
Abstract
Understanding the mechanism of proton pumping requires a detailed description of the energetics and sequence of events associated with the proton transfers, and of how proton transfer couples to conformational rearrangements of the protein. Here, we discuss our recent advances in using computer simulations to understand how bacteriorhodopsin pumps protons. We emphasize the importance of accurately describing the retinal geometry and the location of water molecules.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Proton pumps are remarkable proteins that translocate protons across cellular membranes against the electrochemical gradient. The transport of protons is achieved using polar amino acids and water molecules that act as intermediate carriers of the proton. Because transition states are short lived, the pathway followed by the proton in a complex protein environment cannot be followed directly using an experimental approach. Computer simulations can provide detailed information about the structure and energetics of the proton-transfer pathways. Here, we discuss our recent theoretical work on the mechanism of the bacteriorhodopsin proton pump.
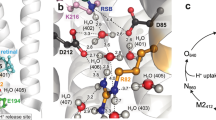
Bacteriorhodopsin is a seven-helical protein found in the plasma membrane of Halobacterium salinarium. The retinal chromophore is covalently bound via a protonated Schiff base to K216. In the low-temperature crystal structures of the bR resting state [1, 2] the Schiff base hydrogen bonds to water molecule w402, which in turn hydrogen bonds to the negatively charged D85 and D212 (Fig. 1). Light-induced photoisomerization of the retinal from all-trans to 13-cis triggers a reaction cycle that consists of several intermediate states (K, L, M, N, O; Fig. 1). The five sequential proton-transfer steps during the reaction cycle (Fig. 1) result in the net transfer of one proton from the cytoplasmic to the extracellular side of the membrane. Except for the first step, which is from the Schiff base to D85 over a distance of ~4 Å, proton transfers in bacteriorhodopsin involve long distances of 10–12 Å (Fig. 1). The time-scales of the transitions associated with proton transfer are on the order of ~10 μs (the first proton-transfer step), or ms. The millisecond timescale associated with the long-distance proton transfers may be required for protein conformational changes. The crystal structures of the reaction cycle intermediates indicate rather small structural rearrangements of the protein in K [3, 4], L [5–9], and early-M [10]; there are structural rearrangements of the protein during the M-to-N [11–15], and N-to-O transitions [16–18]. The passage of the protein through the intermediate conformational states is also accompanied by changes in the number and location of internal water molecules [19, 20].

Proton transfer steps in bacteriorhodopsin. In the bR resting state D96, D115, and the proton release group are protonated [80, 100, 101]. Absorption of light by the retinal chromophore leads to isomerization from all-trans to 13-cis; the 13-cis K state stores 11–14 kcal/mol [61–63]. Step 1—proton transfer from the retinal Schiff base to D85 (the L-to-M transition); step 2—from the proton release group (consisting of water molecules, E194/E204) to the extracellular bulk; step 3—from D96 to the retinal Schiff base (M-to-N); step 4—reprotonation of D96 from the cytoplasm (N-to-O); step 5—from D85 to the extracellular proton release group (O-to-bR). The retinal molecule is all-trans in bR and O, and 13-cis in the K, L, M, and N. The coordinates for preparing the figure were taken from Ref. [1]. X-ray crystal structures have been proposed for all intermediate states of the reaction cycle (see, e.g., Refs. [1–11, 14, 17, 18, 24, 102]; for reviews see, e.g., Refs. [23, 70]). For clarity, amino acids 32-79 containing the second transmembrane helical segment were not included in the figure. Figures 1 and 3a, b were prepared using the Visual Molecular Dynamics software [103]
The coupling between the proton transfer reactions, water location, and protein conformation, is poorly understood. It is not clear, for example, how the triple mutant D96G/F171C/F219L pumps protons in spite of its being locked in a protein conformation similar to that of wild-type M [21]. The local environment may be very important for the proton affinity of the proton transferring groups. For example, relocation of w402 (Fig. 1) such that it no longer hydrogen bonds with D85 can increase the reaction energy for an unproductive proton transfer from D85 back to the retinal Schiff base by ~2 kcal/mol [22]. Relocation of w402 could be brought about by the weakening of the D85:w402 hydrogen bond upon protonation of D85 in early-M (Fig. 1). Indeed, except for the crystal structure of a putative early-M [10], the crystal structures of the M state indicate coordinates for only one [23] or two [7, 11, 24] water molecules in the active site, suggesting that in M water molecules from the active site are either more mobile than before, or have relocated to other sites. Stabilization of the proton on D85 upon relocation of active-site water molecules would allow the protein to sample conformations other than when the Schiff base is protonated. That local chemical reactions and global protein conformational change are coupled had been proposed based on the analysis of structures of bacteriorhodopsin intermediates [13].
Understanding the mechanism of bacteriorhodopsin proton pumping requires a detailed description of the possible pathways of each of the transfer steps. Ideally, the μs–ms dynamics of bacteriorhodopsin embedded in a fluid lipid bilayer would be investigated with a method that allows breaking and forming of chemical bonds. With the remarkable exception of the recent microsecond timescale Molecular Dynamics (MD) simulation of bovine rhodopsin in a hydrated lipid bilayer [25], classical all-atom MD simulations of membrane proteins in lipid bilayers are usually carried out for tens of nanoseconds. The timescale amenable to MD simulations is even shorter if combined quantum mechanical/molecular mechanical (QM/MM) methods [26–28] are used. To overcome these difficulties we used a combination of reaction path calculations, classical and QM/MM MD simulations, classical free energy computations, and QM calculations of active site models.
2 From the crystal structure to proton-transfer paths and assessment of the spectral fingerprints: an overview of the computational methods used
The starting coordinates of the protein are taken from crystal structures. In our computations we used the crystal structures of the bR, K, L, M, and O intermediate states from Refs. [1, 3–5, 7–11, 17, 18]. The crystal structures of the bacteriorhodopsin intermediate states had been solved at low temperatures.
We used the CHARMM software [29] to construct coordinates for the missing internal sidechains, the hydrogen atoms, and for the CH3–CO– and –NH–CH3 neutral caps of the N- and C-termini, respectively. All internal water molecules indicated by the crystal structures were considered in calculations. To assess the role of water molecules on the proton transfer energetics, we have also performed computations in which we considered different numbers and locations of water molecules in the active site. For example, we demonstrated that in the absence of w402 the barrier for proton transfer from the Schiff base to D85 is ~6 kcal/mol lower than when w402 is present [30].
