
Abstract
Rationale
The nicotine discriminative stimulus has been linked to β2-containing (β2*) nicotinic receptors, with little evidence of a role for α7 nicotinic receptors, because nicotine discrimination was very weak in β2 null mutant mice but normal in α7 mutants.
Objectives
As both α7 and β2* nicotinic receptors have been implicated in nicotine-stimulated dopamine overflow, this study focused on the dopamine-mediated element in the nicotine stimulus by examining cross-generalisation between amphetamine and nicotine.
Materials and methods
Male α7 nicotinic receptor null mutant mice and wild-type controls were bred in-house and trained to discriminate nicotine (0.8 mg/kg) or (+)-amphetamine (0.6 mg/kg) from saline in a two-lever procedure with a tandem VI-30 FR-10 schedule of food reinforcement. Dopamine release from striatal slices was determined in parallel experiments.
Results
An α7 nicotinic receptor-mediated component of dopamine release was demonstrated in tissue from wild-type mice using choline as a selective agonist. This response was absent in tissue from null mutant animals. The mutation did not influence acquisition of drug discriminations but subtly affected the results of cross-generalisation tests. In mice trained to discriminate nicotine or amphetamine, there was partial cross-generalisation in wild-type mice and, at certain doses, these effects were attenuated in mutants. Further support for an α7 nicotinic receptor-mediated component was provided by the ability of the α7 nicotinic receptor antagonist methyllycaconitine to attenuate responses to nicotine and amphetamine in wild-type mice.
Conclusions
These findings support the concept of an α7 nicotinic receptor-mediated dopaminergic element in nicotine discrimination, warranting further tests with selective dopamine agonists.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Much progress has been made in identifying the nicotinic receptor subtypes at which the behavioural effects of nicotine originate and in characterising their interactions with other neurotransmitter systems. Nevertheless, evidence remains sparse for the behavioural significance of the widely distributed homomeric α7 nicotinic receptors, as discussed below. The studies described in this paper constitute a new assessment of the possible role of these receptors in drug discrimination, using mutant mice which lack α7 nicotinic receptors and in which the component of striatal dopamine release mediated by these receptors is absent.
The nicotine discriminative stimulus can serve as a sensitive and pharmacologically specific, non-invasive endpoint for studies of its mode of action in the central nervous system (Morrison and Stephenson 1969; Hirschhorn and Rosecrans 1974; Stolerman et al. 1983). Such discriminative properties of drugs are often interpreted as reflecting their subjective effects in humans and they typically allow the construction of steeply ascending, monotonic, dose–response curves.
Converging lines of evidence suggest that heteromeric nicotinic receptors constitute the major target for nicotine as a discriminative stimulus (reviewed by Smith and Stolerman 2008). There is a strong correlation between the affinity of nicotinic ligands at the binding site for tritiated nicotine and their potency in behavioural tests of generalisation with the nicotine stimulus (Reavill et al. 1988); these binding sites were subsequently identified as marking predominantly nicotinic receptors of the α4β2 subtype (Picciotto et al. 1995; Marubio et al. 1999). It was also established that the β2* antagonist dihydro-β-erythroidine (DHβE) potently shifted the dose–response curve for nicotine discrimination to the right in both rats and mice (Stolerman et al. 1997; Gommans et al. 2000). In contrast, the α7 nicotinic receptor antagonist methyllycaconitine (MLA) did not block nicotine discrimination in these species (Brioni et al. 1996; Stolerman et al. 1997; Gommans et al. 2000). Finally, null mutant mice lacking β2* nicotinic receptors were unable to discriminate nicotine at the usual training doses of 0.4–0.8 mg/kg and showed only poor discrimination when retrained at 1.6 mg/kg (Shoaib et al. 2002). In contrast, α7 null mutant mice showed unimpaired acquisition of nicotine discrimination and normal dose–response functions that were indistinguishable from those of wild-type, littermate controls (Stolerman et al. 2004). These findings support the evidence that some other behavioural effects of nicotine relevant to its abuse in tobacco, notably its ability to serve as a positive reinforcer, also involve β2* nicotinic receptors (Picciotto et al. 1998; Maskos et al. 2005).
The possible role of dopamine in the discrimination of nicotine was investigated in the light of several studies in which rats trained to discriminate nicotine showed partial generalisation to amphetamine or to non-selective dopamine agonists such as apomorphine and cocaine (Chance et al. 1977; Reavill and Stolerman 1987; Mansbach et al. 1998). Co-administration of nicotine with amphetamine potentiated the partial generalisation to amphetamine (Reavill and Stolerman 1987). Complete generalisation to amphetamine has occurred in some experiments (Stolerman 1989; Stolerman and Garcha 1989) and the dopamine uptake blocker GBR 12,909 was also fully generalised with nicotine (Gasior et al. 1999). The selective D1 agonists SKF38393 and SKF81297 were partially generalised with nicotine whereas studies with other selective ligands did not support a role for D2, D3 or D4 receptors (Reavill and Stolerman 1987; Mansbach et al. 1998). However, D1 and D2 dopamine antagonists have produced only incomplete and unconvincing blockade, even at doses that greatly reduced overall rates of responding (Reavill and Stolerman 1987; Corrigall and Coen 1994; Mansbach et al. 1998). An inhibitor of dopamine release, ICS-205930, yielded a similar partial blockade of nicotine discrimination (Schechter and Meehan 1991). These experiments suggested that dopamine may have played only a minor role in nicotine discrimination and that no single dopamine receptor subtype was uniquely involved. However, studies on the discrimination of drug mixtures (such as those containing nicotine and a benzodiazepine) have clarified relevant principles; such experiments indicated that clear antagonism is not seen when a drug is used to block only one element in a compound drug stimulus (Mariathasan et al. 1997); this may explain why experiments with antagonists were generally less successful than those with agonists in demonstrating a role for dopamine in nicotine discrimination.
The ability of nicotine to release dopamine, both in vitro and in vivo, is well-established (Wonnacott et al. 2005; Grady et al. 2007). The characteristics of nicotine-induced dopamine release support the view that it is likely to be an important mediator of behavioural effects. Nicotine-induced dopamine release can occur at low in vitro concentrations of nicotine that approximate to those found in the plasma of cigarette smokers (Rowell 1995). It is suppressed in both α4 and β2 null mutant mice (Marubio et al. 2003), implicating α4β2* nicotinic receptors. The subtypes of nicotinic receptors present in cell body areas and terminal fields capable of regulating dopamine release are numerous and complex (see Wonnacott et al. 2005; Grady et al. 2007). For example, heteromeric β2* receptors that contain α4 and/or α6 subunits (plus α5 or β3 subunits) are found at both somatodendritic and presynaptic locations on dopaminergic neurones (Klink et al. 2001; Salminen et al. 2007). Selective antagonists can be used to discriminate between the α4β2* and α6β2* classes of nicotinic receptor: DHβE preferentially inhibits all β2* nicotinic receptors whereas, in contrast, αconotoxin MII (αCTxMII) acts at α6β2* nicotinic receptors (Mogg et al. 2002; Salminen et al. 2007). However, in rats at least, dopamine release can also be increased indirectly by α7 nicotinic receptors on glutamatergic afferents in the ventral tegmental area (Schilström et al. 1998, 2003; Mansvelder and McGehee 2000) and in the striatum (Kaiser and Wonnacott 2000). Both α7 selective agonists such as choline (Alkondon et al. 1997, Barik and Wonnacott 2006) and antagonists such as αbungarotoxin (Kaiser and Wonnacott 2000) have been deployed to implicate this receptor subtype (Alkondon et al. 1997; Barik and Wonnacott 2006).
