
Abstract
The brain integrates complex types of information, and executes a wide range of physiological and behavioral processes. Trillions of tiny organelles, the synapses, are central to neuronal communication and information processing in the brain. Synaptic transmission involves an intricate network of synaptic proteins that forms the molecular machinery underlying transmitter release, activation, and modulation of transmitter receptors and signal transduction cascades. These processes are dynamically regulated and underlie neuroplasticity, crucial to learning and memory formation. In recent years, interaction proteomics has increasingly been used to elucidate the constituents of synaptic protein complexes. Unlike classic hypothesis-based assays, interaction proteomics detects both known and novel interactors without bias. In this trend article, we focus on the technical aspects of recent proteomics to identify synapse protein complexes, and the complementary methods used to verify the protein–protein interaction. Moreover, we discuss the experimental feasibility of performing global analysis of the synapse protein interactome.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Synapse background
The brain is the most complex and dynamically organized organ, with high computation capability. It enables the execution of a wide range of physiological processes and behavior based on integrative neurotransmission among neural systems and across brain regions. The fundamental structural units for neuronal communication are the trillions of chemical synapses. Each synapse consists of a neurotransmitter synthesizing and releasing apparatus, the presynapse, and a transmitter receiving element, the postsynapse. An electric signal arriving at the presynaptic terminal is converted into a chemical message in the form of released transmitters, which diffuse through the synaptic cleft and bind to the receptors in the postsynaptic element. This causes differential ion fluxes into and out of the postsynapse that may result in the propagation of electric signals in the dendrite.
The molecular machinery underlying synaptic function consists of an intricate network of synaptic proteins. It is generally believed that neuronal activity-dependent change of synaptic efficacy is at the basis of learning and memory and is encoded by sequential molecular events at the synapse. Abnormalities in synaptic activity or plasticity, presumably underpinned by aberrant protein expression and/or protein–protein interaction, are known to occur in many, and maybe most, psychiatric disorders. Whereas the most important aspects of synapse function, for example the transmitter receptors, have been studied extensively, the molecular basis of neuroplasticity remains largely unclear. Revealing the mechanisms of synaptic plasticity and synaptic disorders essentially needs to uncover the temporal interactive actions of the multi-protein complexes that shape synapse function. Classic studies focusing on single events fall short of revealing the global changes of the system.
Proteomics has been used to examine the protein constituents of the synapse sub-fractions including synaptic vesicle [1, 2], pre-synapse element [3], and post-synaptic density [4, 5]. Catalogs of sub-synapse proteomes now facilitate construction of a draft of a synapse protein interactome [6, 7], however, understanding the organization of macromolecular protein complexes and sub-synapse structures that drive synapse function requires the identification and quantification of protein constituents of synapse protein complexes.
In the past year we have witnessed a major increase in the application of interaction proteomics on the analysis of brain synapses. Protein complexes have usually been isolated by co-immunoprecipitation approach using antibodies against the bait protein, and then subjected to MS-based proteomics analysis. Tandem affinity purification (TAP), a technique developed a decade ago for the large-scale study of protein–protein interaction in yeast, has since been shown to be applicable to the study of rodent synapse protein complex [8]. From many of these studies meaningful biological conclusions could be drawn. Subsequent physiological studies revealed the roles protein interactions play in driving synapse functions/neuroplasticity. Whereas most of the studies focused on selected (single) bait proteins, an attempt has also been made to assess the experimental feasibility of carrying out a comprehensive analysis of the synapse protein interactome [9].
Proteomics methodologies for analysis of synapse protein complexes
A typical procedure for identifying a protein complex requires affinity-isolation of the complex from a (pre-fractionated) sample extracted in a detergent-containing buffer. The purified protein complex is then subjected to proteomics analysis (Fig. 1). The first proteomics study of a synapse protein complex was reported a decade ago [10]. Throughout the years, interaction proteomics [11] has emerged as a mature method. Continual advances in mass accuracy and sensitivity in mass spectrometry greatly facilitates protein identification. The establishment of large number of proteomics centers has generated easy access to this technology for many neuroscience groups. This has contributed to a sharp increase in proteomics analysis of synapse protein complexes in the past year, as summarized in Table 1.