In calculations of the first proton-transfer step we allowed flexibility of an inner region of ~830 atoms consisting of retinal, at least one layer of amino acids and water molecules around the retinal, and other amino acids and water molecules whose coordinates may change upon proton transfer. Consistent with observations from experiments that in the first half of the reaction cycle there are no significant protein conformational changes [31], our computations indicate changes upon proton transfer that are largely restricted to the active site [22, 30, 32]. Nevertheless, flexibility of the protein is essential for proton transfer [30, 32, 33]. In the presence of a flexible protein environment, computations with a pair of reactant and product states shown to be compatible with active proton pumping indicated an energy barrier of ~12 kcal/mol, and the reaction was almost isoenergetic [30] (the enthalpic barrier estimated from experiments for the first proton-transfer step is ~13 kcal/mol [34]). When only the active site groups (retinal, K216, D85, T89, D212, and w402) were allowed to move, the proton transfer barrier and the reaction energy were ~23 kcal/mol and ~11 kcal/mol, respectively [32, 33]. Flexibility of helix C containing D85 is critical for lowering the energetics of the first proton-transfer step: test computations in which a segment of ten amino acids of helix C containing D85, and water molecules w401 and w406, were allowed to move, indicated a proton transfer barrier of ~19 kcal/mol and a reaction energy of ~6 kcal/mol; these values are intermediate between those obtained with fixed and mobile protein environments [33].
Water molecules were described with the TIP3P model [35]. In the classical free-energy computations, the protein was described with the all-atom parameter set [36] and the retinal chain with the parameter set adjusted in Ref. [37] based on the QM computations from Refs. [38, 39]. In QM/MM computations we used the all-atom parameter set [36] for the aromatic side-chains and the extended atom representation [40] for the remaining groups, or the all-atom parameter set [36] for the entire system.
The QM region depends on the proton transfer step under investigation. For computations of the first proton-transfer step we included in the QM region the retinal, the side-chains of K216, D85, D212, and T89, and water molecule w402 [22]. To investigate the identity of the proton release group (proton transfer step two) we treated with QM the side-chains of E194 and E204, and three water molecules close to E194/E204 [41]. The empty valence of the QM host atoms was satisfied with hydrogen link atoms [28] placed on the Cβ atom in the case of the lysine and glutamate side-chains, and on the Cα atom in the case of the aspartate and threonine residues.
The QM method must be reliable for predicting the retinal ground state properties (bond alternation, twist, proton affinity), for the description of structures and energetics of hydrogen-bonded systems, and for proton transfer energetics. Proper description of the excitation energies is necessary in computations of opsin shifts. In what follows we discuss briefly the applicability of various QM methods for describing the retinal ground- and excited-state properties.
The alternation of the lengths of the single and double bonds of the retinal polyene chain is a key property that has an important effect on, e.g., the retinal torsional barriers and the excited-state properties. Due to the extended and highly correlated π-electron system of the retinal polyene chain, the accurate description of retinal’s ground- and excited-state properties is quite challenging for electronic structure methods. Hartree Fock (HF) and Complete Active Space Self-Consistent Field (CASSCF) methods, which lack dynamic correlation, give a large bond alternation of the single and double bonds of the retinal polyene chain. On the other hand, pure Gradient-Corrected Density Functional methods (DFT-GGA) significantly underestimate the bond length alternation [42, 43]. The underestimation of the bond length alternation by DFT-GGA methods is due to the approximate nature of the exchange functionals in DFT-GGA, such that the polarizability of the extended conjugate systems is overestimated [44]. This overestimation of the polarizability allows an easier distribution of the positive charge of the protonated Schiff base over the π-conjugated system of the retinal; with HF and post-HF methods, the charge is more localized at the Schiff base. Hybrid DFT methods that contain a small fraction of HF exchange (e.g., B3LYP has 20% HF exchange) profit from an error cancellation and, consequently, give reasonable bond length alternations [42, 43]. Nevertheless, the tendency of B3LYP to overestimate the π-electron delocalization leads to a reduced bond alternation in the protonated Schiff base segment, and a more planar retinal geometry [45].
Twisting of the retinal chain can have important effects on the reaction mechanism. The twist can affect the orientation of the Schiff base relative to D85, and the proton affinity of the retinal Schiff base [22, 46, 47]. Whereas pronounced twists of a single/double bond have significant effects on the proton affinity, moderate twisting of both single and double bonds (as usually indicated by the crystal structures) has a rather small effect on the intrinsic proton affinity of the Schiff base [22]. Twisting also affects the excitation energies [48].
To describe the retinal twist, one would ideally use multi-reference methods. But these methods are impractical in studies of retinal in the protein environment. Previous computations on protonated Schiff base models indicated that DFT and HF can be used to describe the torsional properties of the bonds close to the Schiff base [49]. SCC-DFTB also gives a reasonable description of the retinal torsional barriers, within the errors of DFT, whereas the semi-empirical AM1 and PM3 methods, which overestimate the retinal bond alternation, underestimate drastically the torsional barriers of the retinal [50].
The relative proton affinity of the retinal Schiff base and carboxylates largely determine the energetics of proton transfer reactions in which the retinal Schiff base is involved. This relative proton affinity is sensitive to the method and basis set used [22]. Although in general DFT methods give a good description of the proton affinity of small molecules, they overestimate the proton affinity of the protonated Schiff base models [22, 43]. The relatively poor performance of DFT-based methods in describing the proton affinity of protonated Schiff base models is due to DFT’s overestimation of the polarizability of π-conjugated systems discussed above: the positive charge of the Schiff base is too much delocalized over the retinal polyene chain. Due to the neglect of dynamic correlation, HF also overestimates the proton affinity of the retinal Schiff base [22, 43], albeit to a lesser extent than B3LYP.
