
Abstract.
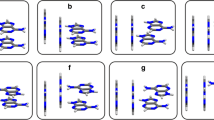
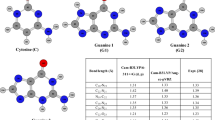
Ab initio calculations with inclusion of correlation effects at the MP2/6-31G* level have been used to predict the interaction energy of stacked cytosine dimer (C/C) as a function of twisting and sliding in the gas phase. Systematic calculations have also been carried out on the solvation free energies of various rotated and translated C/C dimers using a polarized continuum model approach at the HF/6-31G* level with a view to probe the role of various degrees of freedom on the free energy of solvation of the C/C dimer. The interaction energy of the C/C dimer decreases upon changing from a parallel to an antiparallel conformation in the gas phase. The 180°-rotated conformation has been found to be the most stable arrangement when compared to other rotated positions. The rotated and translated dimers exhibit lower solvation free energy than the parallel conformation. The decrease in the dipole moment upon rotation from the parallel to the antiparallel conformation indicates the cancellation of charge distribution upon rotation in the z direction of one cytosine base with respect to the other. The calculation reveals that the present approach could not yield association energy, ΔΔG Asso, in a solvent medium. This may be due to the fact that in the case of floppy molecules the contribution from translational, rotational and vibrational free energies plays a significant role in the calculation of ΔΔG Asso.
Similar content being viewed by others

Author information
Authors and Affiliations
Additional information
Received: 13 December 2001 / Accepted: 25 March 2002 / Published online: 13 June 2002
Rights and permissions
About this article
Cite this article
Amutha, R., Subramanian, V. & Nair, B. Role of twisting and sliding on the solvation of a stacked cytosine dimer: an ab initio study. Theor Chem Acc 107, 343–350 (2002). https://doi.org/10.1007/s00214-002-0351-0
Issue Date:
DOI: https://doi.org/10.1007/s00214-002-0351-0