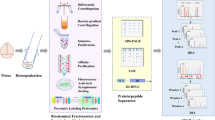
Workflow of immunoprecipitation-based proteomics of synapse protein complexes. Brain tissue is homogenized, and subjected to one or a series of differential centrifugation steps optimized to enrich synapses. After extraction of the sample in a detergent-containing buffer, antibodies against a specific synaptic protein and the protein A/G beads are added and incubated for from 1 h to overnight. The antibody-captured protein complexes can be eluted using different strategies. (1) Acidic buffer elution. This step is often followed by an in-solution digest. (2) SDS sample buffer elution. This step dislodges all the non-covalently bound proteins from the beads. (3) Elution with a high concentration of peptide/protein antigen. This generally gives lowest background proteins. In (2) and (3), the eluted proteins are usually run on an SDS-PAGE and then digested in-gel. In all cases the peptide mixtures are further analyzed by LC–MS–MS
It is self-evident that synaptic protein complexes are often isolated from input enriched in the synapse. In most studies the input was a synaptosome-enriched fraction, or the synaptic plasma membrane. Technical issues may also dictate the type of input sample. For example, Han et al. reported that use of the synaptosome fraction provided a limited amount of materials for affinity purification of protocadherin [12], whereas the crude membrane fraction has high background. To circumvent the problem, the authors used crude membrane protein extract as starting material, and then reduced sample complexity, and thus background, by incorporating sucrose density gradient ultracentrifugation before affinity purification [12]. Whole brain or brain regions have also been used without pre-fractionation for isolation of protein complexes. The purpose of these types of studies was to identify all the interacting proteins of the bait proteins. In such cases, the synapse protein interactors are a subset of the identified protein interactome.
The most widely used detergent for solubilization of protein complexes is Triton X-100, usually in the range 0.5–1%, and up to 4% depending on protein concentration. This non-ionic detergent effectively dissolves membranes, and is mild enough to maintain (most) protein–protein interactions. ComplexioLytes have been used to solubilize receptors and ion channels. Unfortunately the chemical compositions of these detergents are unknown. In some cases, the ionic detergent sodium deoxycholate (DOC) has been used. This is exemplified by the isolation of PSD-95 complex. PSD-95 is partially soluble in Triton X-100; this soluble fraction has been shown to contain multiple protein complexes [9]. Most PSD-95 is embedded in the post-synaptic density, which is not soluble in Triton X-100 [4]. One percent DOC, which has been shown to solubilize core post-synaptic density protein, for example the NMDA receptor [10], was chosen [8] to solubilize PSD-95 directly from mouse forebrain. The complex was then isolated by target affinity tagging (TAP). It should be noted that DOC has been shown to have an adverse effect on protein–protein interactions, resulting in poor recovery of the intact protein complex [21]. The detected proteins may be a small fraction of the proteins that still retain interaction. Alternatively, they may represent secondary interaction to the bait protein that has been stripped from its interactors [22].
The isolated protein complex is commonly fractionated with SDS-PAGE, which separates proteins according to their masses. In some cases the protein pattern of the stained gel from the sample was compared with that of a negative control; protein bands specifically present in the sample lane were digested with trypsin, and analyzed either directly by mass spectrometry or by LC–MS–MS (Table 1). The disadvantage is that proteins existing at levels lower than the detection limit of the staining method would not be selected for further analysis. As current MS sensitivity is at the low femtomole to the upper attomole level, it surpasses the detection limit of silver stained gel, leading to the preferred analysis to cut whole gel lanes into pieces, irrespective of the staining pattern, followed by trypsin digestion and analysis by LC–MS–MS. Comparison of the sample with a negative control then reveals the true protein interactors. The ideal negative control would be an identical analysis, using sample from a (knockout) mouse lacking the bait protein [12], or the use of multiple antibodies directed at the same protein.
Different types of mass spectrometer have been used for protein identification [23]. Because of the high mass accuracy in the MS1 and fast scan rate and high sensitivity in MS–MS, ion trap-FT MS appears to be the preference. Off-line LC MALDI TOF–TOF mass spectrometry has generally been used for iTRAQ reagent-based protein quantitation [7, 18] because it yields intense iTRAQ signature ions and gives a good coverage of peptide b-ion and y-ion series. Other studies have shown that online LC–electrospray MS–MS can also be optimized for iTRAQ reagent based quantitation [24, 25].
Affinity tagging for the analysis of selected protein complex
The classical approach used to isolate a protein complex is to use a specific antibody against the bait protein for co-immunoprecipitation. The success of such an experiment critically depends on the quality of the antibody. To obtain the best result it is not uncommon to use several antibodies against different epitopes of a protein [13] to increase the validity of the study.