We tested the accuracy of various methods and basis sets in describing the relative deprotonation energies of retinal and acetate models in vacuo [22]. The MP2/6-311+G** relative deprotonation energy was best reproduced with B3LYP/6-31G** (3.3 kcal/mol error relative to MP2/6-311+G**), and with Self-Consistent Charge Density Functional Tight Binding SCC-DFTB [51] with a specific parametrization of the Schiff base nitrogen atom (3.6 kcal/mol error) [22]. That SCC-DFTB is reliable for describing the structure and energetics of hydrogen-bonded systems, the ground-state properties of the retinal molecule, and the energetics of proton transfer, has been documented based on a large set of benchmark computations (see, e.g., [22, 30, 41, 50, 52, 53].
For the QM/MM low-energy proton-transfer paths in bacteriorhodopsin, the SCC-DFTB/MM-optimized rate-limiting barrier and reaction energetics agree to within 2 kcal/mol with the energetics computed (without re-optimizing the geometries) with B3LYP/6-31G** [30, 32]. The active-site geometries of a set of conformers with protonated retinal Schiff base/negatively charged D85, and, respectively, with neutral Schiff base/neutral D85, were almost identical when QM/MM optimized using either SCC-DFTB or B3LYP/6-31G** for the QM region; the optimized relative energies agreed to within 2 kcal/mol (0.8 kcal/mol with SCC-DFTB/MM, and 2.6 kcal/mol with B3LYP6-31G**/MM) [22]. The geometry of the twisted 11-cis retinal in the binding pocket of bovine rhodopsin obtained from SCC-DFTB computations was also in good agreement with the experimental NMR data [54].
The SCC-DFTB/MM computations were performed using the implementation of SCC-DFTB in the CHARMM software described in Ref. [52]. B3LYP/MM computations [22, 30, 41] were performed using TURBOMOLE [55] or Q-Chem [56] with CHARMM. Vibrational spectra were computed from the Fourier Transform of the auto-correlation function of the dipole moment extracted from SCC-DFTB/MM MD trajectories [41]. The QM computations on gas phase models of the retinal [22], and of the retinal binding pocket [32] were performed using GAUSSIAN [57] or SCC-DFTB [51].
To compute reaction paths we used the Conjugate Peak Refinement algorithm (CPR, [58]) as implemented in the TreK module of CHARMM. CPR provides a minimum-energy path connecting the reactant and product states (which are stationary points of the potential energy surface); the energy maxima along the CPR path are first-order saddle points, and the highest-energy saddle point gives the rate-limiting energy barrier of the reaction [58]. The path segments between the stationary points of the optimized CPR paths were further refined using the Syncronous Chain Minimization algorithm [59]. The use of CPR and SCM for computing proton transfer paths in bacteriorhodopsin, and the potential inaccuracies of approaches based on simplistic choices of the reaction coordinate, are discussed in detail in Refs. [22, 60].
3 Retinal geometry
The geometry of the retinal is very important for the proton-pumping cycle of bacteriorhodopsin. The retinal twist is used by bacteriorhodopsin to store energy upon photoisomerization [3, 61–65]. Retinal is directly involved in two proton-transfer reactions, the deprotonation of the Schiff base by proton transfer to the extracellular D85, and re-protonation from the cytoplasmic D96. Control of the retinal conformation is also necessary for the efficiency of the O-to-bR step of the reaction cycle [66]. An accurate description of the retinal geometry in each of the reaction steps is thus necessary for understanding the proton pumping mechanism.
The bond alternation of the retinal polyene chain is strongly influenced by protonation of the Schiff base [46] (black, red and magenta curves in Fig. 2c, d), by the interaction with the negatively charged counterion [54, 67, 68] (black and blue/cyan curves in Fig. 2), and by pronounced twists around double bonds [38]. The bond alternation is reduced by protonation of the Schiff base [46] (Fig. 2c, d) and increased due to the interaction with the counterion [54, 67–69] (black, blue, and light blue curves in Fig. 2c). The influence of the twist and Schiff base interactions on the retinal bond alternation are not captured by the crystal structures of the bacteriorhodopsin intermediates, which indicate similar same bond alternation regardless of the retinal’s protonation state and interaction with the protein environment (Fig. 2a, b).

The retinal bond alternation. a, b The pattern of bond alternation is similar in crystal structures with different retinal twists and protonation states. The Schiff base is protonated in bR, K, and L states (a), and deprotonated in M (b). The retinal bonds (given in Å) were measured from the following crystal structures: bR resting state—1C3W [1] and 1CWQ [11]; twisted K—1M0K [3]; L-state models with different retinal geometries—1UCQ [7] and 2NTW [9]; M-state models with different retinal geometries—1M0M [10] and 1CWQ [11]. c, d Retinal bond alternation from QM and QM/MM computations on protonated (c) and unprotonated (d) retinal models. The computations using B3LYP/6-31G** illustrate how the Schiff base protonation and the environment affect the bond alternation (compare blue curves in c and d and, respectively, black and blue curves in c). The QM optimizations of the retinal Schiff base model in the gas phase are discussed in Ref. [22]. The QM/MM optimizations using B3LYP/6-31G** or SCC-DFTB to describe the QM region were performed on a set of reactant and product states of the first proton-transfer step (protonated Schiff base/negatively charged D85, and neutral Schiff base/neutral D85, respectively) [22]
It had been argued that for the first proton transfer step to occur, the retinal Schiff base must remain formally oriented towards the extracellular side in L [70]. There are two crystal structures proposed for L in which the retinal Schiff base is oriented towards the extracellular side [8, 9] (Fig. 3b). Although retinal is a flexible molecule and it samples a relatively wide range of conformations during dynamics [6, 38, 71, 72], it is unclear if all geometries proposed in the L-state crystal structures [5–9] are indeed sampled during the reaction path. The significant energy barrier for retinal isomerisation from 15-trans to 15-syn [32, 46] makes it unlikely that a 13-cis, 15-syn geometry [8] could be reached during the K-to-L transition [33]. The conclusion from computations that sampling of 15-syn retinals prior to retinal deprotonation is unlikely [33] is consistent with spectroscopy indicating that in L retinal is 13-cis, all-trans [73, 74]. In Ref. [9] the retinal C12–C13 = C14–C15 dihedral angle is −107.9°, which is close to the 90°-twisted geometry of the cis–trans isomerisation path, but on the all-trans side of the retinal isomerization curve.

