
Abstract
Sulfonated polyether (ether) ketone or sulfonated PEEK (sPEEK) membranes are one possible candidate for proton-transfer membranes in hydrogen fuel cells. Reaction with hydroxy radicals is expected to be a significant source of degradation of these membranes during fuel cell operation. In this work, the reactivity of the sPEEK polymer molecule with OH radicals is studied by M062X hybrid density functional calculations of the energetics of several reaction paths in a water environment as modeled by polarized continuum model calculations. Reactants, products, encounter minima and transition states are optimized for a reaction pathway in which OH addition is followed by acid-catalyzed water elimination which cationizes the polymer, degradation is expected to follow this reaction as the unstable cation then undergoes bond-breaking or other reactions. Two pathways for this acid-catalyzed cationization, one in which a water molecule plays the role of an additional co-catalyst, are reported. Further calculations explore reaction pathways in which addition of OH to the polymer is followed by bond breaking reactions which would break the polymer chain or the bond between the polymer and sulfonyl groups. Examination of the free energy barriers to all these reactions, relative to reactants, suggests that these direct bond-breaking reactions may compete somewhat with acid-catalyzed water elimination following OH addition.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Fuel cells of particular interest for automotive applications are proton-exchange membrane fuel cells (PEMFCs) [1, 2]. A key component of such devices is the proton exchange membrane (PEM) [1], a semipermeable membrane which allows protons produced at the anode to migrate through to the cathode of the fuel cells [2]. Currently, proton exchange membranes are typically perfluoroalkylsulfonic acid (PFSA) membranes [1, 2], especially Nafion [2,3,4,5]. However, fluorinated membranes and fluorinated polymers in general present environmental hazards upon degradation [6,7,8].
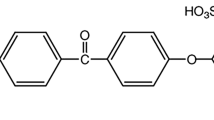
One of several proposed non-fluorinated alternatives to PFSA membranes is sulfonated aromatic membranes, such as the polymer sulfonated polyether(ether) ketone, or sulfonated PEEK (sPEEK) [2, 6, 7], displayed in Fig. 1.

Repeating unit of SPEEK polymer
Both PFSA and sulfonated aromatic membranes such as sPEEK are expected to be subject to degradation during fuel cell operation through reaction with OH [1, 7, 9, 10], H [1, 6, 9, 11], and OOH [1, 9, 11] radicals or H2O2 [12, 13] molecules. In the case of Nafion this degradation has been studied experimentally [9, 12, 14, 15] and there have also been recent computational works on the degradation reactions of Nafion with OH and H radicals [5, 16, 17] with H2O2 [12], and most recently with a hypothesized H3O radical [18].
The formation of OH radicals within a fuel cell, as stated in references [1, 5, 9], is believed to occur as a result of reactions resulting from the presence of O2, H2, and a platinum metal catalyst. Reference [8] notes that H2O2 may be formed from a reaction of O2 and H+ at the cathode, and that H2O2 molecules could react at the metal surface, or possibly on traces of transition metal cations, to produce two OH radicals.
Reference [5] presents computations of the barrier and energy of the reaction of H2O2 on a platinum catalyst surface to form OH radicals. In reference [5], the decomposition of H2O2 to form OH radicals is considered as one step in a multi-step mechanism for the production of OH radicals from reactions of H2 and O2 gases at the metal surface. This mechanism and a similar mechanism involving the production of an OOH intermediate (as opposed to H2O2) are noted to have low energetic barriers. The authors of reference [5] conclude that the metal surface plays an important role in production of OH radicals within fuel cells.
The degradation of sPEEK in the presence of OH radicals is expected to begin with the formation of an adduct of OH with aromatic rings within the polymer. Reference [1] notes that the rate of reaction of OH radicals with aromatic rings is extremely high, and estimates that the rate of OH reaction with the aromatic rings of sPEEK in a water-swollen coiled polymer would be orders of magnitude higher than the unzipping reactions of OH radicals in water-swollen Nafion. Reference [1] thus states that the aromatic rings will be subject to OH attack. Following formation of the adduct, an acid catalyzed water elimination reaction is expected to occur. This process is expected to proceed through of OH addition to the aromatic ring, followed by a protonation of the OH adduct, then elimination of water to leave a cationized aromatic ring [1, 10, 19, 20]. This degradation process for aromatic rings has been hypothesized by experimentalists attempting to study the degradation of sulfonated polymers in the presence of OH with EPR methods. Huber and Roduner [10] studied the reactions of a number of sulfonated aromatic model molecules with OH radicals. EPR experiments observe the formation of benzyl radicals in aqueous solution in the presence of acid. Reference [10] suggests this phenomenon initiates from an attachment of OH to the aromatic ring, followed by protonation, then followed by elimination of a water molecule to leave an aromatic cation. The benzyl radical is then formed by elimination of a proton from a methyl group attached to the aromatic ring.
This proposed mechanism is consistent with a mechanism reported for the production of benzyl radicals in reactions of toluene [19] and other methylbenzenes [20]. In the case of sPEEK, this process is expected to cationize the membrane [1]. In the case of sPEEK, there are no methyl groups attached to the aromatic rings, so the formation of benzyl radicals is not anticipated. Reference [1] states that cationization of the membrane is expected to lead to chain-breaking, cross-linking, or further hydroxylation reactions.
More recent experiments have used spin-trapping methods to explore the reactions of sPEEK with OH directly both in the in situ environment of an operating sPEEK-membrane hydrogen fuel cell [11] and in ex situ [21] experiments. Ex situ experiments found evidence of membrane degradation, detecting both phenoxy and phenyl radical products from sPEEK exposure to OH. In situ experiments observed no membrane degradation products; in reference [21], the authors note that run times for in situ experiments were short, and suggest such phenoxy and phenyl radical products would appear if longer fuel cell run times had been used.