Recently, TAP tagging methodology with a gene-targeting approach in mice has been used, with PSD-95 as a model [8]. First, a knockin mouse was generated with a TAG tag of a poly-histidine affinity tag (HAT) and a triple FLAG tag, in tandem attached at the C-terminal of a PSD-95 construct. A TEV-protease cleavage site separated the HAT and FLAG tags. Second, it was confirmed that the expression level and synapse localization of the TAP-tagged PSD-95 were similar to those of endogenous PSD-95 and that the knocked-in gene did not alter the synapse physiology. Third, the soluble PSD-95 was captured by a FLAG antibody covalently coupled to beads. The beads were incubated with TEV that cut the TEV-protease cleavage site between the HAT and FLAG tags. The released PSD-95 containing HAT tag was recovered by use of an Ni2+-NTA column that binds HAT, and subsequently eluted with imidazole. The PSD-95 complex was separated by SDS-PAGE, trypsin digested, and analyzed by LC–MS–MS. Compared with single-step purification, tandem purification results in fewer contaminants. It also detected more low-abundance proteins that could have been masked by the high-abundance proteins in a single purification. Hundreds of proteins were identified. They were connected into a protein network, using protein–protein interaction data from manually-curated interaction data and the UniHi database. Annotation of the clusters with human disease associations revealed that multiple disorders map to the network, with a significant correlation of schizophrenia within the glutamate receptor clusters. However, the in-vivo affinity tag approach involves a tedious procedure to generate the knockin mouse, and to control for the normal functioning of the mutated protein. This approach is therefore applicable to only a limited set of synapse proteins for in-depth analysis.
A similar TAP approach has been used to identify the interacting proteins of metabotropic glutamate receptor 1 transfected in Madin–Darby canine kidney cells [26]. Several of the identified interactors were shown to associate with the native receptor in the brain by co-immunoprecipitation and western blotting.
Other tags may be used for affinity isolation of a protein complex. For example, affinity purification with anti-GFP antibody from the brain extract of a PCDH-γ-GPF knockin mice has been used for isolation of the PCDH-γ protein complex [12].
Validating protein–protein interactions
A general problem in affinity proteomics is distinguishing specific interactors from contaminants. For example, mitochondrial proteins, proteins involved in metabolism, and cytoskeletal proteins are often detected across IP studies. These proteins are highly abundant. They may not be completely removed during washing steps, and are, therefore, identified subsequently. To address this problem, we have used iTRAQ reagent for comparative analysis of an AMPA receptor complex isolated from wild type and knockout mice [18]. Proteins enriched in the wild type mice as indicated by high iTRAQ ratio in the wild type mice sample represent true interactors of the AMPA receptor. Other studies compared the proteins identified from the affinity purification with the specific antibody and the negative control, respectively. Proteins with high relative peptide query score were regarded as specific interactors [13, 14].
Reversed immunoprecipitation is generally used as an independent method to verify protein–protein interaction data (Table 1), i.e. immunoprecipitation of the interactor co-immunoprecipitates the original bait protein. In studies that identified large numbers of proteins without subsequent verification of the interactions, in particular for those in which a negative control was not included, some of the reported proteins may be contaminants [8, 9, 15].
In principle, yeast two hybrid assay can also be used to probe the binary protein–protein interaction; but today it is sparsely used to confirm the interaction detected by interaction proteomics.
Single proteins might be involved in multiple events in the synapse. Hence, they might be contained in different protein complexes. Therefore, affinity isolation of a bait protein might capture multiple protein complexes, which are characterized simultaneously by proteomics without distinguishing proteins residing in distinct protein complexes. How can an identified protein be assigned to multiple protein complexes? Blue native polyacrylamide gel electrophoresis (BN-PAGE) separates stable protein complexes according to their sizes with good resolution up to around 1,200 kDa. BN-PAGE is a method of choice to confirm and/or differentiate protein constituents from protein complexes, as exemplified by the analyses of AMPA receptor complexes. VandenBerghe et al. [27], solubilized a cerebellar extract and pre-fractionated on a 10–50% glycerol gradient; the fractions were then analyzed by BN-PAGE. The AMPA receptors containing subunits 2/3 migrated as two distinct high-molecular-weight complexes. The well established AMPA receptor interactor, stargazing, co-migrated with one of them, suggesting the presence of an additional AMPA receptor complex. A recent study [13] revealed that this distinct AMPA receptor complex contains cornichon. BN-PAGE in conjunction with antibody shift and western blotting analysis confirmed the mutual exclusive existence of stargazing and cornichon in these two AMPA receptor complexes.