Geometry of the active site prior to the first proton transfer step. a, b comparison of L-state X-ray structural models with the bR resting state. The following color codes are used: green bR resting state [1, 103]; iceblue 1VJM [6]; magenta 1UCQ [7]; blue 2NTW [9]; mauve 1O0A [8]. The Schiff base nitrogen atom is shown as a sphere. The numbers on the right-hand side of the figure indicate the distance (in Å) between the proton donor and acceptor groups measured as follows. For structures in which the Schiff base and D85 are connected via w402, the first number gives the distance between the Schiff base nitrogen and w402, and the second number gives the distance between w402 and the D85 carboxyl oxygen. For L-state models in which there is no water molecule between the Schiff base and D85, the number gives the distance between the Schiff base nitrogen and the closest D85 carboxyl oxygen. The retinal Schiff base is oriented to the cytoplasm in the L-state models from panel A, and towards the extracellular side in b. (The Schiff base N atom is indicated by the small spheres.) c Average geometry of the retinal binding pocket computed from the last 100 ps of QM/MM MD simulation at 300 K discussed in Ref. [76]. The numbers on the right-hand side of panel C give the distance between the Schiff base nitrogen and wB, and between wB and the D85 carboxyl oxygen; c was prepared using Molscript2.1.2 [104] and Povray3.1 g [105]
Our computations indicate that prior to the first proton-transfer step the retinal Schiff base likely re-orients towards the cytoplasmic side [30]. A cytoplasmic orientation of the retinal prior to deprotonation is supported by the crystal structures of the L-state models from Refs. [5–7] (see iceblue and purple structures in Fig. 3a), FTIR experiments [19], and by the good agreement between the bR-L dipole moment change obtained from experiments and that computed with the L-state model structure from Ref. [7] (magenta structure in Fig. 3a) [75]. Importantly, detailed computations of the energetics of the first proton-transfer step indicate that the cytoplasmic-oriented retinal is consistent with a directional proton pumping [22, 30, 32, 33, 63].
The stabilization of a conformer poised for proton transfer could involve the interaction between the protonated Schiff base and a water molecule. NMR indicates that in L the retinal is twisted and the Schiff base interacts strongly with the counterion; the interacting partner could be a polarized water molecule, or one of the negatively charged carboxylate groups of D85/D212 [65]. The structure of an L-like state that would explain the NMR data [65] is, however, not clear (Fig. 3a, b). Our free energy computations indicated that the presence of a water molecule on the cytoplasmic side of the retinal Schiff base as indicated in Ref. [7] (magenta structure in Fig. 3a) is thermodynamically allowed [76]. Reaction path computations in the presence of the cytoplasmic water molecule (labeled here as wB) indicate a low-energy conformer in which wB bridges the protonated Schiff base and the negatively charged D85 via hydrogen bonding (Fig. 3c); the low-energy conformer was also prefered in QM/MM MD simulations at 300 K [76].
The geometry of the Schiff base, D85, and wB in the low-energy conformer depicted in Fig. 3c suggests proton transfer from the Schiff base to D85 via wB. This is indeed the lowest-energy proton transfer path, with a rate-limiting energy barrier of 7–10 kcal/mol [76].
The importance of the bridging water molecule is suggested by the observation that in its absence direct hydrogen bonds between the Schiff base and D85 form only transiently ([6]; Bondar and Smith, unpublished results). Moreover, the persistence of a hydrogen bond between the Schiff base and wB after proton transfer [76] is consistent with the observation from NMR that in early M the deprotonated Schiff base hydrogen bonds with a hydroxyl group [65]. In the absence of the cytoplasmic water molecule the deprotonated Schiff base can interact with the hydroxyl group of T89 [30], albeit less optimally than with the water molecule.
4 Role of internal water molecules
An exhaustive search of the proton transfer paths from the Schiff base to D85 computed with various crystal structures and geometries of the active site demonstrated that water molecules are key determinants of the energetics of proton transfer in bacteriorhodopsin [30, 32, 76]. The location of water molecules can even determine the path followed by the proton. In the presence of w402, w401, and w406 in the active site, three paths were found isoenergetic within the error of the computations—via T89, directly to D85, and via D212/w402 [30]. When w402 is absent, and there is instead a water molecule bridging the Schiff base and D85 (Fig. 3c), proton transfer is via the water molecule with a rate-limiting barrier of 7–10 kcal/mol [76]. The presence of a water molecule on the cytoplasmic side of the Schiff base stabilizes strongly the cytoplasmic-oriented Schiff base relative to the geometry twisted towards D212, such that proton transfer on the D212 side can no longer occur [76].
Water molecules can also contribute to the suppression of proton transfer from D85 back to the retinal Schiff base, as required for a directional proton pumping. The crystal structures of M indicating only one [23] or two water molecules [11, 24] in the D85/D212 region, as compared to three water molecules in the bR resting state [1, 2], suggest that in M the active-site water molecules may have an enhanced mobility, or may relocate to other sites. To assess the role of water molecules on the energetics of proton transfer from D85 back to the retinal Schiff base we performed QM/MM calculations of proton transfer paths with various numbers of active-site water molecules, and QM computations on gas-phase models of the D85/water interactions. The computations indicated that relocation of one or two water molecules from the active site can help stabilize the proton on D85 [22].
The significant effect of water molecules on the energetics of the retinal Schiff base proton transfer reactions discussed above illustrates the importance of accurate information on the location of water molecules in the various intermediates of the proton pumping cycle. An important aspect when discussing the internal water molecules of bacteriorhodopsin is that there might be differences in the location and number of water molecules in cryo-trapped crystal structures and in physiological conditions. Mobile and low-occupancy internal water molecules are difficult to solve crystallographically [77]. The low temperatures used to cryo-trap bacteriorhodopsin intermediate states could influence the information on internal water in crystal structures: the equilibrium water occupancy may be lower at cryogenic temperatures than at room temperature [78]. Classical MD simulations on the bR and M intermediate states at 300 K revealed that the number of diffusive water molecules transiently visiting bacteriorhodopsin is significantly higher than in the crystal structures [79]. It is also not clear whether, on the microsecond timescale of the K and L intermediates, internal water molecules perturbed by retinal’s photoisomerization reach equilibrium with the bulk; the presence and residence times of a specific water molecule in the K/L states could depend not only on its free energy at the K/L location(s), but also on the activation energy associated with the relocation of the water molecule [76].