Computational investigation of the degradation reactions of sPEEK in the presence of OH radicals has been reported in two works. Panchenko [7] has examined the thermodynamic feasibility of some reactions of non-fluorinated sulfonated aromatic membranes with OH radicals, focusing on model molecules for the polymers sPEEK and PSU (polyethersulfone). The calculations studied the thermodynamics of two types of reactions: abstraction of H atoms in sPEEK and PSU by OH groups, and also on the attachment of OH groups to some of the carbon atoms on the aromatic rings present, followed by reactions with O2 molecules. This work implemented the B3LYP functional with 6-31G(d) and 6-311 + G(d, p) basis sets and used the polarizable continuum model (PCM) to incorporate effects of solvation in water. In some cases, sulfonic acid groups were modeled in both protonated and deprotonated form.
The second work [6] studied the reactions of sPEEK with H radicals in a solvated environment with M062X/6-311 + G(2d,2p) optimizations, followed by M062X/6-311 + G(3df,2p) singlepoint calculations performed on optimized structures. To represent sPEEK, this work employed the model molecule sPEEK1, one of the model molecules originally implemented in reference 7. This molecule is displayed in Fig. 2. As seen in the figure, the carbon atoms of the aromatic ring are numbered 1–6. These carbon atoms are sites for radical attachment as well as proton attachment, as seen in reference 6 and in this work.

Model molecules for sPEEK
This study found that addition of H radicals to carbon atoms at sites 1 and 4 of SPEEK1 produces adduct structures in which the C–OCH3 linkage, the portion of the model corresponding to the ether bridge in the sPEEK polymer, becomes extremely fragile; in fact, breaking of these bonds following addition are found to be slightly exergonic processes. Transition state optimizations found that these bond breaking reactions have low free energy barriers; only 4–8 kcal/mol relative to the adducts, and below the relative free energy of the sPEEK1 + H reactants. Similarly, the addition of H radical to the carbon at site 6 produces an adduct structure in which C–S bond breaking is slightly exergonic and in which the barrier to bond breaking is small (~ 7 kcal/mol).
The addition–elimination reactions of sPEEK1 with H suggest similar reactions might occur upon OH addition to sites 1–6 in the aromatic ring of that molecule. In the case of addition at sites 1 and 4, the expected result is phenoxy radicals such as those detected in the ex situ spin trap experiments [21]. Addition of OH radicals at site 6 might also be expected to produce HSO3 radicals in the same fashion as addition of H radicals. It may be speculated that such reactions may compete with the acid-catalyzed elimination reaction in the phenoxy radicals.
One of the goals of this work is the computation of the reaction pathway, including thermodynamics and barrier heights, for OH addition followed acid-catalyzed water elimination in sPEEK as proposed in reference 1. The aromatic ring of the model molecules is thus assumed as the initial attack site. This work provides the first computational density-functional determination of this mechanism.
Another goal of this work is computation of barriers and thermodynamics for addition–elimination reactions analogous to those reported for sPEEK and H radicals in reference 6. These reactions to C–O chain breaking and C–S bond breaking reactions following addition of OH at sites 1,4 and 6. Thus, the aromatic ring is assumed as the initial site of OH attack for all reaction mechanisms in this work. With the computation of the thermodynamics of and barriers to these reactions, the relative competitiveness of addition–elimination reactions with respect to acid-catalyzed water elimination reactions may be assessed.
These computations employ solvated density functional M062X/6-311 + G(2d,2p) calculations of the potential energy surface of the reactions of sPEEK model molecules with OH radicals and H3O+, with singlepoint calculations with a larger basis set at all optimized geometries, to provide improved energetics. The chosen methods provide complete consistency with the calculations implemented within reference 6.
In this manuscript, all species optimized with these solvated density functional M062X/6-311 + G(2d,2p) calculations will be named in bold. Cartesian coordinates for all these structures are provided, in the order mentioned in this article, in the Supplementary Information for this article (see Online Resource 1).
The model molecules include SPEEK1 as noted above, as well as additional molecules SPEEK1PH1 and SPEEK1PH4 also displayed in Fig. 2. The latter two replace –OCH3 moieties at sites 1 and 4 with phenyl rings. Model molecules SPEEK1PH1 and SPEEK1PH4 provide more realistic polymer models and hence improved thermodynamics and barriers for the computations of C–O bond breaking reactions at sites 1 and 4. The following, second section of this work describes the methods of these calculations in detail. The third section, Results, describes the formation of sPEEK hydroxyl radicals and discusses all the optimized reaction pathways in detail, displaying energetics with respect to OH-sPEEK adduct and protonated OH-sPEEK adduct molecules. The fourth section, Discussion and Conclusion, then summarizes the results in terms of reactions on the sPEEK + OH + H3O+ potential energy surface, with one of the models SPEEK1, SPEEK1PH1 and SPEEK1PH4 assuming the role of sPEEK in calculation of the energetics.
2 Computational methods
The Gaussian 16 [22] electronic structure packages were used for all calculations.
As discussed above, reference 6 computed reactions of the sPEEK1 model molecule with H radicals in a solvated environment. This work calculates structures with M062X/6-311 + G(2d,2p) optimizations, with improved energetics provided by M062X/6-311 + G(3df,2p) singlepoint calculations performed on optimized structures. The recently developed M062X hybrid density functional of Zhao and Truhlar [23] was chosen because this functional was originally recommended for computation of systems where thermochemistry, kinetics, and noncovalent interactions are important [23, 24], making it suitable for the reaction thermodynamics and reaction barrier heights. It has been shown to provide good performance in calculating bond dissociation energies for reactions involving radicals [25]. It has very recently been implemented to calculate the reaction pathways, including transition states, of reactions of OOH and OH radicals in aqueous solution [26,27,28]
Within reference 6, additional calculations of the enthalpies of a number gas-phase reactions involving radicals also illustrated that M062X/6-311 + G(3df,2p) singlepoint calculations following M062X/6-311 + G(2d,2p) optimizations can very accurately reproduce the energetics of a number of reactions involving breaking of C–O and C–C bonds to form H radicals as well as radicals of carbon and oxygen atoms such as CH3, OCH3 and OH radicals.
Thermochemistry, kinetics, and noncovalent interactions are also important within this present work, and accuracy in the computed energetics of reactions involving OCH3 and OH radicals is also desirable. Therefore, the use of the functional and basis sets chosen in reference 6 continues in this work.