The putative involvement of α7 nicotinic receptors in the regulation of dopamine release has suggested a need for a further examination of the role of these receptors and dopamine in nicotine discrimination. Other reports have implicated α7 receptors in several behavioural effects of nicotine, including potentiation of brain stimulation reward (Panagis et al. 2000), self-administration (Markou and Paterson 2001) and in signs of nicotine withdrawal (Nomikos et al. 1999). All of these in vivo reports have relied upon the antagonist MLA as a selective tool for investigating the role of α7 nicotinic receptors; unfortunately, this substance also has quite potent antagonist activity at α6β2* nicotinic receptors that are also found on dopamine neurones (Klink et al. 2001; Mogg et al. 2002). Cohen et al. (2003) confirmed earlier reports that MLA does not block discrimination in rats trained with a 0.4-mg/kg dose of nicotine; interestingly, they found that MLA partially blocked generalisation to nicotine in rats trained to discriminate (+)-amphetamine (0.5 mg/kg). Thus, testing the effects of nicotine in amphetamine-trained rats may have unmasked a possible α7 nicotinic receptor-mediated component in the nicotine stimulus.
We have attempted a novel approach using α7 null mutant mice in conjunction with amphetamine to emphasise the role of dopamine in a discriminative response to nicotine. If generalisation from nicotine to amphetamine is a consequence primarily of the dopamine-releasing properties of nicotine, then from Cohen et al. (2003), it can be hypothesised that this release involved α7 nicotinic receptors. In comparison with simply the response to nicotine in rats trained with nicotine, the dopamine-mediated component of the response to nicotine may be enhanced when cross-generalisation between amphetamine and nicotine is tested. Thus, although α7 null mutant mice showed normal discrimination performance when nicotine was used for training (Stolerman et al. 2004), it was hypothesised that they would show weaker generalisation from amphetamine to nicotine. Different groups of α7 null mutant and wild-type mice were, therefore, trained to discriminate either nicotine or amphetamine and dose–response curves for both of these drugs were obtained in all animals. Tests for antagonism by MLA were also carried out to determine whether the interesting results of Cohen et al. (2003) in rats could be reproduced in mice. We also took advantage of a comparison of wild-type and null mutant mice to confirm the contribution of α7 nicotinic receptors to dopamine release from striatal slices in vitro.
Materials and methods
Subjects
The animals were descendants of mice constructed by Orr-Urtreger et al. (1997) and were bred from heterozygotes purchased from The Jackson Laboratory, USA (B6.129S7-Chrna7 tm1Bay, stock no. 003232). These were at backcross generation N8. Heterozygous pairs were used for all subsequent breeding. A total of 21 α7 male mutant mice and 22 male wild-type littermate control mice were used in the behavioural studies. Tissues from a further 10 mutant and 10 wild-type mice were used in the ligand-binding and the dopamine release studies. After weaning and ear-punching at about 3 weeks of age, genotypes were determined by PCR using the primers IMR1002 (CCT ggT CCT gCT gTg TTA AAC TgC TTC), IMR1003 (CTg CTg ggA AAT CCT Agg CAC ACT TgA g) and IMR1004 (gAC AAg ACC ggC TTC CAT CC). Mice were about 11 weeks old at the start of experiments and were housed individually in a room at a controlled temperature (21°C) with a 12-h light–dark cycle (light from 0730 to 1930 hours). The studies complied with local ethical requirements and were carried out in accordance with the Animals (Experimental Procedures) Act, 1986 under licence from the UK Home Office.
Apparatus
Six standard operant conditioning chambers for mice (MED Associates, Georgia, VT, USA) contained in ventilated, sound-attenuating housings were used for drug discrimination experiments. Each chamber was equipped with two ultra-sensitive levers, one on each side of a recess in which a dispenser could deliver 25 mg pellets of food. System control, data acquisition and storage were accomplished with Arachnid software (Paul Fray, Cambridge) running under RISC OS. Dopamine release from striatal slices was determined by filtration, using a Millipore vacuum manifold for 96-well filter plates (model MABVN1250, Millipore, Hertfordshire, UK).
Drug discrimination: training procedure
The procedure was based on Stolerman et al. (1999) and Shoaib et al. (2002). Mice were placed on a restricted diet to maintain them at 85% of their free-feeding weights. Water was available ad libitum. Initially, mice were trained to respond for food pellets delivered automatically under a variable-time 30-s schedule; during this stage of the procedure, no response levers were present in the chambers. One lever was then inserted into each chamber and training to lever-press began, initially on a continuous reinforcement schedule and then under fixed-ratio schedules that increased progressively to FR-10. The duration of each session was 15 min. After this response was acquired, the original lever was removed and a single lever was inserted on the opposite side of each chamber. The mice were again trained to perform under the FR-10 schedule of food presentation. A variable interval (VI) component was then introduced into the schedule. Under this tandem schedule of reinforcement, the tenth bar press after a randomly determined, variable interval of time was reinforced (tandem VI FR schedule). The VI component of the schedule makes it difficult for subjects to distinguish between training sessions and extinction tests, thus allowing the use of a graded index of discriminative response in the latter (Stolerman 1991); the FR component strengthens drug-induced stimulus control (Stolerman 1989). Initially, the mean value of the VI component was 15 s; after five sessions, this was increased to the final value of 30 s (range 7–53 s). Responses during the intervening periods were recorded but not reinforced. One wild-type mouse ceased responding during this period; attempts to reinstate lever-pressing were not successful and it was removed from the study.
Once a baseline of tandem VI-30s FR-10 responding on either lever was obtained, discrimination training commenced. At this point, both levers were present during training. The 21 mutant and the 21 wild-type mice were each divided into two groups. One group of mutant and one group of wild-type mice were trained to discriminate nicotine (0.8 mg/kg s.c.) from saline (n = 10 per group). Different groups of mutant and wild-type mice were trained to discriminate amphetamine (0.6 mg/kg s.c.) from saline (n = 11 per group). These doses were based on previous work (Beardsley et al. 2001; Stolerman et al. 1999). For half of the mice in each group, presses on the left lever were reinforced after drug injections and presses on the right lever were reinforced after saline injections; these arrangements were reversed in the remaining animals. Each mouse had a unique order of drug and saline sessions and the sequence of injections was random except that there were never more than three drugs or saline sessions in succession. Nicotine and amphetamine were administered 10 min prior to training sessions. The duration of the sessions was 15 min.
Drug discrimination: extinction tests
Discrimination performance appeared to have stabilised after 60 training sessions and, subsequently, dose–response (generalisation) tests were carried out. Ten minutes after injection of nicotine or amphetamine, mice were placed in the chambers for 5 min with both levers present but no reinforcers available. In the final experiment, a separate injection of MLA or saline was given 30 min before tests. Mice were tested on Tuesdays and Fridays with saline training sessions on Monday and Thursday and drug training on Wednesday of each week. In each experiment, different treatments were tested once each in every mouse in unique random sequences.
