
Abstract
The study is focused on the CO2 decomposition on small (6-, 7-, 8- and 13-atomic) Cu clusters. The mechanism of the reaction was investigated by means of the molecular dynamics approach on the DFT level of theory. We have determined that there are two possible transition-state structures for the C–O bond dissociation—one where the reaction occurs on the wall of the cluster and the other occurring on the edge formed by two Cu atoms. The reaction depends also on the charge present on the Cu cluster that could be formed in the photoactivation and charge separation on the support.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The CO2 utilization has attracted enormous interest in the recent years and still continues to attract it. The main reason for this is the demand for sustainable fuels solutions, which can be an alternative for increased fossil fuels consumption [1]. The term CO2 reduction used frequently in this context can mean either the reduction of the CO2 emissions into the atmosphere, or the chemical reduction of the CO2 molecule to the value-added products.
The reduction of the CO2 emission to the atmosphere is usually dealt with by means of the CCS (carbon capture and storage) [2]. However, a far more interesting approach is to make use of the CO2 as the feedstock for the production of value-added products. This approach allows in essence to fully recycle the waste CO2 and utilize it again for the zero net emission scheme [3]. In this aspect, two most important pathways are investigated: First is the hydrogenation to formic acid or methanol, which is usually carried out in the liquid phase and catalyzed homogeneously [4]. An important feature of the CO2 hydrogenation to formic acid (FA) is its reversibility, which opens the way to the easy and effective hydrogen storage [5].
The other option is the CO2 reduction to CO which can be subsequently used, for example, as a feedstock for the Fischer–Tropsch process of fuels synthesis, often together with CO2 still present in the CO + H2 syngas [6]. These processes usually make use of the heterogeneous cobalt-based catalysts and are carried out in the gas phase in elevated temperatures [7,8,9].
The major obstacle in the effective CO2 reduction is the unfavorable thermodynamics of the reaction. The potential of reduction of the CO2 to CO2 − (i.e., the transfer of the first electron to the CO2 molecule) amounts to 1.9 eV, and it is the least thermodynamically favorable step in the hydrogenation [10]. This generally requires the use of highly active, noble metal-based catalysts that exhibit reasonably high activity in the reduction process. Examples include the use of Pd compounds in the CO2 hydrogenation to methanol [11] or the already mentioned Ru- or Ir-based pincer complexes in homogeneous hydrogenation to formic acid [5, 12]. The high cost of these catalysts is the reason for the research of the equally active catalysts based on the more abundant compounds. Lesser activity can be compensated by changing the conditions of the reaction, by, for example, enforcing the electron transfer by the electric current.
The copper has been reported to be unique among the electrocatalysts in CO2 hydrogenation process as it is most selective to hydrocarbons—CH4 and C2H4—than any other metal [10]. The mechanism of the Cu activity in the electrochemical reduction of CO2 has been theoretically explored by the group of Norskov [13], and the experimental results of Koper and co-workers [14] provided more insight into the mechanism of this selectivity.
Similarly to the electric current being the driving force of the process, it has been proposed that the charge redistribution upon the photoactivation can also lead to the activity of supported non-noble metal clusters in the CO2 reduction [15]. Numerous semiconductors have been studied in such a reaction, even as early as 1979 [16] but even after relatively long time of research, currently reported activity of supported Cu is still relatively small [17]. The most efficient catalysts contain noble metals, for example, Ru [18] or Pt [19].
It is suggested that the major limitation is the dissociation of the C–O bond in the CO2 molecule. The dissociative adsorption has been studied on clean and stepped copper surfaces [20, 21] as well as on the surface-resembling copper clusters [22]. The bonding energy was found to increase with increasing surface coverage, which might suggest strong interactions between the adsorbed species.
The interactions of the reactants with the active site depend greatly on the geometry of the latter, and this effect will be even more pronounced in case of supported clusters. The stability of the clusters under reactive conditions is still a matter of the debate. The ReaxFF study on the stability of the cobalt clusters was carried out by Zhang et al. [23]. The authors found that there is a strong correlation between the size of the cluster and its ability to reconstruct in the reactive environment. Studies on copper growth on the ZnO support have been carried out by Cheng et al. [24] by means of molecular dynamics simulation. The clusters have been determined as the most stable form when supported on ZnO, contrary to the Cu (111) surface, where monolayers have been observed.