Unrestricted density functional M062X [23], calculations with the 6-311 + G(2d,2p) basis set (M062X/6-311 + G(2d,2p)) optimized all reactants, products, intermediates, and transition states, and determined reaction energetics. For the initial M062X/6-311 + G(2d,2p) calculations, frequency calculations characterized each structure as a minimum or transition state, and provided enthalpy and free energy corrections to the base energy. The connectivity of all transition states to reactants and products was determined by Intrinsic Reaction Coordinate (IRC) calculations.
Single-point M062X/6-311 + G(3df,2p) calculations at optimized geometries provided improved energetics. For M062X/6-311 + G(3df,2p)//M062X/6-311 + G(2d,2p) calculations, the enthalpy and free energy corrections are taken as those found by M062X/6-311 + G(2d,2p) frequency calculations. For all calculations, solvent (water) modeling is provided by the integral equation formalism polarized continuum model [29] (IEFPCM or PCM) as implemented in the Gaussian program. The free Avogadro [30] program was used to visualize optimized structures and also visualize computed imaginary vibrational modes in transition state structures.
The potential energy surface generated is thus compatible with recent work on the model sPEEK1 molecule with hydrogen atoms [6].
3 Results
3.1 Addition of OH to model molecule SPEEK1
The solvated M062X/6-311 + G(d,p) optimizations show that the addition of OH radical (OH) to sites 1–6 of model molecule SPEEK1 is thermodynamically spontaneous and occurs without reverse barrier. Table 1 shows the enthalpies and free energies of formation of adducts at each of the six sites. While enthalpies and free energies of addition of H radicals to SPEEK1 were shown to diverge by up to ~ 8.4 kcal/mol [6], the enthalpies and free energies for the formation of OH adducts are more similar, varying by only 2.4 kcal/mol in the case of the enthalpy and 1.4 kcal/mol in the case of the free energy. Table 1 also shows the enthalpy and entropy of addition provided by solvated B3LYP/6-311 + G(d) calculations as shown in reference 7. In general, the computations of this work predict more exothermic and exergonic additions than reference 7.
3.2 Elimination following addition of OH at site 1
The SPEEK1OH1 adduct forms at site 1 with enthalpies and free energies relative to SPEEK1 + OH as found in Table 1. A C–O bond breaking transition state SPEEK1OH1TS appears at a small enthalpy and free energy relative to the adduct, and connects the adduct to intermediate SPEEK1OH1-INT, at lower enthalpy and free energy to the adduct. This structure is a hydrogen-bonded complex of methanol to a molecule in which –OH replaces –OCH3 at site 1 and the H has been abstracted from the sulfonyl group at site 6, leaving an unpaired electron. The overall reaction leading to the separated products is slightly exothermic (− 3.4 kcal/mol) and exergonic (− 15.1 kcal/mol), as summarized in Table 2. Figure 3 displays optimized structures on the reaction pathway.

Optimized structures for elimination reaction following addition of OH to the carbon atom at site 1 of sPEEK1
The optimized structure for this radical is named SPEEK1OH1-P. The final products, methanol (CH3OH) and SPEEK1OH1-P are found without reverse barrier to the intermediate and are at an enthalpy of − 2.9 (− 3.4) relative to the adduct. Figure 3 displays optimized structures on the pathway of this reaction. The elimination/hydrogen abstraction process reported here is also shown to occur at site 1 following addition of H radicals at site 1 [6]; namely, the elimination process in that reaction also produces methanol and a species with an unpaired electron on the –SO3 group.
M062X/6-311 + G(2d,2p) calculations also computed the addition of OH to site 1 of SPEEK1PH1 to form SPEEK1PH1OH1, and determined a transition state SPEEK1PH1OH1TS for breaking of the C–O bond at site 1. As is also shown in Table 2, replacement of a methyl group for a phenyl group at site 1 of the model molecule produces a transition state with an enthalpy of only 3.7 kcal/mol relative to the reactant, which is 1.0 kcal/mol lower than in the case of the methyl substituent. The free energy of the transition state is also lowered by phenyl replacement of the methyl group, 3.7 kcal/mol as opposed to 4.7 kcal/mol. Phenyl replacement at site 1 might be anticipated to produce a similar lowering of barriers by ~ 1.0 kcal/mol to reaction for the bond-breaking reaction following addition of H radical at site 1 of SPEEK1 as described in reference 6. The barrier relative to the sPEEK-H would then be 6.3, rather than 7.3, kcal/mol [6].
IRC calculations for this transition state confirm the connectivity of SPEEK1PH1OH1TS–SPEEK1PH1OH1 in one direction of the reaction; in the other direction, IRC calculations connect the transition state to SPEEK1PH1OH1-INT. This structure is a hydrogen-bonded complex of separated products phenol (PHENOL) and SPEEK1OH1-P.
Table 2 summarizes the binding enthalpy and free energy of the OH adduct to SPEEK1 and SPEEK1PH1, as well as the relative enthalpy and free energy of the transition state and products.
3.3 Elimination following addition of OH at site 4
Table 1 shows the enthalpy and free energy of formation of the SPEEK1OH4 adduct from SPEEK1 + OH. Energetics of structures for a C–O bond breaking reaction are shown in Table 3. A C–O bond breaking transition state SPEEK1OH4TS appears at an enthalpy of 19.3 kcal/mol and free energy of 18.9 kcal/mol relative to the adduct. This transition state lies at a much higher enthalpy or free energy relative to the adduct than is the case for bond breaking at site 1.
IRC calculations connect SPEEK1OH4TS–SPEEK1OH4 in one direction and to SPEEK1OH4-INT in the other, the latter is an intermediate complex in which an OCH3 radical is hydrogen-bonded to the sulfonyl hydrogen on the phenolic product molecule. Intermediate SPEEK1OH4-INT lies at an enthalpy of ~ 5.0 kcal/mol relative to the adduct. As indicated by the structure of the intermediate exit complex, no hydrogen abstraction from the sulfonyl group takes place during the addition–elimination reaction at site 4, and the final products include a CH3O radical (CH3O) and a phenolic molecule in which OH has replaced OCH3 substituent at site 4 (SPEEK1OH4-P). The final separated products SPEEK1OH4-P + methoxy (CH3O) are found at an enthalpy of 9.7 relative to the adduct; G is − 2.1 relative to the adduct, for a slightly exergonic reaction.