Radioligand binding
P2 membranes were prepared from individual mouse brains by homogenisation and differential centrifugation as previously described (Davies et al. 1999). The final pellet was resuspended in 2.5 mL/mg original weight in ice-cold 50 mM phosphate buffer and stored at −20°C until assay. Protein concentration was estimated by using a colourimetric protein dye reagent (Bradford 1976).
125I-αbungarotoxin (125I-αBgt) binding to mouse brain membranes was carried out according to Whiteaker et al. (2000) with some modifications. 125I-αBgt binding was performed on 250 μg of membranes in a final volume of 200 μL of phosphate buffer supplemented with 0.1% BSA, (pH 7.4) and 125I-αBgt to give a final concentration of 10 nM. Non-specific binding (approximately 15–20% of total binding) was determined in the presence of 1 mM nicotine. Samples were incubated for 3 h at 37°C, then 1 mL of buffer was added to each tube and incubation at 37°C continued for a further hour. Samples were then transferred at 4°C for 30 min before filtration through Gelman glass fibre grade A (GFA) filters, pre-soaked overnight in 0.3% polyethyleneimine (PEI) and 4% milk powder, using a Brandel cell harvester. Filters were washed with ice-cold phosphate buffered saline and counted for radioactivity using a Packard 1600 Tricarb scintillation counter; counting efficiency for 125I-αBgt was 60%. Each assay was conducted in triplicate.
Studies on [3H]-epibatidine binding were carried out as previously described (Sharples et al. 2000) using 150 μg of brain membranes in a final volume of 1 mL (NaCl 118 mM, KCl 4.8 mM, CaCl2 2.5 mM, MgSO4 2 mM, Hepes 20 mM, Tris 20 mM, phenylmethylsulphonylfluoride 0.1 mM and 0.01% sodium azide, pH 7.4). The final concentration of [3H]-epibatidine was 500 pM. Non-specific binding (approximately 5–10% of total binding) was determined in the presence of 1 mM nicotine. Samples were incubated for 1.5 h at room temperature, followed by 30 min at 4°C. Then samples were filtered through Gelman GFA filters, pre-soaked overnight in 0.3% PEI, using a Brandel cell harvester. Filters were washed three times with ice-cold PBS and counted for radioactivity as described above (counting efficiency 45%). Each assay was conducted in triplicate.
[3H]dopamine release from striatal slices
Assessment of [3H]dopamine release from striatal slices was performed using a 96-well assay (Anderson et al. 2000; Jacobs et al. 2002). For each experiment, two mice were killed by cervical dislocation. Brains were rapidly removed and striata dissected and transferred to ice-cold Krebs buffer (KB: NaCl 118 mM, KCl 2.4 mM, CaCl2 2.4 mM, KH2PO4 1.2 mM, MgSO4·7H2O 1.2 mM, NaHCO3 25 mM, d-glucose 10 mM, ascorbic acid 1 mM, gassed with 95% O2 and 5% CO2 for at least 1 h at 37°C; pH was adjusted to 7.4). Tissue was chopped using a McIlwain tissue chopper to give 150 μm prisms and rinsed twice in warm KB. Slices were loaded with [3H]dopamine by incubation for 30 min at 37°C with 50 nM [3H]dopamine in 2.5 mL KB supplemented with 10 μM pargyline (to prevent [3H]dopamine degradation). After five washes in KB containing 10 μM pargyline and 0.5 μM nomifensine (to prevent [3H]dopamine reuptake), slices were resuspended in 5 mL KB and distributed into one half of a 96-well filter plate. Thus, striatal slices from wild-type and mutant mice could be compared in parallel on the same filter plate. Tissue was incubated for 5 min with buffer in the presence or absence of antagonist. Buffer was removed by filtration (basal values) and replaced with buffer (70 μL) containing agonist and/or antagonist. After a further 5 min at 37°C, samples were filtered again and buffer containing released [3H]dopamine was collected in a 96-well Packard Optiplate™ (Perkin Elmer, Belgium). Microscint™ (170 μL per well) was added to Optiplates and each well was counted for 1 min using a Microbeta liquid scintillation counter (Wallac 1450 Microbeta Trilux, Perkin Elmer, Finland); counting efficiency 30%. To determine the amount of tritium remaining in the slices, each filter of the 96-well filter plate was removed, transferred to scintillation vials containing 4 mL of Optiphase™ and counted for 1 min using a Packard 1600 Tricarb liquid scintillation counter (counting efficiency 45%). The amount of [3H]dopamine released was expressed as a percentage of total radioactivity taken up by the slices prior to stimulation (i.e. amount of tritium released+tritium remaining in the tissue), giving a fractional release of [3H]dopamine. Each experiment was performed in six replicates and repeated at least three times.
Data analyses
For drug discrimination data, two- or three-factor repeated-measure analyses of variance (ANOVA) were carried out, followed when appropriate by multiple comparisons with control data by means of Dunnett and Tukey tests, and by t tests with the Bonferroni correction. The total number of responses served as an index of response rate. Data are presented as the means±SEM. The percentage of mice that first pressed the correct lever ten times was used to assess performance during acquisition of nicotine discrimination; drug stimulus generalisation in dose–response studies was assessed from the number of presses on the drug-appropriate lever expressed as a percentage of total presses (a minimum of 10 responses in total was required for calculating this index). Percentage data were subjected to arcsine transformation to normalise distributions, and statistical tests were performed with Unistat 5.6 (Unistat, London). Stolerman et al. (2006) presented a preliminary report on the behavioural studies described in this paper.
For [3H]dopamine release, data are presented as the means±SEM from at least four experiments. Statistical significance was determined using Student’s unpaired t test (Sigma Stat, Jandel scientific, Ekhrath, Germany).
Drugs
Nicotine bitartrate (BDH, Poole, Dorset, UK), amphetamine sulphate and MLA were dissolved in isotonic saline. The pH of nicotine solutions was adjusted to 7 with NaOH. All injections were given subcutaneously in a volume of 1 mL/100 g and all doses were calculated as those of the base. (+)-Amphetamine sulphate, choline, DHβE and MLA were obtained from Sigma Aldrich (Poole, Dorset, UK); αCtxMII was from Tocris Cookson (Avonmouth, Somerset, UK). [125I]αBungarotoxin (256 Ci/mmol) and [3H]-epibatidine (54 Ci/mmol) were obtained from Amersham International (Bucks, UK).
Results
Nicotinic receptor modulation of dopamine release from mouse striatal slices
The absence of assembled α7 nicotinic receptors in α7 null mutant mice was confirmed by the lack of specific binding of [125I]-αbungarotoxin to brain membranes (0.7 ± 2.7 fmol/mg protein, n = 9), whereas wild-type littermates showed 102 ± 10 fmol [125I]-αbungarotoxin bound per milligram of protein (n = 10), comparable to levels of α7 nicotinic receptors previously reported (Orr-Urtreger et al. 1997). Levels of [3H]-epibatidine binding sites (reflecting heteromeric β2* nicotinic receptors, predominantly high-affinity α4β2 receptors) were unchanged in α7 mutant mice compared with wild-type controls (64.3 ± 2.6 and 61.9 ± 3.4 fmol/mg of protein, respectively).