The aim of the work is to determine the activity of the neutral and charged Cu clusters in the CO2 reduction. Particularly, we intend to unravel the role of particular cluster geometries that are most active in this process and elucidate the role of the stability of the cluster on the C–O bond dissociation barrier height.
2 Computational details
The calculations have been done with the Quickstep module [25] of the CP2K program [26] based on the density functional theory (DFT) methodology. This method uses two electron density descriptors: localized Gaussian and plane wave (GPW) basis sets. The double-zeta MOLOPT basis set with polarization function (DZVP) [27] and the complementary plane wave basis set have a cutoff of 400 Rydberg for electron density. The valence electron–ion interaction is based on the norm-conserving and separable pseudopotentials of the analytical form derived by Goedecker, Teter and Hutter (GTH) [28], and the generalized gradient-corrected approximation of Perdew, Burke and Ernzerhof (PBE) is adopted for the exchange–correlation energy functional [29]. The PBE functional has been reported to yield the binding energies well comparable to the experimental values. For more discussion on the applicability of this functional to similar systems, reader is referred to the paper of Hellstrom et al. [30].
The periodic boundary conditions were switched on by using a cubic simulation box of 18 Å sides for all Cu clusters. Spin polarization was included in the calculations, and no symmetry constraints were imposed in structural relaxations.
The dissociation of the C–O bond was studied by means of the constrained MD simulations. The NVT ensemble has been selected, with the temperature set to 300 K and controlled by Nose–Hoover thermostat [31]. The timestep used in simulations was 1.0 fs. The constraint applied was the distance between the C and O atoms of the CO2 molecule, initially set to 1.2 Å, with the growth rate of 0.00015 Å/fs. Upon reaching the limit of 3.5 Å, the constraint was released, and the simulation continued unconstrained until reaching the total time of 15 ps. We have generated 10 MD trajectories for each neutral and charged cluster of a different initial composition, thus carrying out a total of 80 MD simulations.
3 Results and discussion
3.1 Geometries of Cu clusters
The computational model obviously needs to take into account many available geometries of the Cu clusters. The stability of these systems is a subject to environment conditions and interactions with other species, especially due to the fact the energy differences between the particular geometries are reasonably small.
Similar stability studies have been performed before for Cu clusters by Petranovski et al. [32] with the CIS method and Li et al. [33] with real space pseudopotential approximation. These studies are in agreement with the minima computed in the present work by means of the DFT, with the exception of CIS studies of Cu7 cluster [32], where the same geometry is the most stable for both charged and neutral system. Additionally, Li et al. [33] found that several geometries exist with similar electronic energy for anionic Cu6 and Cu7 clusters.
Table 1 shows the geometries and the energies expressed in eV with respect to the most stable cluster. The most stable of the Cu6 clusters is the planar D3h system (6A), as described by Jaque and Torro-Labbe [34]. The energy of the double-capped tetrahedron structure with C2v symmetry (6C) is less stable by 0.24 eV. This structure is, however, the ground state for the anionic system with a charge of −1, and it is more stable than D3h system (6A) by as much as 0.45 eV. Out of the Cu6 clusters, one more should be noted—the “incomplete-D6h” (6D) is equally stable to double-capped tetrahedron when it bears no charge, but when a −1 charge is present, it becomes least stable out of the investigated clusters with the relative energy of 0.59 eV. In addition, we can conclude that the 6B structure is relatively stable in both anionic and neutral form.
The Cu7 clusters follow similar pattern; however, contrary to the Cu6 clusters, the relative energy differences for anionic cluster are much smaller, on the border of the accuracy of the DFT method. The most stable neutral cluster is the one with the highest symmetry—D5h (7A). The cluster with C3v symmetry that is built out of three condensed tetrahedra (7B) is only less stable by 0.20 eV. The least symmetric cluster—capped bipyramid with a square base (7C)—is at the same time least stable out of investigated clusters, but still the energy differences with respect to the ground-state cluster are only 0.33 eV.
