
Abstract
Determining the charge on an atom in molecules and materials is a long-standing open issue. Quantitative analysis with clear physical picture of charge transfer, electrostatic and covalent contributions to bond energy is highly desirable to advance the understanding of both charge and ionicity. However, the consensus among computations from quantum mechanics to semiempirical methods and state-of-the-art experimental measurement on fractional charge remains elusive. In this work, we generalize Born–Haber thermochemical cycle to compute fractional charge on an atom by delicately balancing the interplay of all interacting components, including charge transfer, electrostatic–covalent interactions and bond energy in molecules. The fractional charge of an atom in solid materials can be also computed straightforwardly with this scheme.


Similar content being viewed by others

Change history
25 June 2020
In the original publication of the article, in the Appendix section, under the heading.
References
Tokura Y, Nagaosa N (2000) Orbital physics in transition-metal oxides. Science 288:462–468
Torrance JB, Metzger RM (1989) Role of the Madelung energy in hole conductivity in copper oxides: difference between semiconductors and high-Tc superconductors. Phys Rev Lett 63:1515–1518
Ohta Y, Tohyama T, Maekawa S (1991) Charge-transfer gap and superexchange interaction in insulating cuprates. Phys Rev Lett 66:1228–1231
Chen XJ, Su HB (2005) Electronic mechanism of critical temperature variation in RBa2Cu3O7-δ. Phys Rev B 71:094512
Bayly CI, Merz KM, Ferguson DM, Cornell WD, Fox T, Caldwell JW, Kollman PA, Cieplak P, Gould IR, Spellmeyer DC (1995) A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc 117:5179–5197
Oostenbrink C, Villa A, Mark AE, Van Gunsteren WF (2004) A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J Comput Chem 25:1656–1676
Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M (1983) CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem 4:187–217
Jorgensen WL, Tirado-Rives J (1988) The OPLS potential functions for proteins. Energy minimizations for crystals of cyclic peptides and Crambin. J Am Chem Soc 110:1657–1666
Rappé AK, Casewit CJ, Colwell KS, Goddard WA, Skiff WM (1992) UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J Am Chem Soc 114:10024–10035
Mayo SL, Olafson BD, Goddard WA (1990) A generic force field for molecular simulations. J Phys Chem 94:8897–8909
Van Duin ACT, Dasgupta S, Lorant F, Goddard WA (2001) ReaxFF: a reactive force field for hydrocarbons. J Phys Chem A 105:9396–9409
Gale JD (1997) GULP: a computer program for the symmetry-adapted simulation of solids. J Chem Soc, Faraday Trans 93:629–637
Coppens P (1997) X-ray charge densities and chemical bonding. International Union of Crystallography, Chester
Zuo JM, Spence JCH (2017) Advanced transmission electron microscopy, imaging and diffraction in nanoscience. Springer, Berlin
Wu L, Zhu Y, Vogt T, Su HB, Davenport JW, Tafto J (2004) Valence-electron distribution in MgB 2 by accurate diffraction measurements and first-principles calculations. Phys Rev B 69:1–8
McClellan AL (1963) Tables of experimental dipole moments. W.H. Freeman Co, New York
Albrecht F, Repp J, Fleischmann M, Scheer M, Ondráček M, Jelínek P (2015) Probing charges on the atomic scale by means of atomic force microscopy. Phys Rev Lett 115:076101
Mulliken RS (1955) Electronic population analysis on LCAO-MO molecular wave functions. I. J Chem Phys 23:1833–1840
Cox SR, Williams DE (1981) Representation of the molecular electrostatic potential by a net atomic charge model. J Comput Chem 2:304–323
Bayly CI, Cieplak P, Cornell WD, Kollman PA (1993) A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J Phys Chem 97:10269–10280
Bader RFW (1991) A quantum theory of molecular structure and its applications. Chem Rev 91:893–928
Pauling L (1960) Nature of the Chemical Bond, 3rd edn. Cornell University Press, New York
Phillips JC (1970) Ionicity of the chemical bond in crystals. Rev Mod Phys 42:317–356
Delre G (1958) A simple MO–LCAO method for the calculation of charge distributions in saturated organic molecules. J Chem Soc. https://doi.org/10.1039/JR9580004031