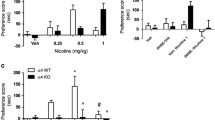
[3H]dopamine release elicited by stimulation of β2* nicotinic receptors was examined in wild-type and α7 nicotinic receptor null mutant mice, using a relatively low concentration of nicotine (1 μM) in the presence or absence of αconotoxin MII or DHβE. In both groups of animals, fractional release of [3H]dopamine in response to 1 μM nicotine was similar (wild-type = 8.0 ± 1.1%; α7 null mutant = 8.7 ± 1.1%; Fig. 1). In both cases, this response was inhibited by about 60% by αCTxMII (200 nM) and almost totally abolished by DHβE (10 μM). Thus, α7 nicotinic receptor null mutant mice did not show any difference in [3H]dopamine release modulated by β2* nicotinic receptors compared with wild-type mice.

Comparison of [3H]dopamine release from striatal slices prepared from wild-type (a) and α7 nicotinic receptor null mutant mice (b). [3H]dopamine release was determined by filtration and expressed as a percentage of the total [3H]dopamine present in the tissue (fractional release). Nicotine (1 μM) was applied in the absence (black bar) or presence of αCTxMII (200 nM, light grey bars) or DHβE (10 μM, open bars); antagonists were applied 5 min before nicotine and remained throughout the stimulation. α7 nicotinic receptors were stimulated by application of choline (1 mM; Cho; dark grey bars). Values are the mean±SEM from four independent experiments. **p < 0.01, ***p < 0.001 significantly different from corresponding nicotine control; §§§p < 0.001 significantly different from wild-type choline response, Student’s t test
In contrast, there were marked differences in the responses to the selective α7 nAChR agonist choline (1 mM) between the two groups of mice. Choline evoked [3H]dopamine release from striatal slices derived from wild-type mice; this was about 60% of the response to 1 μM nicotine (Fig. 1a). In tissue from α7 null mutant mice, choline failed to elicit any response (Fig. 1c), consistent with the absence of 125I-αBgt binding sites and α7 nicotinic receptors.
Acquisition of drug discriminations
Initial accuracy was close to the chance level of 50% for both amphetamine- and nicotine-trained subjects. Data for each group were examined by means of two-factor ANOVA in which the factors were genotype and drug treatment. Figure 2 shows that, as training proceeded, accuracy increased progressively to final levels of approximately 90% for all groups (F(4,67) = 27.3 and F(4,76) = 32.6 for nicotine and amphetamine-trained subjects, respectively; p < 0.001 in both cases). There were no overall differences due to genotype (F(1,18) = 2.10 and F(1,20) < 1 for nicotine and amphetamine). In the case of nicotine-trained subjects (Fig. 2a), there was also no interaction between the training and genotype factors (F(4,67) = 1.22). For training with amphetamine, there was a trend towards a significant interaction (F(4,76) = 2.47, p = 0.052), but the pattern over time was not orderly (Fig. 2c) and not even the largest difference between the genotypes (at trial block 4) was significant (t(19) = 2.04, p = 0.28 after Bonferroni correction for multiple comparisons). No variation in ambient conditions occurred that could account for the relatively poor discrimination performance of the wild-type mice at trial block 4.

Progressive increases in discrimination accuracy during acquisition in mice trained to discriminate nicotine (0.8 mg/kg) (a) or amphetamine (0.6 mg/kg) (b) from saline. For each drug, results are shown for wild-type (solid symbols) and α7 null mutant mice (open symbols). Data are shown as percentages of sessions in which rats selected the correct lever for five successive blocks of 10 sessions each (means±SEM, n = 10–11). Dashed horizontal line indicates chance level responding (50%)
Dose–response curves for training drugs
The results for tests with nicotine (0.1, 0.2, 0.4, 0.8 and 1.2 mg/kg) and saline in mice trained to discriminate nicotine are presented first. The percentage of drug-lever responding increased with the dose of nicotine for both genotypes (F(5,81) = 48.6, p < 0.001) and there was no overall phenotypic difference (F(1,18) < 1). Inspection of Fig. 3a suggests that the dose–response curve for nicotine may have been slightly steeper in wild-type than in mutant mice but the genotype × dose interaction was not significant (F(5,81) = 1.80). Figure 3c shows that administering nicotine in doses of 0.8–1.2 mg/kg reduced the overall rate of responding (F(5,90) = 21.9, p < 0.001) and this effect was also similar in both wild-type and mutant subjects (main effect of genotype F(1,18) < 1; interaction F(5,90) < 1).

Dose–response curves for nicotine and amphetamine in wild-type (solid symbols) and α7 null mutant mice (open symbols) trained to discriminate these drugs from saline (n = 10). a shows responding on the drug-appropriate lever expressed as a percentage of the total numbers of responses on both levers for nicotine-trained mice whereas b shows comparable data for amphetamine-trained mice. c and d show the total numbers of responses on both levers. All data are shown as the means±SEM from 5-min extinction tests. Data for drug-lever responses in the tests with 1.2 mg/kg of nicotine are for six mice only due to response suppression in other animals
Test results for amphetamine (0.05, 0.1, 0.2, 0.4 and 0.6 mg/kg) and saline are shown in Fig. 3b for mice trained to discriminate 0.6 mg/kg of amphetamine. There were orderly increases in the percentage of drug-appropriate responding (F(5,100) = 88.4, p = 0.001) and there was no difference between the genotypes (interaction F(5,100) < 1). From Fig. 3d, it can be seen that the dose of amphetamine did not influence the response rate.
Mice trained with nicotine: dose–response curves for amphetamine
In the first of two experiments, amphetamine tested in doses of 0.2, 0.4, 0.6 and 1.0 mg/kg increased the percentage of drug-appropriate responding in a dose-related manner to a maximum of approximately 60%. To aid clarity of interpretations, two separate two-factor ANOVA were carried out; the first ANOVA was on the data for drug-appropriate responding in the control tests with nicotine and saline, whereas the second ANOVA was on the data for saline and the four doses of amphetamine. In each case, the factors were genotype and drug treatment.
Considering the control data first, nicotine greatly increased drug-appropriate responding (F(1,17) = 264, p < 0.001). Inspection of Fig. 4a indicates that drug-appropriate responding was 79.9 ± 5.8% in the mutant mice compared with 92.8 ± 3.0% in the wild-types, but this potential difference was not adequately supported by the statistical analysis (for genotype × drug interaction, F(1,17) = 4.13, p = 0.058). There was also no overall effect of genotype (F(1,17) = 1.66). The second ANOVA confirmed the partial generalisation to amphetamine (F(4,65) = 9.86, p < 0.001), but there was no genotype × dose interaction (F(4,65) = 1.23). In Fig. 4a, a trend towards a stronger effect of amphetamine (0.6 mg/kg) in the wild-type mice can be discerned. Neither nicotine nor amphetamine significantly altered response rates and this measure was also insensitive to genotype (Fig. 4c).