M062X/6-311 + G(2d,2p) calculations also computed the addition of OH to site 1 of SPEEK1PH4 to form SPEEK1PH4OH4, and determined a transition state SPEEK1PH4OH4TS for breaking of the C–O bond at site 4. IRC calculations were carried out to establish the connectivity of this transition state to SPEEK1PH4OH4 in one direction and to sPEEK1PH4OH4-INT, a hydrogen bonded complex of the final products, PHENOXY and SPEEK4OH4-P.
Table 3 summarizes the binding enthalpy and free energy of the OH adduct to site 4 of SPEEK1 and SPEEK1PH4, the relative enthalpy and free energy of the transition state and products. Figure 4 displays optimized structures on the reaction pathway.

Optimized structures for reactions following OH addition to sPEEK1 at site 4 (top) and site 6(bottom)
Table 3 shows that substitution of phenyl for the OCH3 moiety at site 4 lowers the enthalpy and free energy barriers represented by the transition state by ~ 10 kcal/mol. Phenyl replacement at site 4 might be anticipated to produce a similar lowering of barriers by ~ 10 kcal/mol to reaction for the bond-breaking reaction following addition of H radical at site 4 of SPEEK1 as described in reference 6. The free energy barrier to C–O bond breaking relative to the sPEEK-H adduct would then be lowered from 15.8 kcal/mol [6] to merely 5 kcal/mol.
3.4 Elimination following addition of OH at site 6
3.4.1 Elimination of the HSO3 radical
Table 1 shows that the formation of the SPEEK1OH6 adduct from SPEEK1 + OH is exothermic and exergonic. The enthalpy and free energy of the bond breaking transition state SPEEK1OH6TS are displayed in Table 4. IRC calculations connect this transition state to SPEEK1OH6 in one direction and to a hydrogen-bonded intermediate SPEEK1OH6-INT in the other. This intermediate is as an HSO3 radical hydrogen bonded to the OH group on the phenolic product molecule. The separated products are the HSO3 radical HSO3 and the phenol product SPEEK1OH6-P. The C–S bond breaking reaction is slightly exothermic and occurs spontaneously. The barrier to this reaction is very small relative to SPEEK1OH6, as noted in Table 4. This result is similar to the calculated free energy of the barrier to C–S breaking relative to the adduct formed when an H radical attached at site 6; in that case, the free energy barrier was found to be only 2.4 kcal/mol [6]. Figure 4 displays optimized structures on the reaction pathway.
3.4.2 Thermodynamics of other elimination reactions following OH addition at site 6
Reference 6 suggests that in the case of OH radical addition at site 6, H2SO4 (H2SO4) elimination might occur; the other product would be the sPEEK molecule in which the C–S bond at site 6 is broken, leaving an unpaired electron at C6; this molecule is referred to as sPEEK1OH6-P2. Additional reactions following OH addition at site 6 might involve the production of HSO4 (HSO4) producing a sPEEK1 molecule in which –SO3H is replaced by –H, referred to as sPEEK1OH6-P3. Another possibility would be elimination of H2SO3 (H2SO3), the other product being a sPEEK1 molecule in which a phenoxy radical replaces –SO3H at site 6, referred to as sPEEK1OH6-P4. Table 5 summarizes the enthalpy and free energy of the reactions discussed above.
3.5 Protonation and water elimination following OH addition at site 3
3.5.1 Structure and thermodynamics of protonated sPEEK1OH3 complexes; partial charge of likely protonation sites for sPEEK-OH adducts
Site 3 of SPEEK1 is chosen for computational investigation of the water elimination oxidative process suggested for the degradation of sPEEK membranes [1, 10]. Table 1 notes that the SPEEK1OH3 moiety forms spontaneously and without barrier. The M062X/6-311 + G(2d,2p) geometry optimizations do not locate a minimum corresponding to addition of a proton to the hydroxyl oxygen attached to site 3. The nearest sites for protonation to the hydroxyl group are the carbons at site 2 (SPEEK1OH3H2+) and site 4 (SPEEK1OH3H4+). Protonation at these sites (corresponding to transfer of a proton from an optimized solvated hydronium, H3O+ ) to form the protonated moiety and an optimized solvated water molecule (H2O) is both exothermic and exergonic in the case of site 3, as shown in Table 6. In contrast, addition to site 4 is slightly endothermic and endergonic, as also shown in Table 6.
The difference in the free energy of protonation may be a function of the partial Mulliken charge on the aromatic carbons adjacent to the site of attachment of the OH adduct. The optimized M062X/6-311 + G(2d,2p) SPEEK1OH3 structure exhibits a Mulliken charge of − 0.16453 on site number 2 and a Mulliken charge of + 0.316640 on site 5. The exergonic addition of proton at site 2 may be a function of the partial negative charge at site 2 to form SPEEK1OH3H2+ , while the partial positive charge at site 4 may correspond to the endergonicity of protonation at that site.
Table 7 summarizes the partial charges on adjacent aromatic carbons for adducts sPEEK1OH1 through sPEEK1OH6. All adducts other than sPEEK1OH2 exhibit a negative Mulliken charge on at least one aromatic carbon adjacent to the attachment site of the OH radical. An additional solvated M062X/6-311 + G(2d,2p) calculation of the OH adduct of benzene, BZ-OH, provides a partial negative charge of − 0.101056 on sites 2 and 6, the sites adjacent to the attachment site (referred to as site 1).
This work presents acid-catalyzed water elimination reactions occurring following protonation of sPEEK1OH3 at site 2.