Gasteiger J, Marsili M (1980) Iterative partial equalization of orbital electronegativity—a rapid access to atomic charges. Tetrahedron 36:3219–3228
Hammarstrom LG, Liljefors T, Gasteiger J (1988) Electrostatic interactions in molecular mechanics (MM2) calculations via PEOE partial charges I. Haloalkanes. J Comput Chem 9:424–440
Sanderson RT (1983) Electronegativity and bond energy. J Am Chem Soc 105:2259–2261
Applequist J, Carl JR, Fung KK (1972) Atom dipole interaction model for molecular polarizability. Application to polyatomic molecules and determination of atom polarizabilities. J Am Chem Soc 94:2952
Došen-Mićović L, Jeremić D, Allinger NL (1983) Treatment of electrostatic effects within the molecular mechanics method. 1. J Am Chem Soc 105:1716–1722
Došen-Mićović L, Jeremić D, Allinger NL (1983) Treatment of electrostatic effects within the molecular-mechanics method. 2. J Am Chem Soc 105:1723–1733
Warshel A, Levitt M (1976) Theoretical studies of enzymic reactions: dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J Mol Biol 103:227–249
Gao JL, Habibollazadeh D, Shao L (1995) A polarizable intermolecular potential function for simulation of liquid alcohols. J Phys Chem 99:16460–16467
Warshel A, Sharma PK, Kato M, Parson WW (2006) Modeling electrostatic effects in proteins. Biochim Biophys Acta Proteins Proteom 1764:1647–1676
Ji CG, Mei Y, Zhang JZH (2008) Developing polarized protein-specific charges for protein dynamics: MD free energy calculation of pK(a) shifts for Asp(26)/Asp(20) in thioredoxin. Biophys J 95:1080–1088
Rappé AK, Goddard WA (1991) Charge equilibration for molecular dynamics simulations. J Phys Chem 95:3358–3363
Naserifar S, Goddard WA (2018) The quantum mechanics-based polarizable force field for water simulations. J Chem Phys 149:174502
Pearson RG (1963) Hard and soft acids and bases. J Am Chem Soc 85:3533–3539
Pearson RG (1966) Acids and bases. Science 151:172–177
Parr RG, Pearson RG (1983) Absolute hardness: companion parameter to absolute electronegativity. J Am Chem Soc 105:7512–7516
Drago RS, Wayland BB (1965) A double-scale equation for correlating enthalpies of lewis acid-base interactions. J Am Chem Soc 87:3571–3577
Evans RS, Huheey JE (1970) Electronegativity, acids, and bases—III calculation of energies associated with some hard and soft acid-base interactions. J Inorg Nucl Chem 32:777–793
Vanhooyd G (1971) Calculation of bond energies in diatomic molecules. Theor Chim Acta 22:157
Born M (1919) Die Elektronenaffinitaet der Halogenatome. Verhandl Deut Phys Ges 21:679–685
Haber F (1919) Betrachtungen zur Theorie der Warmetonung. Verhandl Deut Phys Ges 21:750–768
Fajans K (1919) Die Elektronenaffinität der Halogenatome und die Ionisierungsenergie der Halogenwasserstoffe. Verhandl Deut Phys Ges 21:714–722
Heinz H, Suter UW (2004) Atomic charges for classical simulations of polar systems. J Phys Chem B 108:18341–18352
Glasser L, von Szentpaly L (2006) Born–Haber–Fajans cycle generalized: linear energy relation between molecules, crystals, and metals. J Am Chem Soc 128:12314–12321
Perdew JP, Parr RG, Levy M, Jose L, Balduz J (1982) Density-functional theory for fractional particle number: derivative discontinuities of the energy. Phys Rev Lett 49:1691–1693
Cohen AJ, Mori-Sánchez P, Yang W (2007) Development of exchange-correlation functionals with minimal many-electron self-interaction error. J Chem Phys 126:10–15
Cohen AJ, Mori-Sánchez P, Yang W (2008) Insights into current limitations of density functional theory. Science 321:792–794
Sen KD (2013) Chemical hardness (structure and bonding). Springer, Berlin
Born M, Mayer JE (1932) Zur Gittertheorie Der Ionenkristalle. Zeitschrift für Phys 75:1–18
Pearson RG, Gray HB (1963) Partial ionic character of metal-chlorine bonds. Inorg Chem 2:358–363
Rittner ES (1951) Binding energy and dipole moment of alkali halide molecules. J Chem Phys 19:1030–1035
Welch DO, Lazareth OW, Dienes GJ, Hatcher RD (1976) Alkali halide molecules: configurations and molecular characteristics of dimers and trimers. J Chem Phys 64:835–839
Birkholz M (1992) The crystal energy of pyrite. J Phys: Condens Matter 4:6227–6240