Dose–response curves for amphetamine in wild-type and α7 null mutant mice trained to discriminate nicotine (0.8 mg/kg) from saline (n = 9–10). Solid symbols show results for wild-type mice; open symbols show results for mutants. a and b show percent responding on the drug-appropriate lever in two separate experiments in the same animals. c and d show total numbers of responses on both levers. Data above C show performance after administration of saline (circles) or 0.8 mg/kg of nicotine (squares). All data are shown as the means±SEM from 5-min extinction tests
In order to investigate further the trends seen in the preceding experiment, a partial replication of the study was carried out in the same mice; all saline and nicotine control tests were repeated, whereas amphetamine was tested in doses of 0.6 and 1.0 mg/kg only. In the control tests, the effect of nicotine on drug-appropriate responding was significant (F(1,16) = 350, p < 0.01), as was the genotype × nicotine interaction (F(3,47) = 5.93, p < 0.05). The slightly greater effects of nicotine in the wild-type mice accounted for the interaction and confirmed the corresponding trend seen in the previous experiment. A separate analysis of the data for tests with saline and the two doses of amphetamine confirmed the dose-related partial generalisation apparent in Fig. 4b (F(2,31) = 33.4, p < 0.001) and this effect was greater in the wild-type than in the mutant mice (genotype F(1,16) = 5.01, p < 0.05; interaction F(2,31) = 4.75, p < 0.02). Again, neither nicotine nor amphetamine significantly altered the response rates (Fig. 4d). In view of the fact that the response to amphetamine was genotype-dependent in the second study only, an additional ANOVA was carried out on the pooled data for only the two doses of amphetamine that were tested in both studies (i.e. 0.6 and 1.0 mg/kg). This ANOVA showed a genotype-dependent effect for amphetamine (F(1,17) = 5.98, p < 0.05).
Mice trained with amphetamine: dose–response curves for nicotine
In the first of two experiments, nicotine tested in doses of 0.2, 0.4, 0.6 and 0.8 mg/kg increased the percentage of drug-appropriate responding to a maximum of 40.1 ± 11.4% in wild-type mice. This partial generalisation was not seen in the mutants for which the corresponding score was 6.1 ± 1.3% (Fig. 5a). Separate two-factor ANOVA were carried out on the data for drug-appropriate responding in the control tests with amphetamine and saline and on the data for nicotine and saline. In the control tests, amphetamine greatly increased drug-appropriate responding (F(1,17) = 391, p < 0.001) but there were no significant effect due to genotype or genotype × drug interaction. The ANOVA confirmed that nicotine increased drug-appropriate responding (F(4,67) = 8.10, p < 0.001) and that the scores for wild-type and mutant mice differed significantly (genotype F(1,17) = 5.84, p < 0.05; interaction F(4,67) = 5.70, p < 0.001). It is also apparent from Fig. 5c that the largest doses used of nicotine reduced response rates (F(3,51) = 9.76, p < 0.001) but there was no difference between genotypes (F(1,17) = 1.19) and no genotype × drug interaction (F(3,51) = 1.10).

Dose–response curves for nicotine in wild-type and α7 nicotinic receptor null mutant mice trained to discriminate amphetamine (0.6 mg/kg) from saline. Solid symbols show results for wild-type mice; open symbols show results for mutants. a shows percent responding on the drug-appropriate lever for 10 wild-type and nine mutant mice; b shows comparable results for 10 wild-type and eight mutant mice. c and d show total numbers of responses on both levers. Data above C show performance after administration of saline (circles) or 0.6 mg/kg of amphetamine (squares). All data are shown as the means±SEM from 5-min extinction tests. Data for percent drug-lever responses in the tests with 0.8 and 1.2 mg/kg of nicotine are for only eight wild-type and four mutant mice, respectively, due to response suppression in other animals
A second study of generalisation to nicotine was carried out to extend the observations to a larger dose and thus obtain more complete data for dose–response relationships. All saline and amphetamine control tests were repeated, whereas nicotine was tested in doses of 0.8 and 1.2 mg/kg only. In the control tests (Fig. 5b), amphetamine increased drug-appropriate responding (F(1,16) = 638, p < 0.001), but neither the main effect of genotype nor the genotype × nicotine interaction was significant. Amphetamine was also without significant effect on response rates. Separate analysis of the data for tests with saline and the two doses of nicotine confirmed the partial generalisation in the wild-type mice (drug effect F(2,24) = 10.6, p < 0.001), but the trend towards a greater effect in the wild-type than in the mutant mice was not significant (for genotype F(1,16) < 1; interaction F(2,24) = 1.92). Figure 5d shows that, as in the previous experiment, nicotine reduced response rates (F(2,24) = 10.6, p < 0.001). As the response to nicotine was smaller in the mutant mice in the first study only, an additional ANOVA was carried out on the pooled data for the 0.8-mg/kg dose of nicotine that was tested in both studies. This ANOVA showed a genotype-dependent effect for nicotine (F(1,19) = 9.73, p < 0.01).
Discrimination studies with methyllycaconitine
The results for mice trained to discriminate nicotine are considered first. MLA (10 mg/kg) attenuated the discriminative stimulus effect of nicotine but did not block it completely (Fig. 6a). Performance after administering only MLA did not differ from that after saline. One-factor repeated-measure ANOVA confirmed the significant effect of drug treatments (F(5,40) = 9.80, p < 0.001). Figure 6c shows that drug treatments also influenced response rates (F(5,40) = 3.51, p = 0.01). The number of responses after administering nicotine was lower than after saline (Tukey test, p < 0.051), although this effect was not apparent when MLA was co-administered with nicotine; the difference between performance in the presence and absence of MLA was not significant (p = 0.061).

Discriminative stimulus and response rate-reducing effects of nicotine and amphetamine after administration of the nicotinic α7 receptor antagonist methyllycaconitine (MLA) to wild-type mice. Results (means±SEM) are shown for tests with saline (Sal), nicotine (Nic) and amphetamine (Amp) in nine mice trained with nicotine (a) and 11 mice trained with amphetamine (b). Grey bars saline control pretreatment, black bars MLA 10 mg/kg. ***p < 0.001 when compared with saline controls; §§p < 0.01 and §§§p < 0.001 for comparisons of performance in the presence and absence of MLA. Data for percent drug-lever responses after administering nicotine without MLA to amphetamine-trained mice are for nine animals only due to response suppression in other animals
Mice trained to discriminate amphetamine showed a moderate increase in drug-appropriate responding after administration of nicotine (0.6 mg/kg) and MLA blocked this effect. MLA also attenuated the response to amphetamine (Fig. 6b). Nicotine (but not amphetamine) reduced response rates, and MLA blocked this effect (Fig. 6d). One-factor ANOVA confirmed the differences due to drug treatments for both discriminative effects (F(5,48) = 18.0, p = 0.001) and response rates (F(5,50) = 7.16, p = 0.001).
Discussion
The present studies provide the first evidence that deletion of the gene coding for the α7 subunit of neuronal nicotinic receptors may affect the characteristics of the nicotine discriminative stimulus. The finding that cross-generalisation between nicotine and amphetamine was impaired, albeit to a small extent, in the α7 null mutant mice supports the view that the dopamine-mediated element in the nicotine discriminative stimulus may be mediated through α7 receptors. Ligand-binding and dopamine release studies were also compatible with this view, as were the behavioural results with the nicotinic receptor antagonist MLA. Other findings, such as the negligible effect of the mutation on acquisition of nicotine discrimination and the partial cross-generalisation between amphetamine and nicotine, support previous observations in mice, rats and primates.