3.5.2 Barrier to protonation of sPEEK1OH3
The mechanism of the acid catalyzed elimination reaction studied in this work requires that the SPEEK1OH3 hydroxylated aromatic ring is protonated by a hydronium molecule. The protonation of aromatic rings by hydronium has been the subject of theoretical investigation [31]. Earlier gas-phase ab initio calculation [31] of the benzene-hydronium potential energy surface finds that a transition state for protonation connects a benzene-hydronium encounter complex to an exit complex for protonated benzene to water. The energy change from separated reactants to separated product is found to be − 10.5 kcal/mol, and the transition state for the protonation process is found to be energetically lower than both the reactant and the product, connecting in one direction to an encounter complex of hydronium and benzene, and in the other direction to an exit complex of protonated benzene and water.
M062x/6-311 + G(2d,2p) optimizations locate an encounter complex of SPEEK1OH3 with the hydronium molecule (H3O+), PEC-1. (see Fig. 5). A protonation transition state from PEC-1 to a complex of water and SPEEK1OH3H2+ is not presented in this work. A series of constrained geometry optimizations [32] in which the C–H internuclear distance shown in Fig. 6 is fixed at values between 1.66811 angstroms, the optimized distance C–H distance for structure SPEEK1OH3H2+ , and values near 1 angstrom. All other nuclear coordinates are permitted to optimize. The resulting energies from these calculations are plotted in Fig. 6. Online Resource 2 of the Supplementary Information provides a table displaying the C–H internuclear distances and energies of the resulting structures relative to PEC-1. Online Resource 2 also provides Z-matrices for the structures resulting from the constrained optimizations.

Optimized structures of hydronium-SPEEK1 encounter complex PEC-1 and sPEEK1 + -water exit complex PEX-1. The C–H separation in PEC-1 is 1.668 angstroms

Plot of electronic energy as a function of C–H separation. The top shows the maximum at 1.65000 angstroms relative to PEC-1, the bottom plot shows the progression of energies from 1.66811 angstroms to 1.09569 angstroms (PEC-1 to PEX-1)
Figure 6 displays an extremely flat potential energy surface. Of the points plotted, the maximum potential energy relative to PEC-1 is found at a C–H distance of 1.65000; the relative energy is only ~ 0.07 kcal/mol greater than PEC-1. At distances smaller than 1.65000, the potential energy decreases to values below the electronic energy of PEC-1. The smallest C–H distance for constrained optimization represented in this figure is 1.14235 angstroms; when this geometry is made a starting point for a full optimization, a complex of SPEEK1OH3H2+ and water is optimized. This complex, PEX-1, has a C-H internuclear separation of only 1.09569 angstroms. The electronic energy is 21.7 kcal/mol lower than that of PEC-1.
Figure 6 suggests that little or no energetic barrier exists to protonation of SPEEK1OH3.
3.5.3 Elimination of water
SPEEK1OH3H2+ may eliminate water to form a cationized SPEEK1 molecule (SPEEK1+). Elimination occurs through the transition state H2O-ETS as displayed in Fig. 7. IRC calculations establish the connectivity of this transition state to SPEEK1OH3H2+ and to the SPEEK1 cation SPEEK1+ and a water molecule in the other. The thermodynamics of the reaction pathway are displayed in Table 8. As seen in Table 8, the barrier to the reaction is extremely high relative to the protonated hydroxyl-sPEEK complex.

Structures of protonated sPEEK-hydroxy adduct SPEEK1OH3H2+ , and transition states, intermediates, and product for water elimination reactions
Water elimination after protonation may also occur via a second process involving a reaction of SPEEK1OH3H2+ with an explicit water molecule; note that this process lies on the potential energy surface of the reactions of sPEEK model molecules with OH radicals and H3O+.
The resulting reaction may be written.
SPEEkK1OH3H2+ + H2O → SPEEK1+ + 2H2O.
The transition state H2O-ETS-2 connects to a hydrogen-bonded complex of the reactants, water and SPK1OH3H2+ , referred to as WEC-1. In the other direction, this transition state connects to minimum WEC-2, which exhibits one water molecule hydrogen bonded to a second water; the second water interacts with an SPEEK1+ molecule via a donative interaction between a lone pair on the water and the half-empty aromatic bonding orbital on the cation. This is a metastable structure with enthalpy and free energy greater than the final products; the final products can be reached by transition state WEC-2-TS, which connects WEC-2 to a hydrogen bonded exit complex of a SPEEK1 cation and two water molecules, referred to as WEX-1. The separated products may be reached from WEX-1 lie at an enthalpy of − 8.2 and free energy of − 9.1 relative to SPK1OH3H2+ + H2O. Figure 7 displays optimized structures on the reaction path. The energetics of this reaction relative to SPEEK1OH3H2+ + H2O are displayed in Table 9.
While hydronium catalyzes this elimination reaction by protonation, the water molecule acts as the co-catalyst for hydrogen atom transfer from a carbon atom to the OH group. The participation of water as a co-catalyze produces a relative barrier of 23.1 kcal/mol versus 39.4 kcal/mol, or a lowering of the barrier by 16.3 kcal/mol. A similar lowering of a hydrogen-transfer barrier appears in a computational study of the transfer of a hydrogen atom from sulfur to oxygen within the thioformic acid molecule in an gas-phase environment [33]; here, a barrier to transfer of 33 kcal/mol is lowered by 21 kcal/mol when a water molecule is included to facilitate the transfer.
4 Discussion and conclusion
The thermodynamic formulation of transition state theory [34, 35] expresses rates of reaction in terms of the equilibrium between reactants and transition state, typically presenting rate constants which depend exponentially on the negative of the free energy difference between reactants and transition state. In this formulation, the lower the free energy of a transition state is, relative to the reactants, the faster the reaction might be expected to proceed.
Figure 8 displays a reaction energy diagram for the reactions discussed in this work. In this diagram, M062X/6-311 + G(3df,2p)//M062X/6-311 + G(2d,2p) free energies of optimized structures are reported relative to the M062X/6-311 + G(3df,2p)//M062X/6-311 + G(2d,2p) free energy of reactants, which consist of OH, H3O+ , and one of the model molecules presented in Fig. 2. For reactions following addition at site 6, and for the acid-catalyzed water elimination reactions, sPEEK1 is the model molecule; in the case of the chain-breaking reactions at site 1 or 4, results are presented relative to the corresponding phenyl-substituted species sPEEK1PH1 or sPEEK1PH4, respectively.