Interestingly, this least symmetric cluster is most stable one when anionic (−1 charge) systems are considered, but the energy differences between the clusters are very small—0.10 and 0.14 eV for D5h and C3v clusters, respectively—and as such should be considered small enough to be distinguished with the DFT method. This implies that the interconversion between the charged clusters is very easy, provided that there are no high kinetic barriers. Although the transition-state structures have not been calculated for the needs of the present study, we do not expect them to be high, and the results presented in the second section of this manuscript confirm this assumption.
For Cu8 clusters, we have observed even smaller differences in the energy of the charged systems. The most stable is the highest symmetry cluster, which is also the most stable when no charge is present. The double-capped square bipyramid is less stable by only 0.10 eV, which again should be considered less than the accuracy of the DFT method. The other two investigated models—incomplete D3h (8C) and anti-prism (8D), could not be optimized in the anionic state and converged to the stable ones instead. That confirms the assumption of easy interconversion of clusters.
For Cu13, only two systems have been investigated. The high symmetry D5h (8B) turned out to be less stable in neutral and anionic forms by 0.34 and 0.39 eV, respectively. That implies that the distorted cluster is most stable, contrary to all smaller clusters investigated.
To conclude, the transformation between structures is easy, especially for the anionic Cu7 and Cu8 clusters, where the relative energies do not exceed 0.15 eV. For anionic Cu6 cluster, such transformation needs at least 0.45 eV and can occur easier for neutral cluster, where only 0.24–0.29 eV is required to overcome energetic minimum.
Interestingly, the Cu13 cluster is not influenced by the charge it bears, and for both neutral and anionic systems, the same amount of energy (0.34 and 0.39 eV, respectively) is needed to trigger the transformation.
It has to be clarified that these results disregard the interactions with other species, and all clusters are considered to be in the gas phase. The interactions with the support fall beyond the scope of this study mainly due to the plethora of systems able to support small Cu clusters. In the next section, we will focus on the possibility of the interaction CO2 molecule to trigger the transformation between different clusters.
3.2 CO2 dissociative adsorption
The CO2 molecule is very thermodynamically stable, and therefore, its interaction with Cu clusters is expected to be weak. On the other hand, copper-based catalysts are known to catalyze the CO2 hydrogenation [22]; however, the dissociative adsorption on clean Cu surfaces is still under debate [20]. The interactions with cluster will obviously differ from the clean surfaces, for two reasons—the small size of the clusters does not allow it to have fully metallic character and the undercoordination of the atoms of the clusters provides the binding sites for the CO2 molecule.
For the needs of the present study, we have carried out the constrained MD simulations, where the constraint was set to gradually increase the C–O bond length in the CO2 molecule. That generated a very reactive oxygen species that was able to strongly interact with the Cu cluster and acted as the factor triggering the potential interconversion to another geometry. In most cases, the geometry of the cluster changed to the most stable shape during the equilibration part of the MD run, and only upon the interaction with the reactive oxygen species, other conversions have been observed.
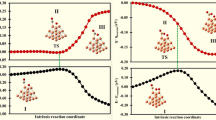
Out of the ten concurrent simulations for Cu6 clusters, three main different pathways have been identified and are shown in Fig. 1 (the plots that were consistent with one another have been omitted for clarity). In two of them, a significant increase in the energy is initially observed. This is the result of the partial dissociation of the CO2 molecule from the cluster, while constrained C–O distance is increasing. In four cases, we have observed the complete dissociation of the CO2 from the Cu cluster that caused increase in the total energy of the system by more than 2 eV due to the reactive oxygen being formed away from the cluster. These geometries have been disregarded as physically unfeasible. Evidently, the immobilization of the CO2 molecule next to the reactive site is a crucial factor, and the reaction can be greatly facilitated in such conditions—for example, in confined space of a microporous support.

Two alternative pathways of CO2 dissociation on neutral Cu6 cluster (green and red) and anionic Cu −6 cluster (orange)
The alternative pathways of CO2 dissociation on Cu6 cluster differ in the orientation of the bond being cleaved. The anionic system (curve marked with orange color in Fig. 1) displays slightly higher energy barrier for the C–O bond dissociation than two pathways for the neutral systems. The cleaved C–O bond is aligned in parallel to the bridge site between two Cu atoms. Preferentially, the lower energy pathways are possible for neutral systems, where the O atom is located on the hollow site formed by three Cu atoms. Interestingly—in both preferred anionic and neutral pathways, the Cu6 cluster has identical geometry, that is, however, different from its geometry in the beginning of the MD run. Therefore, we conclude that only the position of the dissociating CO2 molecule is responsible for the favorable kinetics of the reaction; the preferred position depends in this case on the total charge of the cluster.
