porcine crude heparin. Anal Bioanal Chem 399, 757–762 (2011). https://doi.org/10.1007/s00216-010-4362-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-010-4362-8