The demonstration that potential interactors exist in close proximity at the sub-cellular level is often used as supporting evidence of their in-situ interaction. Immunocytochemistry is routinely applied for co-localization studies, but the spatial resolution is too low to verify protein–protein interaction. Fluorescent resonance energy transfer enables in-cell visualization of protein pairs [28] at a distance of typically less than 10 nm. This method requires the transfection, usually in a cell culture, of the potential interacting protein pair, one with a donor chromophore and the other with an acceptor chromophore. The disadvantages are that this method detects the transfected proteins rather than the endogenous proteins, and that the presence of the chromophores may affect protein interaction. Recently, an antibody-based proximity ligation assay (PLA) technology was developed to study protein–protein interaction in situ [29, 30]. The sample is incubated with two target-specific primary antibodies from two different host species. After washing, two PLA probes, each with a species-specific secondary antibody with a short DNA strand, are added. If the two proteins are in close proximity, and in the presence of the added circle-forming DNA oligonucleotides, the DNA strands from the two antibodies hybridize to form a circular DNA. This DNA can then be amplified and detected with fluorescent probes. As antibodies against synaptic proteins are widely available, this technique should be applicable for specific analysis of interactions in the synaptic protein interactome. A proof-of-principle experiment for high-content high-throughput analysis of protein–protein interaction using PLA on an automatic fluorescence microscope, cellomics, was recently demonstrated [31].
Reproducibility and overlap between studies
Interaction proteomics studies (Table 1) often generate useful protein–protein interaction information giving better insight into the regulation of the bait proteins. A recurring question is: how complete is the proteomics analysis? AMPA receptor research has provided a good test case. The AMPA receptors are the most studied synaptic proteins, because of their central role in controlling synapse function and plasticity. In recent months several novel AMPA receptor interactors have been reported, including cornichon [13], CKAMP44 [18] and GEF-H1 [17]. These proteins modulate the properties of AMPA receptor channel in distinct manners, and play important roles in synapse plasticity. Why have these new interactors not been detected previously? Cornichon is a small protein, <20 kDa, and may run together with the front on a 10% SDS-PAGE; small proteins are often ignored in routine analysis. In another study [18] not involving the use of SDS-PAGE, the total trypsin digest of the sample was fractionated by LC and analyzed by MALDI TOF–TOF MS. The MS mass range of 850–2700 Da covers only the mass of a single tryptic peptide of cornichon. This peptide was masked by other co-eluting peptides, and not characterized. CKAMP44 was identified with a number of peptides [18]. However, this novel protein is not annotated in SwissProt, and would not have been identified if this database had been used. GEF-H1 is another novel AMPA receptor interactor. The affinity isolation procedure [17] deviated from the other procedures in that it consisted of a wheat germ agglutinin chromatography step to enrich membrane proteins before they were subjected to large-scale immunoprecipitation. Furthermore, it is not unlikely that the co-localization of the AMPA receptor and GEF-H1 to the dendritic spine, and thus their interaction, is regulated by neuronal activity [32]. Finally, the differences may be because of trivial technical issues such as under-sampling in the mass spectrometer, and the interference of tags or epitopes required for enrichment with binding of members in the protein complex.
Towards global analysis of the synapse protein interactome
Synapses are rather simple, well-defined structures with probably two thousand different types of proteins present (excluding the synaptic mitochrondrial genome and specific posttranslational modifications). Considering the similar complexity of, for instance, yeast, a systems biology analysis of the synapse seems feasible. The prerequisite is that a large number of high-quality antibodies against synaptic proteins are readily available. Commercial antibodies are generally too expensive to be used for global co-immunoprecipitation experiments. Recently, a cooperative venture was established between the University of California at Davis, the National Institutes of Health, and Antibodies Inc. It now provides >200 low-cost, good quality monoclonal antibodies against diverse classes of synapse proteins. In a recent study, antibodies from this venture have been used to examine the protein complexes of PSD-95, CASK, KCNQ2, Kv4.2, CaVβ4, and GIT1 [9]. A simple synapse network was constructed, demonstrating the complexity of the protein interaction.