Su HB, Welch DO, Wong-Ng W (2004) Strain effects on point defects and chain-oxygen order-disorder transition in 123 cuprate compounds. Phys Rev B 70:054517
Ewald PP (1921) Die Berechnung optischer und elektrostatischer Gitterpotentiale. Ann Phys 369:253–287
Jackson JD (1999) Classical electrodynamics, 3rd edn. Wiley, Hoboken
Condon EU, Odishaw H (1958) Handbook of physics. McGraw-Hill, New York
Ransil BJ, Sinai JJ (1967) Toward a charge-density analysis of the chemical bond; the charge-density bond model. J Chem Phys 46:4050–4074
Menéndez M, Martín Pendás A, Braïda B, Savin A (2015) A view of covalent and ionic bonding from maximum probability domains. Comput Theor Chem 1053:142–149
Brewer L, Brackett E (1961) The dissociation energies of gaseous alkali halides. Chem Rev 61:425–432
Pearson RG (1985) Absolute electronegativity and absolute hardness of Lewis acids and bases. J Am Chem Soc 107:6801–6806
Molina JJ, Lectez S, Tazi S, Salanne M, Dufrêche JF, Roques J, Simoni E, Madden PA, Turq P (2011) Ions in solutions: determining their polarizabilities from first-principles. J Chem Phys 134:014511
Ignatiev VD (2005) Sizes of atoms and ions and covalency of bonding in molecules and crystals. J Struct Chem 46:744–751
Lippert E (1958) The strengths of chemical bonds, von T. L. Cottrell. Butterworth Scientific, Oxford
Dietrich H (1958) Tables of interatomic distances and configuration in molecules and ions, herausgeg. von A. D. Mitchell und L. C. Cross. Special publication no. 11. Wiss. Herausgeber: L. E. Sutton. The Chemical Society
Huber KP, Herzberg G (1979) Molecular spectra and molecular structure IV. Constants of diatomic molecules. Van Nostrand Reinhold Co., New York
Pauling L (1932) The nature of the chemical bond. IV. The energy of single bonds and the relative electronegativity of atoms. J Am Chem Soc 54:3570–3582
Sanderson RT (1975) Interrelation of bond dissociation energies and contributing bond energies. J Am Chem Soc 97:1367–1372
Fajans K (1923) Die naturwissenschaften. Naturwissenschafte 11:165–172
Klemperer W, Norms WG, Büchler A, Emslie AG (1960) Infrared spectra of lithium halide monomers. J Chem Phys 33:1534–1540
Klemperer W, Rice SA (1957) Infrared spectra of the alkali halides. I. Lithium halides. J Chem Phys 26:618–624
Pearson EF, Gordy W (1969) Millimeter- and submillimeter-wave spectra and molecular constants of LiF and LiCl. Phys Rev 177:52–58
Catlow CRA, Stoneham AM (1983) Ionicity in solids. J Phys C: Solid State Phys 16:4321–4338
Madden PA, Wilson M (1996) ‘Covalent’ effects in ‘ionic’ systems. Chem Soc Rev 25:339–350
Walsh A, Sokol AA, Buckeridge J, Scanlon DO, Catlow CRA (2017) Electron counting in solids: oxidation states, partial charges, and ionicity. J Phys Chem Lett 8:2074–2075
Walsh A, Sokol AA, Buckeridge J, Scanlon DO, Catlow CRA (2018) Oxidation states and ionicity. Nat Mater 17:958–964
Shimakawa Y, Lufaso MW, Woodward PM (2018) Negative and positive thermal expansion-like volume changes due to intermetallic charge transfer based on an ionic crystal model of transition-metal oxides. APL Mater 6:086106
Salanne M, Marrocchelli D, Merlet C, Ohtori N, Madden PA (2011) Thermal conductivity of ionic systems from equilibrium molecular dynamics. J Phys: Condens Matter 23:102101
Cooley JA, Promkhan P, Gangopadhyay S, Donadio D, Pickett WE, Ortiz BR, Toberer ES, Kauzlarich SM (2018) High seebeck coefficient and unusually low thermal conductivity near ambient temperatures in layered compound Yb2−xEuxCdSb2. Chem Mater 30:484–493
Li N, Bediako DK, Hadt RG, Hayes D, Kempa TJ, Von Cube F, Bell DC, Chen LX, Nocera DG (2017) Influence of iron doping on tetravalent nickel content in catalytic oxygen evolving films. Proc Natl Acad Sci 114:1486–1491
Callejas JF, Read CG, Roske CW, Lewis NS, Schaak RE (2016) Synthesis, characterization, and properties of metal phosphide catalysts for the hydrogen-evolution reaction. Chem Mater 28:6017–6044
Grimaud A, Hong WT, Shao-Horn Y, Tarascon JM (2016) Anionic redox processes for electrochemical devices. Nat Mater 15:121–126
Luo K, Roberts MR, Guerrini N, Tapia-Ruiz N, Hao R, Massel F, Pickup DM, Ramos S, Liu YS, Guo J, Chadwick AV, Duda LC, Bruce PG (2016) Anion redox chemistry in the cobalt free 3d transition metal oxide intercalation electrode Li[Li0.2Ni0.2Mn0.6]O2. J Am Chem Soc 138:11211–11218