The most energetically stable product of the reaction is the cluster similar to the most stable standalone Cu6 D3h cluster (6A), but with the oxygen atom located in one vertex, and the Cu atom in between O and C atoms that were initially bonded. The interconversion between the less thermodynamically stable geometries has been observed for three different systems, so we can assume it is a facile process.
The dissociation of the CO2 on the anionic Cu6 cluster occurs only in one way, as all generated trajectories were consistent with each other. The cleaved bond is aligned in parallel to the bridge between two Cu atoms, similarly to the less favored pathway in neutral Cu6 cluster. Interestingly, the final product is significantly different than for neutral cluster and is geometrically equivalent to the most stable anionic Cu6 cluster (6C); however, the reaction is endothermic by approximately 0.5 eV. The kinetic barriers for the reaction on anionic Cu6 are comparable to the case of neutral Cu6 and lie in the range of 1.0 and 1.5 eV depending on the run.
Figure 2 shows the charge distribution among the three atoms of CO2 molecule along the trajectory shown in red in Fig. 1. Initially, the sum of the charges is zero, which represents neutral, isolated CO2 molecule. The moiety gains slightly neutral charge of approximately −0.1 e upon the activation and coordination to the edge of the cluster. This charge is mostly localized on the oxygen atom bound to the cluster, while the changes on the CO moiety are minor. At this step of the trajectory (approximately 2000th step), the cluster reorganizes, and the activated CO2 molecule coordinates to the triangular wall of the cluster. This leads to significant decrease in the charge on CO moiety (both C and O atoms) as well as O atom being dissociated. The later is, however, part of the steady decrease in the charge since the beginning of the run, which seems not to be affected by the change in the cluster structure.

Changes in Mulliken charges on carbon (gray), oxygens (orange and red) atoms and their sum (green) in neutral Cu6 cluster
Interestingly, the final charge on the C atom is nearly the same as it was in the beginning of the run. At the same time, it can be clearly seen the divergence of the charges on the oxygen atoms—from initial −0.15 e, dissociating oxygen forms a strong bond with Cu cluster gaining negative charge of −0.50 e and the other oxygen becomes almost neutral (−0.05 e).
The charge distribution in the case of anionic cluster follows a similar pattern. Charges on both oxygen atoms diverge in the same way, and from initial −0.25 e, Cu-bound oxygen gains more negative charge of −0.60 e and the C-bound one settles at −0.10 e. At the same time, the charge on the C atom varies only slightly at approximately 0.2 e. The sum of the charges on the atoms from CO2 molecule is approximately −0.5 e during the whole run.
The most significant difference for the case of neutral Cu7 cluster is the weaker interaction of the reactants. As much as seven out of ten trajectories generated show the dissociation of the CO2 from the cluster, which together with the increase in the C–O bond length causes significant raise of the total energy of the system. This is visible in Fig. 3—the blue line, which is representative for the dissociation on the neural Cu7 cluster, shows the increase in energy of almost 2 eV. The associated geometry is shown next to the maximum and displays the fully dissociated CO2–Cu7 system. For this reason, we have been able to identify only one pathway that leads via the intermediate on the hollow site of the triangle formed by Cu atoms. The geometry of the cluster slightly differs for those runs; however, the effect of the geometry on the total energy is minor.

Energy profiles for a CO2 dissociating on the neutral (blue) and anionic (orange) Cu7 clusters
Two distinct products have been identified for the Cu7 clusters. More energetically stable is the product with CO bound to the bridge site of the 7B cluster, causing the overall reaction to be thermoneutral. Less stable is the distorted 7A product, where the oxygen atom causes the cleavage of two Cu–Cu bonds in the cluster, which accounts for its lower stability.