Large-scale proteomics analyses of synapse protein complexes are now in progress (the EU 7th-framework health, SynSys, and Eurospin initiatives). The workflow for the analysis is shown in Fig. 2. The objectives of this large-scale study spanning four years are to identify the protein network underlying synapse function and to translate this into a realistic dynamical model of the synapse. Certainly, a challenge in the coming years is the further development of data-analysis tools that include proper means of tagging identified protein–protein interactions with a probability or fidelity score, and the visualization tools that give biological insight of the (dynamics of) the synapse protein interactome.

Work flow for global analysis of synaptic protein complexes. Global analysis of the synaptic protein interactome requires hundreds of high-quality antibodies. We routinely use antibodies that are applicable for western blot analysis, and use this analysis for quality control of the IP experiment. IP is performed in detergent-solubilized samples, thus the efficiency of extraction of the protein of interest by the selected detergent must be checked beforehand. This step can be coupled with analysis of sub-cellular localization in term of enrichment of the protein in different synaptic sub-domains (for example the synaptosome, microsome, synaptic membrane, postsynaptic density, and lipid raft). Next, a test IP with Triton X-100-extracted complexes is performed followed by immunoblotting analysis to determine the recovery of the protein. On the basis of this result the immunoisolation procedure is optimized by changing the amounts of input material, antibody, and/or the protein A/G beads. Proteins enriched in the PSD and lipid rafts are partly, or not, soluble in buffers containing Triton X-100. These proteins are then extracted with stronger detergents (for example, DOC). After optimization, an IP followed by LC–MS–MS analysis is performed, with several biological replicates to estimate the sample variation
Outlook
Proteomics analyses of synapse protein complexes continue to identify the associated proteins; functional studies will answer how these proteins form a functional unit. The TAP technique will be applied to studies of several key synapse proteins. A major new development is the global analysis of synapse protein complexes, using hundreds of antibodies raised across different classes of synapse proteins. This will be supplemented by large-scale studies with BN-PAGE and proximity ligation immunocytochemistry to confirm protein–protein interactions. To develop interaction proteomics analysis to a new level of biological relevance one major focus will be to provide a dynamic model of the interactome based on challenges of the synapse network using well-defined stimuli (e.g. synaptic stimulation) and using a collection of mouse mutants with (null) mutations for synaptic genes. Experiments using biologically relevant stimuli will yield information on the dynamic behavior of the protein network and it will allow us to study the spatio-temporal pattern of the synaptic protein interactome rather than solely describing static protein interaction within a cell. These experiments will be crucial to understanding the meaning of protein complexes in the context of signal transduction and synaptic plasticity. In addition, interactome analysis of synaptic mutants will reveal changes in the network that can be used to provide models of synaptic (dys)function of human diseases. We expect that within five years a comprehensive synapse protein interactome will be constructed, and the dynamics of the protein complexes will have started to be mapped. This will provide insight into synapse function, and understanding of how synaptic complex dysfunction leads to various brain disorders.
References