Reaction energy diagram showing M062X/6-311 + G(3df,2p)//M062X/6-311 + G(2d,2p) free energies for optimized structures in this work. Optimized structures are shown as numbers, a key to which number corresponds to which structure appears at the bottom of the diagram. All energetics shown are M062X/6-311 + G(3df,2p)//M062X/6-311 + G(2d,2p) free energies relative to the M062X/6-311 + G(3df,2p)//M062X/6-311 + G(2d,2p) free energy of reactants, which consist of OH, H3O+ , and sPEEK*, where sPEEK* is one of the model molecules presented in Fig. 2. This appears at the point of 1 in the diagram. Structures for the addition–elimination reaction following OH addition at site 1 appear as structures 2–5, here the identity of sPEEK* for the purpose of relative energetics is sPEEK1PH1. Structures 6–9 are optimized structures corresponding to the elimination reaction following OH addition at site 4, here the identity of sPEEK* for the purpose of relative energetics is sPEEK1PH4. Structures 10–13 are optimized structures corresponding to the elimination reaction following the addition of OH at site 6, here the identity of sPEEK* for the purpose of relative energetics is sPEEK1. Structures on the pathway for acid-catalyzed water elimination following OH addition at site 3 to form sPEEK1OH3 (14) appear as 14,15,16, and 17, the product sPEEK1+ + 2H2O; structures 18–22 are optimized structures for acid-catalyzed water elimination with water co-catalyst, also leading to 17, the product sPEEK1+ + 2H2O. The addition of a proton from H3O+ to sPEEK1OH3 to form sPEEK1OH3H2+ and H2O is shown as barrierless as discussed in Sect. 3.5.2
Table 10 summarizes computed M062X/6-311 + G(3df,2p)//M062X/6-311 + G(2d,2p) free energies of products and transition states for reactions studied in this work. As in Fig. 8, all energetics in this table are reported relative to the free energy of reactants, which consist of OH, H3O+ , and one of the model molecules presented in Fig. 2. Again, sPEEK1 is the model molecule for reactions following addition at site 6 and for the acid-catalyzed water elimination reactions; for the chain-breaking reactions at site 1 or 4, results are presented relative to sPEEK1PH1 or sPEEK1PH4, respectively.
This work presents the first density functional computation of the acid-catalyzed water elimination reaction of hydroxyl radicals with the sPEEK polymer. This reaction, like all others computed in this work, proceeds from the addition of an OH radical to a carbon on the aromatic ring of the polymer. Figure 8 and Table 10 show that the reaction is the most exergonic of the reactions studied by at least 14.4 kcal/mol. However, direct elimination of water after protonation at an aromatic carbon adjacent to the hydroxy addition site must proceed through a transition state with a free energy greater than that of the reactants by 7.5 kcal/mol. The participation of a water molecule acting as a co-catalyst to effect the transfer of the H atom from the adjacent carbon creates a much lower barrier to the reaction; the free energy of the transition state with the water co-catalyst is 8.8 kcal/mol lower than the sPEEK1 + OH + H3O+ reactants.
The main chain of the sPEEK polymer contains ether-bridged aromatic rings; some of which contain sulfonyl groups for the purpose of proton exchange. Chain breaking elimination reactions would be predicted to result from the addition of OH radicals to aromatic carbons bonded to the oxygen atoms of the ether bridge. In the event that no -SO3H groups are adjacent to these carbons, the best model for the addition–elimination reaction is provided by the computed reaction pathway for the reaction of OH with sPEEK1PH4. While the chain breaking reaction is exergonic, the free energy of the transition state for this reaction is only slightly lower than that of the reactants at − 1.7 kcal/mol. If an adjacent –SO3H group is present, a chain-breaking reaction may occur with transfer of an H atom from –SO3H to the oxygen atom; the best model for that process in this work is the computed reaction of OH with sPEEK1PH1; the free energy relative to reactants of the transition state for this reaction, is substantially lower, − 10.1 kcal/mol. Addition elimination reactions producing –SO3H are modeled by the addition of OH to sPEEK1 at site 6; this also has a low relative free energy, − 11.3 kcal/mol.
The acid-catalyzed water elimination reaction, co-catalyzed by water, has a transition state with a low free energy relative to reactants. Calculations of protonated adducts of SPEEK1OH3 have produced results suggesting that the exergonicity of protonation of sPEEK-OH adducts adjacent to the site of OH attachment may correspond to negative partial charges on the adjacent carbon atoms. The distribution of partial charges in OH adducts displayed in Table 7 would then suggest that many aromatic carbon sites on the sPEEK polymer, including those with no sulfonyl groups in the aromatic ring (as modeled by BZ-OH), are likely sites for OH addition followed by protonation at an adjacent carbon atoms with a negative partial charge. The reaction hence might be expected to be a large fraction of the degradation reactions of the sPEEK polymer with OH. Direct addition–elimination chain breaking reactions may only happen at sites where aromatic carbons attach to bridging ether oxygens, and the free energy of the barrier to reaction is greater than that of reactants; this might be expected to be an extremely small fraction of such degradation reactions.
Reactions such as the one reported for sPEEK1 and sPEEK1PH1, in which a hydrogen atom is transferred from a sulfur to an oxygen, or addition elimination reactions producing –SO3H, following addition at site 6, have very low free energy barriers, lower than that of the acid-catalyzed, water co-catalyzed elimination reaction, and these reactions might be expected to compete somewhat with a predominant acid-catalyzed, water co-catalyzed elimination reaction.
Additional computational study might further elucidate mechanisms for other degradation reactions. The absence of observation of HSO3 product in the experiments described in references [10, 11] is puzzling in light of the extremely low free energy for the barrier to formation of this product following addition of OH radicals to site 6 reported in this work and in the case of H radicals as discussed in reference 6. One possible explanation may be the presence of a competing reaction that produces oxo-acids of sulfur; these closed-shell sulfur compounds would be undetectable by EPR or ESR methods [10, 11] implemented in the experiments described.