The absence of α7 nicotinic receptors in the mutant animals was indicated by the dramatic reduction in binding of 125I-αBgt, in agreement with the findings of Orr-Urtreger et al. (1997). Importantly, levels of heteromeric receptors defined by the binding of [3H]-epibatidine were unchanged. This is consistent with the conservation of β2* nicotinic receptor-mediated dopamine release in the α7 mutants, as suggested by the block of nicotine-stimulated dopamine release by αCTxMII and DHβE. The specificity of these antagonists is consistent with contributions of heteromeric α6β2* and α4β2* receptor subtypes (Klink et al. 2001; Salminen et al. 2007). Choline was used to selectively activate α7 nicotinic receptors (Alkondon et al. 1997). In agreement with previous findings for rat striatal slices (Kaiser and Wonnacott 2000; Barik and Wonnacott 2006), choline revealed a component of dopamine release in wild-type mice that can be attributed to α7 nicotinic receptors (Fig. 1). The absence of choline-stimulated dopamine release in striatal tissue from mutant mice confirms that α7 nicotinic receptors also enhance the release of this transmitter. Thus, the behavioural studies are built upon the preceding neurochemical background that supports important roles for both heteromeric nicotinic receptor subtypes and for homomeric α7 receptors in the regulation of dopamine overflow.
The lack of significant differences between wild-type and α7 null mutant mice in the acquisition of nicotine discrimination (Fig. 2) and in the dose–response and most of the other control data for nicotine (Figs. 3 and 4) confirm earlier results for α7 mice (Stolerman et al. 2004). The response after administration of the training dose of nicotine to the mutant mice was lower than that in the wild-type animals when it was administered as an active control in one experiment (Fig. 4b). In the previous studies involving training mice at two different doses of nicotine followed by dose–response tests, there was no instance of a significant difference between the genotypes with respect to responses to nicotine. Thus, in the experiments as a whole, there was no detectable difference between the genotypes with respect to their ability to discriminate nicotine from saline. There were also no differences between the effects of nicotine on overall rates of responding in the two genotypes in any experiment, confirming previous reports in studies both of nicotine discrimination and of its effects on responding under a fixed-ratio schedule of food reinforcement (Stolerman et al. 2004; Naylor et al. 2005). Therefore, the studies with mutant mice did not provide support for a role of α7 receptors in regulating rates of responding.
The present findings go beyond global assessments of the entire stimulus complex produced by nicotine by using amphetamine as a tool to investigate the role of stimulus elements mediated through dopamine and other monoamines. The findings of partial generalisation between amphetamine and nicotine in the wild-type animals extend to mice the similar observations made previously in rats and monkeys (Chance et al. 1977; de la Garza and Johanson 1983; Mansbach et al. 1998; Reavill and Stolerman 1987). Complete cross-generalisation between nicotine and psychomotor stimulants is rarer but has occurred in some experiments (Gasior et al. 1999; Stolerman and Garcha 1989). These behavioural observations, together with results from studies with selective dopamine agonists, antagonists and reuptake inhibitors, are typically explained as a consequence of the shared ability of nicotine and amphetamine to stimulate dopamine overflow (reviewed by Smith and Stolerman 2008). Characteristics of cross-generalisation between nicotine, amphetamine and the nicotinic agonist cytisine have also been interpreted as evidence for a dopamine-mediated element in the nicotine discriminative stimulus (Chandler and Stolerman 1997; Di Chiara 2000).
Comparisons of cross-generalisation between nicotine and amphetamine in the α7 null mutants test the role of α7 nicotinic receptors in the dopamine-mediated element in the nicotine stimulus. Cross-generalisation with amphetamine was significantly impaired in two experiments (Figs. 4b and 5a), whereas in other experiments there were similar but non-significant trends (Figs. 4a and 5b). Thus, the experiments suggested some weakening of cross-generalisation in the α7 null mutant mice, although the effect was apparent at only some of the drug doses tested.
Additional support for a role for α7 receptors in nicotine discrimination comes from the experiments with the antagonist MLA, which attenuated the discriminative stimulus effect of nicotine in wild-type mice (Fig. 6a). The doses used of MLA were reported previously as sufficient to occupy receptors in rat brain (Turek et al. 1995; Lockman et al. 2005). The weakened response to nicotine in animals trained to discriminate nicotine was unexpected and not seen in previous studies in either rats or mice (Brioni et al. 1996; Stolerman et al. 1997; Gommans et al. 2000). The reason for this difference is not known although it may be related to changes in protocols such as the use of a 0.6-mg/kg training dose of nicotine and a VI 30 s component in the tandem schedule of food reinforcement in the present studies compared with a 1.2-mg/kg dose and a VI 60 s interval in Gommans et al. (2000). MLA also weakened the partial generalisation to nicotine in wild-type mice trained to discriminate amphetamine (Fig. 6c), an effect predicted by the hypothesis that α7 receptors play a greater role in dopamine-mediated stimulus elements than in the nicotine stimulus as a whole. This finding extends to mice the observations of Cohen et al. (2003) on rats.
Interestingly, MLA also partly blocked the discriminative response to the training dose of amphetamine (Fig. 6b). This finding was unexpected because the concept underlying the studies assumed that amphetamine-stimulated dopamine release occurs downstream from the α7 nicotinic receptors. The block by MLA could be explained if tonic activity of nicotinic inputs to the dopamine system modulate amphetamine-induced dopamine overflow; a possible basis for such an effect resides with cholinergic cells in the pedunculopontine tegmental nucleus that project to the midbrain dopamine neurones of the ventral tegmental area (Oakman et al. 1995). However, the specificity of the dose used of MLA was not tested by determining its effects on responses to other training drugs in mice. MLA can also weaken the discriminative properties of Δ9-tetrahydrocannabinol in rats (Solinas et al. 2007). As noted above, MLA also has relatively potent antagonist activity at α6β2* nicotinic receptors (Klink et al. 2001; Mogg et al. 2002), although its potency in vivo may be limited by poor brain penetration in subjects exposed chronically to nicotine (Lockman et al. 2005).
The observations reported in this paper provide limited support for the hypothesis that α7 receptors have a minor role in the dopamine-mediated element of nicotine discrimination, whereas previous studies implicated only heteromeric nicotinic receptors such as α4β2*. The main new findings are (1) release of dopamine induced by the α7 agonist choline was absent in α7 null mutant mice; (2) in wild-type mice, there was partial generalisation between nicotine and amphetamine, both in animals trained to discriminate nicotine and in those trained to discriminate amphetamine; (3) cross-generalisation involving nicotine and amphetamine was weakened in the mutant mice; (4) MLA impaired the discriminative stimulus effect of nicotine in wild-type mice. Thus, dopamine-related effects appear to be more prominent in stimulus elements shared between nicotine and amphetamine than in the entire stimulus complex produced by nicotine. The following limitations need to be noted. Firstly, studies comparing selective dopamine agonists in wild-type and α7 null mutant mice are needed; the influence of amphetamine on monoamine neurotransmitters is not limited to the dopamine system, although the results of tests for a role of noradrenaline in nicotine discrimination have been negative (reviewed by Smith and Stolerman 2008). Finally, the present behavioural studies with MLA should be regarded as preliminary because only one dose of the antagonist was used and the attenuation of nicotine discrimination conflicted with some previous observations.