The interactions between CO2 and anionic Cu7 cluster are stronger, and the CO2 molecule tends not to dissociate from the cluster in most cases. Indeed, initially we have observed a slight increase in the energy, but the system quickly settled in one of two major pathways. The pathways are similar to ones observed for Cu6 clusters—dissociation on the bridge site (edge) of the hollow site (wall). The dissociation on the bridge site is less kinetically preferred as the barrier associated with it is in the order of 1.5 eV. More preferred is the dissociation on the wall of the cluster, which is consistent with the similar mechanism observed in case of neutral C6 cluster. The barrier is of the same magnitude as for anionic C6 cluster and amounts to approximately 1.0 eV.
The weak interaction and separation of CO2 and the cluster is also the reason why the charges fluctuate in the initial steps of the trajectory of the neutral Cu7 clusters. Upon the binding of the two, similar charge divergence on two O atoms is observed, as it was the case for Cu6 clusters. The final steps of the simulation are very close to the neutral Cu6 cluster, with similar charges on particular atoms C: 0.35 e, C-bound O: −0.05 e, cluster-bound O: −0.5 e.
For anionic Cu7 cluster, the situation is slightly different. Due to much stronger interaction between the components of the system, CO2 was already activated after the equilibration run (see Fig. 3), and initial charges on the oxygens in the production run have already diverged. The cluster-bound O atom bore a charge of −0.4 e, and the C-bound one—a charge of −0.2 e. The sum of the charges on the atoms belonging to the CO2 molecule remained lower than −0.4 e for the whole duration of the run. The charge on the C atom was close to the 0.2 e, but at the end of the run decreased by 0.1 e. This coincides with the formation of the bond between C and the second Cu atom from the cluster (see right-hand structure in Fig. 3). The charges are shown in Fig. 4.

Changes in Mulliken charges on carbon (gray), oxygens (orange and red) atoms and their sum (green) in anionic Cu7 cluster
The dissociation profiles on the Cu8 clusters are shown in Fig. 5. With respect to the interactions with the charged cluster, the profiles are similar to Cu7 clusters—in both cases CO2 molecule dissociates from the neutral cluster, which combined with constrained increase in the bond length is responsible for the observed increase in the energy of the system in the initial phase. Similarly to the anionic Cu7 cluster, the anionic Cu8 cluster does not display this effect, and CO2 remains in proximity of the cluster.

The energy profiles of the CO2 dissociation on the neutral (blue) and anionic (orange) Cu8 clusters
There are similarities with respect to the geometry of the active site for the dissociation as well. Two possible geometries have been observed—with the dissociation occurring at the edge of the cluster in case of anionic system, and at the triangular wall in case of neutral system. It can be observed that for a neutral system, the energetics of the C–O dissociation is favoured compared to the charged system. However, the dissociation of the CO2 from the cluster is a limiting factor, and despite a barrier of approximately 1 eV, the reaction is unlikely to occur.
The anionic pathway is also less preferred when compared to the anionic Cu7 system. The barrier for C–O dissociation on anionic Cu8 cluster amounts to approximately 1.5 eV, while it is more favorable for anionic Cu7 cluster, where it amounts to 1.0 eV. This is caused by the different geometry of the active site—the dissociation occurring at the edge of the cluster results in the raise of the energetic barrier, as it was already observed for the Cu6 cluster. While theoretically possible, the dissociation at the triangular wall has not been observed in any trajectory involving anionic Cu8 cluster.
The end product of the process in both neutral and anionic Cu8 clusters is similar and consists of a carbonyl bound to a vertex of a cluster and the oxygen atom bound to the hollow site on the triangular wall. The product is energetically more stable for the neutral cluster, where the reaction is thermoneutral, while the negatively charged species is less stable by approximately 0.5 eV.
Figure 6 shows the profile of the Mulliken charges for the neutral Cu8 cluster. The plot displays similar characteristics to neutral Cu6 plot (Fig. 2). Before the CO2 molecule is coordinated to the cluster, the sum of charges on the particular atoms is 0. Upon the formation of the strong bond between the components, the charge is transferred to the C and C-bound O atoms. The total charge remains at approximately −0.2 e until the end of the run. Similar divergence of the charge on two atoms is visible as it is the case for all other simulations (Figs. 2 and 4).