In addition for the reaction producing HSO3 after addition at site 6, Table 10 includes the possible elimination reactions at site 6 as presented in Table 5. Transition states for these reactions are not reported in this work. H2SO3 production, however, is highly exergonic with respect to sPEEK1OH6. The free energy barrier must be at least equal to the relative free energy of sPEEK1OH6, which, as seen in Table 5, is − 12.6 kcal/mol relative to the sPEEK + OH + H3O+ reactants. The products are H2SO3, which as a closed-shell molecule would not be detected by electron resonance experiments, and a phenoxy radical; phenoxy radicals are observed in the experiments in degradation involving sPEEK discussed in reference 11. Future work will investigate the transition state to H2SO3 elimination reactions following addition at site 6.
As seen in Table 5, the reactions producing HSO4 and H2SO4 are endergonic with respect to sPEEK1OH6, meaning that free energy barriers to these reactions relative to the reactants must be at least as high as the free energies of the products. Therefore, reactions that produce closed-shell H2SO4 or open-shell HSO4 might be considered uncompetitive as these free energies of products are higher than the free energy barriers reported in Table 10.
While uncompetitive, more computational information on possible reactions producing H2SO4 may assist in providing more complete information on possible degradation reactions of sPEEK and OH radicals. H2SO4 might be produced in a reaction arising from attack of the S atom by the OH radical. In the case of Nafion, computations showed that OH attack on the S atom of the HSO3 moiety led to a reaction producing H2SO4 [5]. This reaction was found to have a ΔE of − 23 kcal/mol/, the barrier to the reaction was found to be 18.7 kcal/mol relative to the reactants. In the case of sPEEK, the analogous reaction producing H2SO4 and the radical product sPEEK1OH6-P2 is shown in Table 10 to be only mildly exergonic relative to the sPEEK + OH + H3O + reactants. The free energy of any barrier to this possible minor product, as noted above, is necessarily greater than the free energy of the products, but its relative energy with respect to the reactants remains unknown.
The solvated B3LYP calculations discussed in reference 7 find endergonic H-abstraction reactions for the reaction of OH with sPEEK1. Specifically, free energies of H abstraction from site 2 of sPEEK by OH to form water and sPEEK1 radicals are found to have values of approximately − 25 or − 39 kcal/mol, with the more exergonic value occurring in the case of deprotonated sPEEK. H abstraction from the –SO3H group by OH radicals might also occur, possibly with a low barrier to reaction; Reference 5 notes that in the case of Nafion this abstraction reaction is found to be barrierless and exothermic (ΔE = − 5.3 kcal/mol). Future work will confirm the exergonicity of these reactions in neutral and anionic species, as well as determining transition states and barrier heights for H-abstraction reactions.
References
Gubler L, Nauser T, Coms FD, Lai YH, Gittleman CS (2018) Prospects for durable hydrocarbon-based fuel cell membranes. J Electrochem Soc 165:F3100–F3103. https://doi.org/10.1149/2.0131806jes
Fan L, Tu Z, Chan SH (2021) Recent development of hydrogen and fuel cell technologies: a review. Energy Rep 7:8421–8446. https://doi.org/10.1016/j.egyr.2021.08.003
Kundu PP, Dutta K. (2016) Hydrogen fuel cells for portable applications. In: Compendium of hydrogen energy, vol. 4, Hydrogen Use, safety and the hydrogen economy, Chapter 6, pp 111-131, https://doi.org/10.1016/B978-1-78242-364-5.00006-3
Powering Fuel Cells with Nafion™ Membranes. (2023) Nafion. https://www.nafion.com/en/applications/fuel-cells. Accessed 21 Jan 2023
Yu TH, Sha Y, Liu WG, Merinov BV, Shirvanian P, Goddard WA (2011) Mechanism for degradation of nafion in PEM fuel cells from quantum mechanics calculations. J Am Chem Soc 133:19857–19863. https://doi.org/10.1021/ja2074642
Stevens JE, Utterbeck KD, Piatkowski A, Spicer MN (2018) Density functional theory investigation of mechanisms of degradation reactions of sulfonated PEEK membranes with H radicals in fuel cells: addition–elimination bond-breaking reactions in a model molecule. Theor Chem Acc 137:105. https://doi.org/10.1007/s00214-018-2281-5
Panchenko A (2005) DFT investigation of the polymer electrolyte membrane degradation caused by OH radicals in fuel cells. J Membr Sci 278:269–278. https://doi.org/10.1016/j.memsci.2005.11.010
Lohmann R, Cousins IT, DeWitt JC, Glüge J, Goldenman G, Herzke D, Lindstrom AB, Miller MF, Ng CA, Patton S, Scheringer M, Trier X, Wang Z (2020) Are fluoropolymers really of low concern for human and environmental health and separate from other PFAS?: environ. Sci Technol 54:12820–12828. https://doi.org/10.1021/acs.est.0c03244
Danilczuk M, Coms FD, Schlick S (2009) Visualizing chemical reactions and crossover processes in a fuel cell inserted in the ESR resonator: detection by spin trapping of oxygen radicals, nafion-derived fragments, and hydrogen and deuterium atoms. J Phys Chem B 113:8031–8042. https://doi.org/10.1021/jp901597f
Huber G, Roduner H (1998) EPR investigation of HO· radical initiated degradation reactions of sulfonated aromatics as model compounds for fuel cell proton conducting membranes. J Mater Chem 9:409–418. https://doi.org/10.1039/A807129B
Danilczuk M, Schlick S, Coms FD (2013) Detection of radicals by spin trapping ESR in a fuel cell operating with a sulfonated poly(ether ether ketone) (SPEEK) membrane. Macromolecules 46:6110–6117. https://doi.org/10.1021/ma401188u