The evidence developed for a possible role of α7 nicotinic receptors was obtained by the combined use of behavioural cross-generalisation techniques and genetically modified mice to emphasise the possible dopamine-like element in the nicotine stimulus. Studies using other paradigms may benefit if they employ parallel approaches to identify mechanisms that contribute in only minor ways to the behavioural effects of nicotine.
References
Alkondon M, Pereira EF, Cortes WS, Maelicke A, Albuquerque EX (1997) Choline is a selective agonist of alpha7 nicotinic acetylcholine receptors in the rat brain neurons. Eur J Neurosci 9:2734–2742
Anderson DJ, Puttfarcken PS, Jacobs I, Faltynek C (2000) Assessment of nicotinic acetylcholine receptor-mediated release of [3H]-norepinephrine from rat brain slices using a new 96-well format assay. Neuropharmacology 39:2663–2672
Barik J, Wonnacott S (2006) Indirect modulation by α7 nicotinic acetylcholine receptors of noradrenaline release in rat hippocampal slices: interaction with glutamate and GABA systems and effect of nicotine withdrawal. Mol Pharmacol 69:618–628
Beardsley PM, Sokoloff P, Balster RL, Schwartz J-C (2001) The D3R partial agonist, BP 897, attenuates the discriminative stimulus effects of cocaine and d-amphetamine and is not self-administered. Behav Pharmacol 12:1–11
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Brioni JD, Kim DJB, O’Neill AB (1996) Nicotine cue: lack of effect of the a7nicotinic receptor antagonist methyllycaconitine. Eur J Pharmacol 301:1–5
Chance WT, Murfin D, Krynock GM, Rosecrans JA (1977) A description of the nicotine stimulus and tests of its generalization to amphetamine. Psychopharmacology 55:19–26
Chandler CJ, Stolerman IP (1997) Discriminative stimulus properties of the nicotinic agonist cytisine. Psychopharmacology 129:257–264
Cohen C, Bergis OE, Galli F, Lochead AW, Jegham S, Biton B, Leonardon J, Avenet P, Sgard F, Besnard F, Graham D, Coste A, Oblin A, Curet O, Voltz C, Gardes A, Caille D, Perrault G, George P, Soubrié P, Scatton B (2003) SSR591813, a novel selective and partial a4b2 nicotinic receptor agonist with potential as an aid to smoking cessation. J Pharmacol Exp Ther 306:407–420
Corrigall WA, Coen KM (1994) Dopamine mechanisms play at best a small role in the nicotine discriminative stimulus. Pharmacol Biochem Behav 48:817–820
Davies ARL, Hardick DJ, Blagbrough IS, Potter BVL, Wolstenholme AJ, Wonnacott S (1999) Characterisation of the binding of [3H]methyllycaconitine: a new radioligand for labelling α7-type nicotinic acetylcholine receptors. Neuropharmacology 38:679–690
de la Garza R, Johanson C-E (1983) The discriminative stimulus properties of cocaine in the rhesus monkey. Pharmacol Biochem Behav 19:145–148
Di Chiara G (2000) Role of dopamine in the behavioural actions of nicotine related to addiction. Eur J Pharmacol 393:295–314
Gasior M, Shoaib M, Yasar S, Jaszyna M, Goldberg SR (1999) Acquisition of nicotine discrimination and discriminative stimulus effects of nicotine in rats chronically exposed to caffeine. J Pharmacol Exp Ther 288:1053–1073
Gommans J, Stolerman IP, Shoaib M (2000) Antagonism of the discriminative and aversive stimulus properties of nicotine in C57BL/6J mice. Neuropharmacology 39:2840–2847
Grady SR, Salminen O, Laverty DC, Whiteaker P, McIntosh JM, Collins AC, Marks MJ (2007) The subtypes of nicotinic acetylcholine receptors on dopaminergic terminals of mouse striatum. Biochem Pharmacol 74:1235–1246
Hirschhorn ID, Rosecrans JA (1974) Studies on the time course and the effect of cholinergic and adrenergic receptor blockers on the stimulus effect of nicotine. Psychopharmacologia 40:109–120
Jacobs I, Anderson DJ, Surowy CS, Puttfarcken PS (2002) Differential regulation of nicotinic receptor-mediated neurotransmitter release following chronic (−)-nicotine administration. Neuropharmacology 43:847–856
Kaiser S, Wonnacott S (2000) a-Bungarotoxin-sensitive nicotinic receptors indirectly modulate [3H]dopamine release in rat striatal slices via glutamate release. Mol Pharmacol 58:312–318
Klink R, d’Exaerde Ad A, Zoli M, Changeux J-P (2001) Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J Neurosci 21:1452–1463
Lockman PR, Van der Schyf CJ, Abbruscato TJ, Allen DD (2005) Chronic nicotine exposure alters blood–brain barrier permeability and diminishes brain uptake of methyllycaconitine. J Neurochem 94:37–44
Mansbach RS, Rovetti CC, Freedland CS (1998) The role of monoamine neurotransmitter systems in the nicotine discriminative stimulus. Drug Alcohol Depend 52:125–134
Mansvelder HD, McGehee DS (2000) Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron 27:349–357
Mariathasan EA, Stolerman IP, White J-AW (1997) Antagonism of AND and AND–OR drug mixture discrimination in rats. Drug Alcohol Depend 44:31–34
Markou A, Paterson NE (2001) The nicotinic antagonist methyllycaconitine has differential effects on nicotine self-administration and nicotine withdrawal in the rat. Nicotine Tob Res 3:361–373
Marubio LM, del Mar Arroyo-Jimenez M, Cordero-Erausquin M, Léna C, Le Novère N, de Kerchove d’Exaerde A, Huchet M, Damaj MI, Changeux J-P (1999) Reduced antinociception in mice lacking neuronal nicotinic receptor subunits. Nature 398:805–810
Marubio LM, Gardier AM, Durier S, David D, Klink R, Arroyo-Jimenez MM, McIntosh JM, Rossi F, Champtiaux N, Zoli M, Changeux JP (2003) Effects of nicotine in the dopaminergic system of mice lacking the alpha4 subunit of neuronal nicotinic acetylcholine receptors. Eur J Neurosci 17:1329–1337
Maskos U, Molles BE, Pons S, Besson M, Guiard BP, Guilloux JP, Evrard A, Cazala P, Cormier A, Mameli-Engvall M, Dufour N, Cloez-Tayarani I, Bemelmans AP, Mallet J, Gardier AM, David V, Faure P, Granon S, Changeux JP (2005) Nicotine reinforcement and cognition restored by targeted expression of nicotinic receptors. Nature 436:103–107
Mogg AJ, Whiteaker P, McIntosh JM, Marks M, Collins AC, Wonnacott S (2002) Methyllycaconitine is a potent antagonist of alpha-conotoxin-MII-sensitive presynaptic nicotinic acetylcholine receptors in rat striatum. J Pharmacol Exp Ther 302:197–204
Morrison CF, Stephenson JA (1969) Nicotine injections as the conditioned stimulus in discrimination learning. Psychopharmacologia 15:351–360