NMR data on the L-state intermediate at cryogenic temperatures indicated that the energy landscape is rugged; four L substates were distinguished according to their 15 N chemical shifts [65]. A single L intermediate, which with the strongest Schiff base:counterion interactions, persisted when the temperature was increased [65]. Detailed reaction path computations have also indicated that on the potential energy surface of the pre-proton transfer state there are several energy minima distinguished by the Schiff base, counterion, and water interactions [30, 76]. Prolonged MD simulations of bacteriorhodopsin intermediates in hydrated lipid bilayers could provide valuable information on the location and the dynamics of the water molecules during the reaction cycle. A potential important caveat of these simulations is that the need of computational efficiency will make impractical the use of QM/MM, and the classical force fields may not be sufficiently accurate to describe the hydrogen-bonding of the strongly polarized water molecules of the bacteriorhodopsin Schiff base region [76].
5 Long-distance proton transfers
Theoretical investigations of the long-distance proton transfers in bacteriorhodopsin (Fig. 1) are difficult due to the coupling between proton transfer, conformational changes of the protein, and relocation of water molecules. One of the first questions that needed to be addressed for understanding the second proton transfer step (from the extracellular proton release group to the extracellular bulk) was the exact nature of the proton release group. Earlier experiments indicated the involvement of E204 either as the proton release group [80] or as the group that delivers the released proton to E194 [81, 82]. Based on more recent data, it was proposed that the proton is stored in a cluster of water molecules that interacts with protein amino acids [83, 84]. This proposal was apparently supported by the observation of a continuum band in spectra calculated from QM/MM Carr–Parinelo dynamics in which the proton release group was modeled as a dimer of water molecules sharing a proton (Zudel ion, H5O2 +) [85]. However, it remained unclear if a Zundel ion is indeed the proton release group: In Ref. [85] the symmetry of the Zundel ion was maintained with distance constraints, and the negatively charged E194/E204 were described with MM, i.e., they could not compete with the Zundel ion for the proton.
The distance and the relative orientation of E194/E204 in the crystal structure of the bR resting state [1] would suggest that the two glutamate residues are hydrogen bonded (Fig. 1). In test QM/MM computations with E204 protonated, the relative orientation and the distance between E194 and E204 was in good agreement with the crystal structure [41, 86]. In contrast, when the proton release group was modeled as a Zundel ion, the distance between E194 and E204 sampled values of 3–6.5 Å, significantly higher than the hydrogen-bonding distance in the crystal structure [41]. When both the Zundel ion and the negatively charged E194/E204 pair were treated with QM, the proton was attracted by E194/E204 and remained shared by E194/E204 for the entire length of the simulations [41]. This geometry with E194/E204 sharing a proton gives an IR spectrum in reasonable qualitative agreement with experiments [41], bringing support to the initial proposal [87, 88] that the E194/E204 dyad, probably in association with water molecules, may act as the extracellular proton release group. E194/E204 may also be important for controlling the mechanical stability of the extracellular region of bacteriorhodopsin [89]. Further computations are required to understand fully how protonation of D85 leads to deprotonation of the proton release group, and what is the pathway of the proton from the proton release group to the extracellular bulk. It will also be important to better assess the nature and the dynamics of the proton release group by performing prolonged simulations of bacteriorhodopsin in a hydrated lipid bilayer.
6 Assessing spectral fingerprints of the reaction cycle intermediates
The spectral fingerprints of the retinal can be used to validate structural models of the intermediates sampled during the reaction cycle. As discussed above, the accurate description of the electronic structure of retinal is a significant challenge for QM methods. Particularly difficult is the correct prediction of the excited states properties. A first critical test for any QM method is the extent to which it can reproduce the experimental vertical excitation energies of retinals in the gas phase [90], as it determines the starting point from which the blue shift due to the protein environment has to be estimated [42]. If the calculated gas phase value is too high, the underestimation of the blue shift due to the protein environment can lead to an artefactual error compensation as, e.g., in the case of TD-DFT. The experimental value of 2.03 eV is reasonably reproduced by CASPT2 [91] and Spectroscopy Oriented Configuration Interaction (SORCI, [92, 93]) [42], but overestimated by TD-DFT [42].
The absorption spectrum of SRII is blue shifted by 70 nm relative to bacteriorhodopsin [94, 95]. We used SORCI [92, 93] to investigate the molecular origin of the difference in excitation energies of bacteriorhodopsin and sensory rhodopsin II. SORCI gives a spectral shift of 0.29 eV [48], close to the 0.32 eV value from experiments. The calculations suggested that the blue shift likely arises from differences in the neutral amino acids that constitute the retinal binding pocket, and in the extracellular hydrogen-bonded network [48]. The stronger electrostatic interactions of the retinal in sensory rhodopsin II as compared to bacteriorhodopsin lead to a stronger bond alternation, and a larger effect of the protein on the retinal dipole moment [48].
In order to average over the protein fluctuations, absorption spectra should be sampled along MD trajectories. In the case of the bR resting state, however, the presence of the strong hydrogen bonded network in the retinal binding pocket leads to a rather rigid structure. The absorption maximum calculated by sampling the excitation energies using OM2/MRCI along the QM/MM MD trajectory coincides with the vertical excitation energy evaluated for optimized structures [48]. This indicates that optimized structures may lead to meaningful excitation energies, at least as long as there are no significant fluctuations of the atomic coordinates.
The use of fixed point charges for the MM atoms is a limitation in using standard QM/MM methods to compute vertical excitation energies. The dipole moment of retinal changes significantly upon excitation: In Cl− salt, the change in the dipole moment upon excitation of protonated all-trans retinal is 12D [96]; SORCI gives a value of ~10D for bacteriorhodopsin [48]. As a result of the dramatic dipole moment change, the protein polarization is different in the ground and excited states. The effect of the retinal dipole moment change on the protein electrostatic environment is neglected in the standard QM/MM methods. To account for the protein polarization effects, we implemented recently a polarizable force field that allows us to determine an electrostatic protein environment consistent with the ground and excited state dipole moment of the retinal [97]. Compared to the standard QM/MM models, the inclusion of protein polarization effects can lead to red-shifts of ~0.1 eV relative to the computation in which the standard QM/MM approach is employed [97]. These observations indicate that inclusion of protein polarization is important for computing accurate relative and absolute excitation energies of retinal proteins.