Kadirov MK, Bosnjakovic A, Schlick S (2005) Membrane-derived fluorinated radicals detected by electron spin resonance in UV-irradiated nafion and dow ionomers: effect of counterions and H2O2. J Phys Chem B 109:7664–7670. https://doi.org/10.1021/jp044987t
Tsuneda T, Singh RK, Iiyama A, Miyatake K (2017) Theoretical investigation of the H2O2-induced degradation mechanism of hydrated nafion membrane via ether-linkage dissociation. ACS Omega 2:4053–4064. https://doi.org/10.1021/acsomega.7b00594
Ghassemzadeh L, Kreuer KD, Maier J, Muller K (2010) Chemical degradation of nafion membranes under mimic fuel cell conditions as investigated by solid-state NMR spectroscopy. J Phys Chem C 114:14635–14645. https://doi.org/10.1021/jp102533v
Bosnjakovic A, Schlick S (2004) Nafion perfluorinated membranes treated in Fenton media: radical species detected by ESR spectroscopy. J Phys Chem B 108:4332–4337. https://doi.org/10.1021/jp037519c
Coms FD (2008) The chemistry of fuel cell membrane chemical degradation. ECS Trans 16(2):235–255. https://doi.org/10.1149/1.2981859
Ishimoto T, Koyama M (2012) A review of molecular-level mechanism of membrane degradation in the polymer electrolyte fuel cell. Membranes 2:395–414. https://doi.org/10.3390/membranes2030395
Long LC, Coms F, Pivovar B, Dalhke G, Yandrasits M (2022) Role of H3O· radical in the degradation of fuel cell proton-exchange membranes. ACS Phys Chem Au 2:527–534. https://doi.org/10.1021/acsphyschemau.2c00037
Christensen HC, Sehested K, Hart EJ (1973) Formation of benzyl radicals by pulse radiolysis of toluene in aqueous solutions. J Phys Chem 7:983–987. https://doi.org/10.1021/j100627a003
Sehested K, Corfitzen H, Christensen HC, Hart EJ (1975) Rates of reaction of O–, OH, and H with methylated benzenes in aqueous solution. optical spectra of radicals. J Phys Chem 79:310–315. https://doi.org/10.1021/j100571a005
Pinteala M, Schlick S (2009) Direct ESR detection and spin trapping of radicals generated by reaction of oxygen radicals with sulfonated poly(ether ether ketone) (SPEEK) membranes. Polym Degrad Stab 94:1779–1787. https://doi.org/10.1016/j.polymdegradstab.2009.06.009
Gaussian 16, Revision C.01, Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich AV, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark MJ, Heyd JJ, Brothers EN, Kudin KN, Staroverov VN, Keith TA, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ (2016) Gaussian, Inc., Wallingford CT
Zhao Y, Truhlar D (2008) Density functionals with broad applicability in chemistry. Acc Chem Res 41:157–167. https://doi.org/10.1021/ar700111a
Zhao YT, DG, (2008) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testingof four M06-class functionals and 12 other functionals. Theor Chem Acc 120:215–241. https://doi.org/10.1007/s00214-007-0310-x
Zhao Y, Truhlar DG (2008) How well can new-generation density functionals describe the energetics of bond-dissociation reactions producing radicals? Acc Chem Res 41:157–167. https://doi.org/10.1021/ar700111a
Hoa NT, Van LTN, Vo Q (2022) The hydroperoxyl antiradical activity of natural hydroxycinnamic acid derivatives in physiological environments: the effects of pH values on rate constants. RSC Adv 12:15115–15122. https://doi.org/10.1039/D0NJ01567A
Milanović Ž, Dimić D, Žižić M, Milenković D, Marković Z, Avdović E (2021) Mechanism of antiradical activity of newly synthesized 4,7-dihydroxycoumarin derivatives-experimental and kinetic DFT study. Int J Mol Sci 22:13273. https://doi.org/10.3390/ijms222413273
Boulebd H, Mechler M, Hoa NT (2020) Thermodynamic and kinetic studies of the antiradical activity of 5-hydroxymethylfurfural: computational insights. New J Chem 44:9863–9869. https://doi.org/10.1039/D0NJ01567A
Miertuš S, Scrocco E, Tomasi J (1981) Electrostatic interaction of a solute with a continuum. A direct utilization of AB initio molecular potentials for the prevision of solvent effects. Chem Phys 55:117–129. https://doi.org/10.1016/0301-0104(81)85090-2
Avogadro: an open-source molecular builder and visualization tool. Version 1.2.0 (2023) http://avogadro.cc/. Accessed 21 Jan 2023
Kryachko ES, Nguyen MT (2001) Low energy barrier proton transfer in protonated benzene-water complex. J Phys Chem A 105:153–155. https://doi.org/10.1021/jp001956z
Schlegel BH (2011) Geometry optimization WIREs. Comput Mol Sci 1:790–809. https://doi.org/10.1002/wcms.34
Daurete F, Torro-Labe A (2010) The catalytic effect of water on the keto-enol tautomerisation reaction of thioformic acid. Mol Phys 108:1375–1384. https://doi.org/10.1080/00268971003698064
Steinfeld JI, Francisco JS, Hase WL (1989) Chemical kinetics and dynamics. Prentice-Hall, p 322
Laidler KJ, King CM (1983) The development of transition state theory. J Phys Chem 87:2657–2664. https://doi.org/10.1021/j100238a002
Acknowledgements
This work was supported by the National Institutes of Health Common Fund and Office of Scientific Workforce Diversity under three linked awards R5GM118981, TL4GM118983, and 1UL1GM118982 administered by the National Institute of General Medical Sciences. This work was much assisted by helpful discussions with the late Dr. Shulamith Schlick and the late Dr. Marek Danilczuk.
All authors declare no competing interest.
Author information
Authors and Affiliations
Contributions
JES wrote and prepared the manuscript text. Results within the manuscript are the results of computations made by JES, CMP, AP, ZRS, and NO.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stevens, J.E., Pefley, C.M., Piatkowski, A. et al. Density functional theory investigation of mechanisms of degradation reactions of sulfonated PEEK membranes with OH radicals in fuel cells: addition–elimination reactions and acid catalyzed water elimination. Theor Chem Acc 142, 49 (2023). https://doi.org/10.1007/s00214-023-02981-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-023-02981-2