Changes in Mulliken charges on carbon (gray), oxygens (orange and red) atoms and their sum (green) in neutral Cu8 cluster
In case of anionic Cu8, the pattern is similar, with the lower initial total charge on CO2 molecule. Upon activation, the charge is −0.35 e, and after C–O bond cleavage, it drops to approximately −0.5 e, which is the lowest value observed. In the later stage of the run, the cluster rearranges and the CO moiety gains additional charge, which leads to the slight increase in the overall charge to −0.4 e. This value is preserved until the end of the run.
The Cu13 system is the biggest among the investigated ones and is characterized by the lowest activation barrier for the C–O dissociation. This effect is due to the fact that the Cu13 cluster adapts its geometry to maximize the interactions with the CO2 molecule. While for the smaller clusters the reaction occurred at the edge or a triangular wall, the wall of the Cu13 cluster where the CO2 is bound assumes a butterfly conformation. That leads to the binding of all three atoms of CO2 to the active cluster.
The transition-state structure in both anionic and neutral cases is similar, and the bond dissociation occurs at the square wall of the cluster. It has to be pointed out that the size of the cluster allows for such a change in its geometry to form a square site at the wall, which has not been observed for clusters smaller than Cu13. Both—carbon and oxygen atoms—are bound to the two nearest Cu atoms. The activation energy amounts to approximately 1.0 and 0.7 eV for the neutral and anionic system, respectively. The geometry of the final product is again similar to the Cu8 cluster, where the carbonyl is only bound to one Cu atom, contrary to Cu6 and Cu7, where it is bound to two Cu atoms. Interestingly, the reaction is thermoneutral for the neutral case, but slightly exothermic (ΔE = −0.2 eV) for the anionic case. This is because the reshaping of the cluster occurs after the constraint has been released.
The charge profiles shown in Fig. 7, look very similar in case of both neutral and anionic clusters. This is caused by the quick activation of the CO2 molecule during the equilibration run. Thus, the charge is already transferred from the cluster even before the production run. The net charge of the carbon and both oxygen atoms is −0.2 e and varies only slightly during the whole run. The same divergence of the charge on the O atoms has been observed. In both cases, we have also observed the slight increase in the charge on the carbon atom. Interestingly, the net charge transferred to CO2 molecule is independent on the charge of the cluster. This is different than for the smaller clusters, where the additional electron influenced the overall charge transferred.

The energy profiles for the C–O bond dissociation on the neutral (orange) and anionic (green) Cu13 cluster
4 Conclusions
From the comparison of all graphs, it is clear that the desorption of CO2 from the neutral clusters, which is thermodynamically unfavorable, is responsible for the destabilization of all these systems. The situation changes for all negatively charged clusters. Additional electron stabilizes the active complex and explains why neutral copper cluster is not active in CO2 reduction. The support is needed for the charge transfer, adsorption and activation of CO2 molecule.
There are three observed transition-state structures—with the active complex formed by 2, 3 or 4 atoms. Depending on the cluster size, these sites become more stable—such as square site in case of Cu13 cluster, that leads to the lowest activation barrier. The opposite has been observed for the two atomic edge sites, where the activation barrier for the C–O bond dissociation was the highest.
In almost all cases, the product of the reaction is of the same stability as reactant or a little less stable ~0.3 eV. In case of the neutral Cu6 cluster, the CO2 dissociation reaction is exothermic by approximately 0.5 eV.
The charge is transferred mostly to the cluster-bound oxygen atom, which gains the most negative charge already in the initial phase of the run. The carbon atom does not significantly change the charge during the MD runs.