Streltsov SV, Khomskii DI (2016) Covalent bonds against magnetism in transition metal compounds. Proc Natl Acad Sci 113:10491–10496
Sawatzky GA, Geertsma W, Haas C (1976) Magnetic interactions and covalency effects in mainly ionic compounds. J Magn Magn Mater 3:37–45
Frisch M et al (2009) Gaussian 09 (Revision A02). Gaussian Inc., Wallingford CT
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789
Miehlich B, Savin A, Stoll H, Preuss H (1989) Results obtained with the correlation energy density functionals of Becke and Lee Yang and Parr. Chem Phys Lett 157:200–206
Godbout N, Salahub DR, Andzelm J, Wimmer E (1992) Optimization of Gaussian-type basis sets for local spin density functional calculations. Part I. Boron through neon, optimization technique and validation. Can J Chem 70:560–571
Sosa C, Andzelm J, Elkin BC, Wimmer E, Dobbs KD, Dixon DA (1992) A local density functional study of the structure and vibrational frequencies of molecular transition-metal compounds. J Phys Chem 96:6630–6636
Acknowledgements
I am particularly grateful to Weicheng Su for enlightening advice in the early stage of this work and indelible joyful dialogs throughout the years. I also thank Weitao Yang for inspiring discussions on the fractional charge concept, David Welch and Bill Goddard for thought-provoking advice on this intriguing subject, and Kok-Khoo Phua for the generous hospitality at Institute of Advanced Studies, where this work was initially formulated. I would like to gratefully acknowledge fruitful discussions and technical support with Chi Xiong, Feng Zhou, W. Wilwin, Imanuel Rava, Long T. Ta, Terry Lv, Yihua Lu, Luan Q. Le and Ellen Octavia. This work is supported in part by “Key-Area Research and Development Program of Guangdong Province” (Grant No. 2019B010128001), HKUST Grants (IGN17SC04; R9418) and Society of Interdisciplinary Research (SOIRÉE).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Published as part of the special collection of articles derived from the Chemical Concepts from Theory and Computation.
Appendix
Appendix
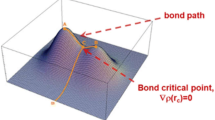
1.1 A graphic solution of Eq. (13)
The Eq. (13) can be solved graphically by plotting algebraic functions at both sides as a function of fractional charge. The point of intersection yields the fractional charge which optimized charge transfer, ionic, polarization and covalent interactions in bond energy. The same scheme can be applied to solve Eq. (11) to compute fractional charge of an atom in solid materials (Fig. 3).

A graphic method to determine fractional charge \(q\) by searching the intersection point of two curves. Both curves represent fractional charge-dependent binding energy of diatomic molecules
1.2 An approximate solution of Eq. (17)
Assuming the total polarization of the anion and cation can be approximated as:
Equation (13) is approximated into the following quadratic one:
The solution of this equation is
The approximate solution in Eq. (24) explicitly highlights that the fractional charge results from the delicate balance among charge transfer, electrostatic–covalent interactions and bond energy in molecules.
1.3 Calculations by density functional theory-based methods
All geometric optimization and energy calculations were performed by Gaussian 09 [89] (RevA.02) using B3LYP functional [90,91,92] and DGDZVP basis set [93, 94]. Optimizations were performed with cut-off values for maximum force of 0.00015, root-mean-square (RMS) force of 0.0001, maximum displacement of 0.0018 and RMS displacement of 0.0012 (all in atomic unit). The bond energy was calculated by Ebond = (EM + EX) − EMX; electron affinity (EA) was calculated by \(\text {EA}=E_{{X}^{-}}-E_X\); and ionization potential (IP) was calculated as IP = \(\text {IP}=E_{{M}^{+}}-E_M\).
Rights and permissions
About this article

Cite this article
Su, H. On fractional charge in molecules and materials. Theor Chem Acc 139, 84 (2020). https://doi.org/10.1007/s00214-020-2580-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-020-2580-5