Naylor C, Quarta D, Fernandes C, Stolerman IP (2005) Tolerance to nicotine in mice lacking alpha7 nicotinic receptors. Psychopharmacology 180:558–563
Nomikos GG, Hildebrand BE, Panagis G, Svensson TH (1999) Nicotine withdrawal in the rat: role of alpha7 nicotinic receptors in the ventral tegmental area. NeuroReport 10:697–702
Oakman SA, Faris PL, Kerr PE, Cozzari C, Hartman BK (1995) Distribution of pontomesencephalic cholinergic neurons projecting to substantia nigra differs significantly from those projecting to ventral tegmental area. J Neurosci 15:5859–5869
Orr-Urtreger A, Goldner FM, Saeki M, Lorenzo I, Goldberg L, De Biasi M, Dani JA, Patrick JW, Beaudet AL (1997) Mice deficient in the alpha7 neuronal nicotinic acetylcholine receptor lack alpha-bungarotoxin binding sites and hippocampal fast nicotinic currents. J Neurosci 17:9165–9171
Panagis G, Kastellakis A, Spyraki C, Nomikos G (2000) Effects of methyllycaconitine (MLA), an α7 nicotinic receptor antagonist, on nicotine- and cocaine-induced potentiation of brain stimulation reward. Psychopharmacology 149:388–396
Picciotto MR, Zoli M, Léna C, Bessis A, Lallemand Y, LeNovère N, Vincent P, Pich EM, Brûlet P, Changeux J-P (1995) Abnormal avoidance learning in mice lacking functional high-affinity nicotine receptor in the brain. Nature 374:65–67
Picciotto MR, Zoli M, Rimondini R, Léna C, Marubio LM, Pich EM, Fuxe K, Changeux J-P (1998) Acetylcholine receptors containing the β2 subunit are involved in the reinforcing properties of nicotine. Nature 391:173–177
Reavill C, Stolerman IP (1987) Interaction of nicotine with dopaminergic mechanisms assessed through drug discrimination and rotational behaviour in rats. J Psychopharmacol 1:264–273
Reavill C, Jenner P, Kumar R, Stolerman IP (1988) High affinity binding of [3H]-nicotine to rat brain membranes and its inhibition by nicotine analogues. Neuropharmacology 27:235–241
Rowell PP (1995) Nanomolar concentrations of nicotine increase the release of [3H]dopamine from rat striatal synaptosomes. Neurosci Lett 189:171–175
Salminen O, Drapeau JA, McIntosh JM, Collins AC, Marks MJ, Grady SR (2007) Pharmacology of alpha-conotoxin MII-sensitive subtypes of nicotinic acetylcholine receptors isolated by breeding of null mutant mice. Mol Pharmacol 71:1563–1571
Schechter MD, Meehan SM (1991) Further evidence for the mechanisms that may mediate nicotine discrimination. Pharmacol Biochem Behav 41:807–812
Schilström B, Nomikos GG, Nisell M, Hertel P, Svensson TH (1998) N-methyl-d-aspartate receptor antagonism in the ventral tegmental area diminishes the systemic nicotine-induced dopamine release in the nucleus accumbens. Neuroscience 82:781–789
Schilström B, Rawal N, Mameli-Engvall M, Nomikos GG, Svensson TH (2003) Dual effects of nicotine on dopamine neurons mediated by different nicotinic receptor subtypes. Int J Neuropsychopharmacol 6:1–11
Sharples CG, Kaiser S, Soliakov L, Marks MJ, Collins AC, Washburn M, Wright E, Spencer JA, Gallagher T, Whiteaker P, Wonnacott S (2000) UB-165: a novel nicotinic agonist with subtype selectivity implicates the α4β2* subtype in the modulation of dopamine release from rat striatal synaptosomes. J Neurosci 20:2783–2791
Shoaib M, Gommans J, Morley A, Stolerman IP, Grailhe R, Changeux JP (2002) The role of nicotinic receptor beta-2 subunits in nicotine discrimination and conditioned taste aversion. Neuropharmacology 42:530–539
Smith JW, Stolerman IP (2008) Recognising nicotine: the neurobiological basis of nicotine discrimination. In Henningfield JE, London ED, Pögün S (eds) Nicotine psychopharmacology: from molecule to behavior. Springer (in press)
Solinas M, Scherma M, Fattore L, Stroik J, Wertheim C, Tanda G, Fratta W, Goldberg SR (2007) Nicotinic α7 receptors as a new target for treatment of cannabis abuse. J Neurosci 27:5615–5620
Stolerman IP (1989) Discriminative stimulus effects of nicotine in rats trained under different schedules of reinforcement. Psychopharmacology 97:131–138
Stolerman IP (1991) Measures of stimulus generalization in drug discrimination experiments. Behav Pharmacol 2:265–282
Stolerman IP, Garcha HS (1989) Temporal factors in drug discrimination: experiments with nicotine. J Psychopharmacol 3:88–97
Stolerman IP, Pratt JA, Garcha HS, Giardini V, Kumar R (1983) Nicotine cue in rats analysed with drugs acting on cholinergic and 5-hydroxytryptamine mechanisms. Neuropharmacology 22:1029–1037
Stolerman IP, Chandler CJ, Garcha HS, Newton JM (1997) Selective antagonism of behavioural effects of nicotine by dihydro-β-erythroidine in rats. Psychopharmacology 129:390–397
Stolerman IP, Naylor C, Elmer GI, Goldberg SR (1999) Discrimination and self-administration of nicotine by inbred strains of mice. Psychopharmacology 141:297–306
Stolerman IP, Chamberlain S, Bizarro L, Fernandes C, Schalkwyk L (2004) The role of nicotinic receptor alpha7 subunits in nicotine discrimination. Neuropharmacology 46:363–371
Stolerman IP, Quarta D, Naylor, CG, Fernandes C (2006) The role of nicotinic alpha7 receptors in the dopamine-mediated component of nicotine discrimination. Paper presented at 68th Annual Scientific Meeting of the College on Problems of Drug Dependence, Scottsdale, Arizona, June. Available at http://www.cpdd.vcu.edu
Turek JW, Kang C-H, Campbell JE, Arneric SP, Sullivan JP (1995) A sensitive technique for the detection of the α7 neuronal nicotinic acetylcholine receptor antagonist, methyllycaconitine, in rat plasma and brain. J Neurosci Methods 61:113–118
Whiteaker P, Jimenez M, McIntosh JM, Collins AC, Marks MJ (2000) Identification of a novel nicotinic binding site in mouse brain using [125I]-epibatidine. Br J Pharmacol 131:729–739
Wonnacott S, Sidhpura N, Balfour DJ (2005) Nicotine: from molecular mechanisms to behaviour. Curr Opin Pharmacol 5:53–59
Acknowledgement
The research was supported by grants from the European Union and the British Medical Research Council.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Quarta, D., Naylor, C.G., Barik, J. et al. Drug discrimination and neurochemical studies in α7 null mutant mice: tests for the role of nicotinic α7 receptors in dopamine release. Psychopharmacology 203, 399–410 (2009). https://doi.org/10.1007/s00213-008-1281-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-008-1281-x