7 Concluding remarks
The advances in computer simulation techniques allow us to investigate at unprecedented levels of detail the mechanisms of proton pumps. Bacteriorhodopsin is an excellent model system to investigate proton pumping, because we have structural information about all intermediate states along the pumping cycle, about the identity of the groups involved in proton transfer, and about the possible roles of the internal water molecules. Challenges specific to bacteriorhodopsin and other retinal-containing proteins are the controversies regarding the details of the retinal geometry from X-ray crystal structures, and the need to use appropriate quantum chemical methods to describe retinal. The dramatic effect of water molecules on the retinal proton transfer reactions [30, 76, 98, 99] highlight the importance of accurate information about the location and dynamics of water molecules in the interior of proton-pumping proteins. Such detailed information can only be derived by using MD simulations to assess the structures provided by X-ray crystallography. Computations are also an extremely valuable tool for interpreting spectral fingerprints from experiments, which in turn can be used to validate structural models.
References
Luecke H, Schobert B, Richter H-T, Cartailler J-P, Lanyi JK (1999) J Mol Biol 291:899. doi:10.1006/jmbi.1999.3027
Belrhali H, Nollert P, Royant A, Menzel C, Rosenbuch JP, Landau EM, Pebay-Peyroula E (1999) Structure 7:909. doi:10.1016/S0969-2126(99)80118-X
Schobert B, Cupp-Vickery J, Hornak V, Smith SO, Lanyi JK (2002) J Mol Biol 321:715. doi:10.1016/S0022-2836(02)00681-2
Edman K, Nollert P, Royant A, Belrhali H, Pebay-Peyroula E, Hajdu J, Neutze R, Landau EM (1999) Nature 401:822. doi:10.1038/44623
Royant A, Edman K, Ursby T, Pebay-Peyroula E, Landau EM, Neutze R (2000) Nature 406:645. doi:10.1038/35020599
Edman K, Royant A, Larsson G, Jacobson F, Taylor T, van der Spoel D, Landau EM, Pebay-Peyroula E, Neutze R (2004) J Biol Chem 279:2147. doi:10.1074/jbc.M300709200
Kouyama T, Nishikawa T, Tokuhisa T, Okumura H (2004) J Mol Biol 335:531. doi:10.1016/j.jmb.2003.10.068
Lanyi JK, Schobert B (2003) J Mol Biol 328:439. doi:10.1016/S0022-2836(03)00263-8
Lanyi JK, Schobert B (2007) J Mol Biol 365:1379. doi:10.1016/j.jmb.2006.11.016
Lanyi JK, Schobert B (2002) J Mol Biol 321:727. doi:10.1016/S0022-2836(02)00682-4
Sass H, Büldt G, Gessenich R, Hehn D, Neff D, Schlesinger R, Berendzen J, Ormos P (2000) Nature 406:649. doi:10.1038/35020607
Sass HJ, Schachowa IW, Rapp G, Koch MHJ, Oesterhelt D, Dencher NA, Büldt G (1997) EMBO J 16:1484. doi:10.1093/emboj/16.7.1484
Kataoka M, Kamikubo H (2000) Biochim Biophys Acta 1460:166. doi:10.1016/S0005-2728(00)00137-7
Schobert B, Brown LS, Lanyi JK (2003) J Mol Biol 330:553. doi:10.1016/S0022-2836(03)00576-X
Radionov AN, Kaulen AD (1999) FEBS Lett 451:147. doi:10.1016/S0014-5793(99)00577-3
Chen D, Wang JM, Lanyi JK (2007) J Mol Biol 366:790. doi:10.1016/j.jmb.2006.12.017
Rouhani S, Cartailler JP, Facciotti MT, Walian P, Needleman R, Lanyi JK, Glaeser RM, Luecke H (2001) J Mol Biol 313:615. doi:10.1006/jmbi.2001.5066
Okumura H, Murakami M, Kouyama T (2005) J Mol Biol 351:481. doi:10.1016/j.jmb.2005.06.026
Morgan JE, Gennis RB, Maeda A (2008) Photochem Photobiol 84:1038. doi:10.1111/j.1751-1097.2008.00377.x
Kandori H (2004) Biochim Biophys Acta 1658:72. doi:10.1016/j.bbabio.2004.03.015
Tittor J, Paula S, Subramaniam S, Heberle J, Henderson R, Oesterhelt D (2002) J Mol Biol 319:555. doi:10.1016/S0022-2836(02)00307-8
Bondar A-N, Suhai S, Fischer S, Smith JC, Elstner M (2007) J Struct Biol 157:454. doi:10.1016/j.jsb.2006.10.007
Luecke H (2000) Biochim Biophys Acta 1460:133. doi:10.1016/S0005-2728(00)00135-3
Takeda K, Matsui Y, Kamiya N, Asachi S, Okumura H, Kouyama T (2004) J Mol Biol 341:1023. doi:10.1016/j.jmb.2004.06.080
Grossfield A, Ptman MC, Feller SE, Soubias O, Gawrisch K (2008) J Mol Biol 381:478. doi:10.1016/j.jmb.2008.05.036
Warshel A (1991) Computer modeling of chemical reactions in enzymes and solutions. Wiley, New York
Singh UC, Kollman P (1986) J Comput Chem 7:718. doi:10.1002/jcc.540070604
Field MJ, Bash PA, Karplus M (1990) J Comput Chem 11:700. doi:10.1002/jcc.540110605
Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M (1983) J Comput Chem 4:187. doi:10.1002/jcc.540040211
Bondar A-N, Elstner M, Suhai S, Smith JC, Fischer S (2004) Structure 12:1281. doi:10.1016/j.str.2004.04.016
Subramaniam S, Lindahl M, Bullough P, Faruki AR, Tittor J, Oesterhelt D, Brown L, Lanyi JK, Henderson R (1999) J Mol Biol 287:145. doi:10.1006/jmbi.1999.2589