References
Andres RJ, Boden TA, Breon FM, Ciais P, Davis S, Erickson D, Gregg JS, Jacobson A, Marland G, Miller J, Oda T, Olivier JGJ, Raupach MR, Rayner P, Treanton K (2012) A synthesis of carbon dioxide emissions from fossil-fuel combustion. Biogeosciences 9:1845–1871. doi:10.5194/bg-9-1845-2012
Pires JCM, Martins FG, Alvim-Ferraz MCM, Simões M (2011) Recent developments on carbon capture and storage: an overview. Chem Eng Res Des 89:1446–1460. doi:10.1016/j.cherd.2011.01.028
Cherubini F, Peters GP, Berntsen T, Stromman AH, Hertwich E (2011) CO2 emissions from biomass combustion for bioenergy: atmospheric decay and contribution to global warming. GCB Bioenergy 3:413–426. doi:10.1111/j.1757-1707.2011.01102.x
Tanaka R, Yamashita M, Nozaki K (2009) Catalytic hydrogenation of carbon dioxide using Ir(III)—pincer complexes. J Am Chem Soc 131:14168–14169. doi:10.1021/ja903574e
Kothandaraman J, Czaun M, Goeppert A, Haiges R, Jones JP, May RB, Prakash GKS, Olah GA (2015) Amine-free reversible hydrogen storage in formate salts catalyzed by ruthenium pincer complex without pH control or solvent change. Chemsuschem 8:1442–1451. doi:10.1002/cssc.201403458
Riedel T, Claeys M, Schulz H, Schaub G, Nam SS, Jun KW, Choi MJ, Kishan G, Lee KW (1999) Comparative study of Fischer–Tropsch synthesis with H2/CO and H2/CO2 syngas using Fe and Co-based catalysts. Appl Catal A 186:201–213. doi:10.1016/S0926-860X(99)00173-8
Melaet G, Ralston WT, Li CS, AlayogluS An K, Musselwhite N, Kalkan B, Somorjai GA (2014) Evidence of highly active cobalt oxide catalyst for the Fischer–Tropsch synthesis and CO2 hydrogenation. J Am Chem Soc 136:2260–2263. doi:10.1021/ja412447q
Melaet G, Lindeman AE, Somorjai GA (2014) Cobalt particle size effects in the Fischer–Tropsch synthesis and in the hydrogenation of CO2 studied with nanoparticle model catalysts on silica. Top Catal 57:500–507. doi:10.1007/s11244-013-0206-z
Chakrabarti D, de Klerk A, Prasad V, Gnanamani MK, Shafer WD, Jacobs G, Sparks DE, Davis BH (2015) Conversion of CO2 over a co-based Fischer–Tropsch catalyst. Ind Eng Chem Res 54:1189–1196. doi:10.1021/ie503496m
Hori Y (2008) Electrochemical CO2 reduction on metal electrodes. In: Vayenas CG, White RE, Gamboa-Aldeco ME (eds) Modern aspects of electrochemistry. Springer, New York. doi:10.1007/978-0-387-49489-0
Jadhav SG, Vaidya PD, Bhanage BM, Joshi JB (2014) Catalytic carbon dioxide hydrogenation to methanol: a review of recent studies. Chem Eng Res Des 92:2557–2567. doi:10.1016/j.cherd.2014.03.005
Filonenko GA, Smykowski D, Szyja BM, Li G, Szczygieł J, Hensen EJM, Pidko EA (2015) Catalytic hydrogenation of CO2 to formates by a lutidine-derived Ru–CNC pincer complex: theoretical insight into the unrealized potential. ACS Catal 5:1145–1154. doi:10.1021/cs501990c
Peterson AA, Abild-Pedersen F, Studt F, Rossmeisl J, Nørskov JK (2010) How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ Sci 3:1311–1315. doi:10.1039/C0EE00071J
Schouten KJP, Kwon Y, van der Ham CJM, Qin Z, Koper MTM (2011) A new mechanism for the selectivity to C1 and C2 species in the electrochemical reduction of carbon dioxide on copper electrodes. Chem Sci 2:1902–1909. doi:10.1039/C1SC00277E
Chang X, Wang T, Gong J (2016) CO2 photo-reduction: insights into CO2 activation and reaction on surfaces of photocatalysts. Energy Environv Sci 9:2177–2196. doi:10.1039/C6EE00383D
Inoue T, Fujishima A, Konishi S, Honda K (1979) Photoelectrocatalytic reduction of carbon dioxide in aqueous suspensions of semiconductor powders. Nature 277:637–638. doi:10.1038/277637a0
Ola O, Maroto-Valer MM (2014) Copper based TiO2 honeycomb monoliths for CO2 photoreduction. Catal Sci Technol 4:1631–1637. doi:10.1039/C3CY00991B
Sasirekha N, Basha SJS, Shanthi K (2006) Photocatalytic performance of Ru doped anatase mounted on silica for reduction of carbon dioxide. Appl Catal B 62:169–180. doi:10.1016/j.apcatb.2005.07.009