Takamori S, Holt M, Stenius K, Lemke EA, Gronborg M, Riedel D, Urlaub H, Schenck S, Brugger B, Ringler P, Muller SA, Rammner B, Grater F, Hub JS, De Groot BL, Mieskes G, Moriyama Y, Klingauf J, Grubmuller H, Heuser J, Wieland F, Jahn R (2006) Cell 127(4):831–846
Burre J, Volknandt W (2007) J Neurochem 101(6):1448–1462
Phillips GR, Florens L, Tanaka H, Khaing ZZ, Fidler L, Yates JR 3rd, Colman DR (2005) J Neurosci Res 81(6):762–775
Li KW, Hornshaw MP, Van Der Schors RC, Watson R, Tate S, Casetta B, Jimenez CR, Gouwenberg Y, Gundelfinger ED, Smalla KH, Smit AB (2004) J Biol Chem 279(2):987–1002
Collins MO, Husi H, Yu L, Brandon JM, Anderson CN, Blackstock WP, Choudhary JS, Grant SG (2006) J Neurochem 97(Suppl 1):16–23
Abul-Husn NS, Bushlin I, Moron JA, Jenkins SL, Dolios G, Wang R, Iyengar R, Ma'ayan A, Devi LA (2009) Proteomics 9(12):3303–3315
Li KW, Miller S, Klychnikov O, Loos M, Stahl-Zeng J, Spijker S, Mayford M, Smit AB (2007) J Proteome Res 6(8):3127–3133
Fernandez E, Collins MO, Uren RT, Kopanitsa MV, Komiyama NH, Croning MD, Zografos L, Armstrong JD, Choudhary JS, Grant SG (2009) Molecular Systems Biology 5:269
Klemmer P, Smit AB, Li KW (2009) J Proteomics 72(1):82–90
Husi H, Ward MA, Choudhary JS, Blackstock WP, Grant SG (2000) Nat Neurosci 3(7):661–669
Wodak SJ, Pu S, Vlasblom J, Seraphin B (2009) Mol Cell Proteomics 8(1):3–18
Han MH, Lin C, Meng S, Wang X (2010) Mol Cell Proteomics 9(1):71–83
Schwenk J, Harmel N, Zolles G, Bildl W, Kulik A, Heimrich B, Chisaka O, Jonas P, Schulte U, Fakler B, Klocker N (2009) Science New York NY 323(5919):1313–1319
Zolles G, Wenzel D, Bildl W, Schulte U, Hofmann A, Muller CS, Thumfart JO, Vlachos A, Deller T, Pfeifer A, Fleischmann BK, Roeper J, Fakler B, Klocker N (2009) Neuron 62(6):814–825
Paulo JA, Brucker WJ, Hawrot E (2009) J Proteome Res 8(4):1849–1858
Zhang W, St-Gelais F, Grabner CP, Trinidad JC, Sumioka A, Morimoto-Tomita M, Kim KS, Straub C, Burlingame AL, Howe JR, Tomita S (2009) Neuron 61(3):385–396
Kang MG, Guo Y, Huganir RL (2009) Proc Natl Acad Sci USA 106(9):3549–3554
von Engelhardt J, Mack V, Sprengel R, Kavenstock N, Li KW, Stern-Bach Y, Smit AB, Seeburg PH, Monyer H (2010) Science 327:1518–1522
de Wit J, Sylwestrak E, O'Sullivan ML, Otto S, Tiglio K, Savas JN, Yates JR 3rd, Comoletti D, Taylor P, Ghosh A (2009) Neuron 64(6):799–806
Ogawa Y, Oses-Prieto J, Kim MY, Horresh I, Peles E, Burlingame AL, Trimmer JS, Meijer D, Rasband MN (2010) J Neurosci 30(3):1038–1048
Fukata Y, Tzingounis AV, Trinidad JC, Fukata M, Burlingame AL, Nicoll RA, Bredt DS (2005) J Cell Biol 169(3):399–404
Jing-Ping Z, Tian QB, Sakagami H, Kondo H, Endo S, Suzuki T (2005) Brain Res 135(1/2):204–216
Domon B, Aebersold R (2006) Science New York NY 312(5771):212–217
Kuzyk MA, Ohlund LB, Elliott MH, Smith D, Qian H, Delaney A, Hunter CL, Borchers CH (2009) Proteomics 9(12):3328–3340
Dayon L, Pasquarello C, Hoogland C, Sanchez JC, Scherl A. J Proteomics 73(4):769–777
Francesconi A, Kumari R, Zukin RS (2009) J Neurochem 108(6):1515–1525
Vandenberghe W, Nicoll RA, Bredt DS (2005) Proc Natl Acad Sci USA 102(2):485–490
Imoukhuede PI, Moss FJ, Michael DJ, Chow RH, Lester HA (2009) Biophys J 96(7):2949–2960
Massinen S, Tammimies K, Tapia-Paez I, Matsson H, Hokkanen ME, Soderberg O, Landegren U, Castren E, Gustafsson JA, Treuter E, Kere J (2009) Hum Mol Genet 18(15):2802–2812
Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, Landegren U (2006) Nature Methods 3(12):995–1000
Leuchowius KJ, Jarvius M, Wickstrom M, Rickardson L, Landegren U, Larsson R, Soderberg O, Fryknas M, Jarvius J (2010) Mol Cell Proteomics 9(1):178–183
Ryan XP, Alldritt J, Svenningsson P, Allen PB, Wu GY, Nairn AC, Greengard P (2005) Neuron 47(1):85–100
Acknowledgements
This study is supported by FP7 EU grant SYNSYS (nr. 242167)
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Li, K.W., Klemmer, P. & Smit, A.B. Interaction proteomics of synapse protein complexes. Anal Bioanal Chem 397, 3195–3202 (2010). https://doi.org/10.1007/s00216-010-3658-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-010-3658-z