Bondar A-N, Fischer S, Smith JC, Elstner M, Suhai S (2004) J Am Chem Soc 126:14668. doi:10.1021/ja047982i
Bondar A-N, Smith JC, Fischer S (2006) Photochem Photobiol Sci 5:547. doi:10.1039/b516451f
Ludman K, Gergely C, Váró G (1998) Biophys J 75:3110. doi:10.1016/S0006-3495(98)77752-5
Jorgensen W, Chandrasekhar J, Madura J, Impey R, Klein M (1983) J Comput Chem 79:926
MacKerell AD Jr, Bashford D, Bellott RL, Dunbrack RL Jr, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WEIII, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M (1998) J Phys Chem B 102:3586. doi:10.1021/jp973084f
Gruia AD, Bondar A-N, Smith JC, Fischer S (2005) Structure 13:617. doi:10.1016/j.str.2005.01.021
Tajkhorshid E, Baudry J, Schulten K, Suhai S (2000) Biophys J 78:683. doi:10.1016/S0006-3495(00)76626-4
Nina M, Roux B, Smith JC (1995) Biophys J 68:25. doi:10.1016/S0006-3495(95)80184-0
Neria E, Fischer S, Karplus M (1996) J Chem Phys 105:1902. doi:10.1063/1.472061
Phatak P, Ghosh N, Yu H, Cui Q, Elstner M (2008) Proc Natl Acad Sci USA 105:19672. doi:10.1073/pnas.0810712105
Wanko M, Hoffmann M, Strodel P, Koslowski A, Thiel W, Neese F, Frauenheim T, Elstner M (2005) J Phys Chem B 109:3606. doi:10.1021/jp0463060
Wanko M, Hoffmann M, Frauenheim T, Elstner M (2006) J Comput Aided Mol Des 20:511. doi:10.1007/s10822-006-9069-8
Champagne B, Perpete EA, Jacquemin D, van Gisbergen SJA, Baerends E-J, Soubra-Ghaoui C, Robins KA, Kirtman B (2000) J Phys Chem A 104:4755. doi:10.1021/jp993839d
Tergesten F, Buss V (1998) J Mol Struct THEOCHEM 430:209
Tajkhorshid E, Paisz B, Suhai S (1999) J Phys Chem B 103:4518. doi:10.1021/jp982625d
Schulten K, Tavan P (1978) Nature 272:85. doi:10.1038/272085a0
Hoffmann M, Wanko M, Strodel P, König PH, Frauenheim T, Schulten K, Thiel W, Tajkhorshid E, Elstner M (2006) J Am Chem Soc 128:10808. doi:10.1021/ja062082i
Paizs B, Tajkhorshid E, Suhai S (1999) J Phys Chem B 103:5388. doi:10.1021/jp982624l
Zhou H, Tajkhorshid E, Frauenheim T, Suhai S, Elstner M (2002) Chem Phys 277:91. doi:10.1016/S0301-0104(01)00695-4
Elstner M, Porezag D, Jungnickel G, Elsner J, Haugk M, Frauenheim T, Suhai S, Seifert G (1998) Phys Rev B 58:7260. doi:10.1103/PhysRevB.58.7260
Cui Q, Elstner M, Kaxiras E, Frauenheim T, Karplus M (2001) J Phys Chem B 105:569. doi:10.1021/jp0029109
Riccardi D, König P, Guo H, Cui Q (2008) Biochemistry 47:2369. doi:10.1021/bi701950j
Sugihara M, Buss V, Entel P, Elstner M, Frauenheim T (2002) Biochemistry 41:15259. doi:10.1021/bi020533f
Alrichs R, Bär M, Häser M, Horn H, Kölmel C (1989) Chem Phys Lett 162:165. doi:10.1016/0009-2614(89)85118-8
Shao Y, Fusti-Molnar L, Jung Y, Kussmann J, Ochsenfeld C, Brown ST, Gilbert ATB, Slipchenko LV, Levchenko SV, O’Neil DPO, Distasio RA Jr, Lochan RC, Wang T, Beran GJO, Besley NA, Herbert JM, Lin CY, van Voornis T, Chien SH, Sodt A, Steele RP, Rassolov VA, Maslen PE, Korambath PP, Adamson RD, Austin B, Baker J, Byrd EFC, Dachsel H, Doerksen RJ, Drew A, Duniets BD, Dutoi AD, Furlani TR, Gwaltney SR, Heyden A, Hirata S, Hsu C-P, Kedziora G, Khalliulin RZ, Klunzinger P, Lee AM, Lee MS, Liang W, Lotan I, Nair N, Peters B, Proynov EI, Piezniazek PA, Rhee YM, Ritchie J, Rosta E, Sherrill CD, Simmonett AC, Subotnik JE, Woodcock HLIII, Zhang W, Bell AT, Chakraborty AK, Chipman DM, Keil FJ, Warshel A, Hehre WJ, Schaefer HFIII, Kong J, Krylov AI, Gill PMW, Head-Gordon M (2006) Phys Chem Chem Phys 8:3172. doi:10.1039/b517914a
Frisch MJ et al (1998) GAUSSIAN 1998. Gaussian Inc, Pitsburgh
Fischer S, Karplus M (1992) Chem Phys Lett 194:252. doi:10.1016/0009-2614(92)85543-J
Choi C, Elber R (1991) J Chem Phys 94:751. doi:10.1063/1.460343
Bondar N, Elster M, Fischer S, Smith JC, Suhai S (2004) Phase Transit 77:47. doi:10.1080/01411590310001622455
Birge RR, Cooper TM (1983) Biophys J 42:61
Birge RR, Cooper TM, Lawrence AF, Masthay MB, Zhang CF, Zidovetzki R (1991) J Am Chem Soc 113:4327
Bondar A-N, Fischer S, Suhai S, Smith JC (2005) J Phys Chem B 109:14786. doi:10.1021/jp0531255
Hayashi S, Tajkhorshid E, Schulten K (2002) Biophys J 83:1281. doi:10.1016/S0006-3495(02)73900-3
Mak-Jurkauskas ML, Bajaj VS, Hornstein MK, Belenky M, Griffin RG, Herzfeld J (2008) Proc Natl Acad Sci USA 105:883. doi:10.1073/pnas.0706156105
Delaney JK, Yahalom G, Sheves M, Subramaniam S (1997) Proc Natl Acad Sci USA 94:5028. doi:10.1073/pnas.94.10.5028
Okada T, Sugihara M, Bondar A-N, Elstner M, Entel P, Buss V (2004) J Mol Biol 342:571. doi:10.1016/j.jmb.2004.07.044
Schreiber M, Buss V, Sugihara M (2003) J Chem Phys 119:12045. doi:10.1063/1.1632898