Feng X, Sloppy JD, LaTempa TJ, Paulose M, Komarneni S, Bao N, Grimes CA (2011) Synthesis and deposition of ultrafine Pt nanoparticles within high aspect ratio TiO2 nanotube arrays: application to the photocatalytic reduction of carbon dioxide. J Mater Chem 21:13429–13433. doi:10.1039/C1JM12717A
Nakamura J, Rodriguez JA, Campbell CT (1989) Does CO2 dissociatively adsorb on Cu surfaces? J Phys Condens Matter 1:SB149. doi:10.1088/0953-8984/1/SB/026
Rasmussen PB, Taylor PA, Chorkendorff I (1992) The interaction of carbon dioxide with Cu(100). Surf Sci 269–270:352–359. doi:10.1016/0039-6028(92)91274-F
Wang GC, Jiang L, Morikawa Y, Nakamura J, Cai ZS, Pan YM, Zhao XZ (2004) Surf Sci 570:205–217. doi:10.1016/j.susc.2004.08.001
Zhang XQ, Iype E, Nedea SV, Jansen APJ, Szyja BM, Hensen EJM, van Santen RA (2014) Site stability on cobalt nanoparticles: a molecular dynamics ReaxFF reactive force field study. J Phys Chem C 118:6882–6886. doi:10.1021/jp500053u
Chenga YT, Lianga T, Nieb X, Choudharya K, Phillpota SR, Asthagirib A, Sinnotta SB (2014) Cu cluster deposition on ZnO: morphology and growth mode predicted from molecular dynamics simulations. Surf Sci 621:109–116. doi:10.1016/j.susc.2013.10.025
VandeVondele J, Krack M, Mohamed F, Parrinello M, Chassaing T, Hutter J (2005) Quickstep: fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput Phys Commun 167:103–128. doi:10.1016/j.cpc.2004.12.014
CP2K version 2.6. CP2K is freely available from http://www.cp2k.org
VandeVondele J, Hutter J (2007) Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J Chem Phys 127:114105. doi:10.1063/1.2770708
Goedecker S, Teter M, Hutter J (1996) Separable dual-space Gaussian pseudopotentials. Phys Rev B Condens Matter Mater Phys 54:1703–1710. doi:10.1103/PhysRevB.54.1703
Perdew J, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77:3865–3868. doi:10.1103/PhysRevLett.77.3865
Hellstrom M, Spangberg D, Hermansson K, Broqvist P (2014) Small Cu clusters adsorbed on ZnO(1010) show even–odd alternations in stability and charge transfer. J Phys Chem C 118:6480–6490. doi:10.1021/jp412694y
Nosé S (1984) A unified formulation of the constant temperature molecular-dynamics methods. J Chem Phys 81:511–519. doi:10.1063/1.447334
Petranovskii V, Gurin V, Machorro R (2005) Spectroscopic observation and ab initio simulation of copper clusters in zeolites. Catal Today 107–108:892–900. doi:10.1016/j.cattod.2005.07.039
Li S, Alemany MM, Chelikowsky JR (2006) Real space pseudopotential calculations for copper clusters. J Chem Phys 125:034311. doi:10.1063/1.2216698
Jaque P, Torro-Labbe A (2002) Characterization of copper clusters through the use of density functional theory reactivity descriptors. J Chem Phys 117:3208. doi:10.1063/1.1493178
Acknowledgements
B. Szyja would like to acknowledge the financial support from the OPUS programme (Project No. 2016/21/B/ST4/03699) Granted by the National Science Centre in Poland. This work was partially supported by statutory activity subsidy from the Polish Ministry of Science and Higher Education for the Faculty of Chemistry of Wroclaw University of Science and Technology (0401/0252/16). The simulations have been carried out at Academic Computing Center TASK in Gdańsk, Wrocław Centre for Networking and Supercomputing WCSS and Academic Computer Centre Cyfronet in Kraków. This research was supported in part by PL-Grid Infrastructure.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Klaja, O., Szczygieł, J., Trawczyński, J. et al. The CO2 dissociation mechanism on the small copper clusters—the influence of geometry. Theor Chem Acc 136, 98 (2017). https://doi.org/10.1007/s00214-017-2129-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-017-2129-4