Fujimoto K, Hayashi S, Hasegawa J, Nakatsujia HJ (2007) J Chem Theory Comput 3:605. doi:10.1021/ct6002687
Lanyi JK (2004) Annu Rev Physiol 66:665. doi:10.1146/annurev.physiol.66.032102.150049
Lemaître V, Yeagle P, Watts A (2005) Biochemistry 44:12667. doi:10.1021/bi0506019
Lau PW, Grossfield A, Feller SE, Pitman MC, Brown MF (2007) J Mol Biol 372:906. doi:10.1016/j.jmb.2007.06.047
Fodor SPA, Ames JB, Gebhard R, van der Berg EMM, Stoeckenius W, Lugtenburg J, Mathies RA (1988) Biochemistry 27:7097. doi:10.1021/bi00418a064
Ames JB, Fodor SPA, Gebhard R, Raap J, van der Berg EMM, Lugtenburg J, Mathies A (1989) Biochemistry 28:3681. doi:10.1021/bi00435a009
Tóth-Boconádi R, Dér A, Taneva SG, Keszthelyi L (2006) Biophys J 90:2651. doi:10.1529/biophysj.105.068817
Bondar A-N, Baudry J, Suhai S, Fischer S, Smith JC (2008) J Phys Chem B 112:14729. doi:10.1021/jp801916f
Levitt M, Park BH (1993) Structure 1:223. doi:10.1016/0969-2126(93)90011-5
Gottschalk M, Dencher NA, Halle B (2001) J Mol Biol 311:605. doi:10.1006/jmbi.2001.4895
Grudinin S, Büldt G, Gordeliy V, Baumgartner A (2005) Biophys J 88:3252. doi:10.1529/biophysj.104.047993
Brown LS, Sasaki J, Kandori H, Maeda A, Needleman R, Lanyi JK (1995) J Biol Chem 270:27122. doi:10.1074/jbc.270.45.27122
Dioumaev A, Richter H-T, Brown LS, Tanio M, Tuzi S, Saitô H, Kimura Y, Needleman R, Lanyi JK (1998) Biochemistry 37:2496. doi:10.1021/bi971842m
Kalaidzidis IV, Belevich IN, Kaulen AD (1998) FEBS Lett 434:197. doi:10.1016/S0014-5793(98)00980-6
Garczareck F, Brown LS, Lanyi JK, Gerwert K (2005) Proc Natl Acad Sci USA 102:3633. doi:10.1073/pnas.0500421102
Garczarek F, Gerwert K (2006) Nature 439:109. doi:10.1038/nature04231
Rousseau R, Kleinschmidt V, Schmitt UW, Marx D (2004) Angew Chem Int Ed 43:4804. doi:10.1002/anie.200453857
Bondar A-N, Smith JC (2009) Isr J Chem (in press)
Zscherp C, Schlesinger R, Tittor J, Oesterhelt D, Heberle J (1999) Proc Natl Acad Sci USA 96:5498. doi:10.1073/pnas.96.10.5498
Essen L-O, Siegert R, Lehman WD, Oesterhelt D (1998) Proc Natl Acad Sci USA 95:11673. doi:10.1073/pnas.95.20.11673
Sapra KT, Doehner J, Renugopalakrishnan V, Padrós E, Muller DJ (2008) Biophys J 95:3407. doi:10.1529/biophysj.108.131904
Andersen LH, Nielsen IB, Kristensen MB, Ghazaly MOA, Haake S, Nielsen MB, Petersen MA (2005) J Am Chem Soc 127:12347. doi:10.1021/ja051638j
Sekharan S, Sugihara M, Buss V (2007) Angew Chem Int Ed 46:269. doi:10.1002/anie.200603306
Neese FJ (2003) J Chem Phys 119:9428
Neese F (2004) ORCA–An ab initio, density functional and semiempirical program package; Version 2.3–Revision 09. Max Planck Institut für Strahlenchemie, Mülheim
Oesterhelt D, Stoeckenius W (1973) Proc Natl Acad Sci USA 70:2853. doi:10.1073/pnas.70.10.2853
Chizhow I, Schmies G, Seidel R, Sydor JR, Lüttenberg B, Engelhard M (1998) Biophys J 75:999. doi:10.1016/S0006-3495(98)77588-5
Mathies R, Stryer L (1976) Proc Natl Acad Sci USA 73:2169. doi:10.1073/pnas.73.7.2169
Wanko M, Hoffman M, Frauenheim T, Elstner M (2008) J Phys Chem B 112:11462. doi:10.1021/jp802408g
Hayashi S, Ohmine I (2000) J Phys Chem B 104:10678. doi:10.1021/jp001508r
Murata K, Fujii Y, Enomoto N, Hata M, Hoshimo T, Tsuda M (2000) Biophys J 79:982. doi:10.1016/S0006-3495(00)76352-1
Metz G, Siebert F, Engelhard M (1992) FEBS Lett 303:237. doi:10.1016/0014-5793(92)80528-O
Gerwert K, Souvignier G, Hess B (1990) Proc Natl Acad Sci USA 87:9774. doi:10.1073/pnas.87.24.9774
Humphrey W, Dalke AK, Schulten K (1996) J Mol Graphics 14.1: 33
Matsui Y, Sakai K, Murakami M, Shiro Y, Adachi S, Okumura H, Kouyama T (2002) J Mol Biol 324:469. doi:10.1016/S0022-2836(02)01110-5
Kraulis PJ (1991) Acta Crystallogr 24:946
Acknowledgments
The work discussed here had been supported in part by the Deutsche Forschungsgemeinschaft (DFG) Consortium on Molecular Mechanisms of Retinal Proteins Action. A.-N.B. is supported by grants GM74637 and GM68002 from the National Institutes of General Medical Sciences. J.C.S. is supported by a Laboratory-Directed Research and Development grant from the US Department of Energy. M.E. is supported by grants from the German Research Foundation (DFG).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Dedicated to Professor Sandor Suhai on the occasion of his 65th birthday and published as part of the Suhai Festschrift Issue.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Bondar, AN., Smith, J.C. & Elstner, M. Mechanism of a proton pump analyzed with computer simulations. Theor Chem Acc 125, 353–363 (2010). https://doi.org/10.1007/s00214-009-0565-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00214-009-0565-5