
Abstract
This current study reports, for the first time, on the potent cytotoxicity of (Z)-3-hexenyl-β-D-glucopyranoside, as well as its cellular and molecular apoptotic mechanisms against Panc1 cancer cells. The cytotoxicity of three compounds, namely (Z)-3-hexenyl-β-D-glucopyranoside (1), gallic acid (2), and pyrogallol (3), which were isolated from C. rotang leaf, was investigated against certain cancer and normal cells using the MTT assay. The cellular apoptotic activity and Panc1 cell cycle impact of compound (1) were examined through flow cytometry analysis and Annexin V-FITC cellular apoptotic assays. Additionally, RT-PCR was employed to evaluate the effect of compound (1) on the Panc1 apoptotic genes Casp3 and Bax, as well as the antiapoptotic gene Bcl-2. (Z)-3-hexenyl-β-D-glucopyranoside demonstrated the highest cytotoxic activity against Panc1 cancer cells, with an IC50 value of 7.6 µM. In comparison, gallic acid exhibited an IC50 value of 21.8 µM, and pyrogallol showed an IC50 value of 198.2 µM. However, (Z)-3-hexenyl-β-D-glucopyranoside displayed minimal or no significant cytotoxic activity against HepG2 and MCF7 cancer cells as well as WI-38 normal cells, with IC50 values of 45.8 µM, 108.7 µM, and 194. µM, respectively. (Z)-3-hexenyl-β-D-glucopyranoside (10 µM) was demonstrated to induce cellular apoptosis and cell growth arrest at the S phase of the cell cycle in Panc1 cells. These findings were supported by RT-PCR analysis, which revealed the upregulation of apoptotic genes (Casp3 and Bax) and the downregulation of the antiapoptotic gene Bcl-2. This study emphasizes the significant cellular potency of (Z)-3-hexenyl-β-D-glucopyranoside in specifically inducing cytotoxicity in Panc1 cells.
Graphical Abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Pancreatic cancer is one of the most fatal malignant tumors, posing a significant challenge in early diagnosis due to the absence of clear signs and symptoms. In advanced stages, pancreatic cancer exhibits a high mortality rate, with approximately 97% of patients expected to succumb within 5 years of diagnosis (Mario et al. 2018; Zhang et al. 2018). Disturbingly, the incidence, prevalence, and mortality rates of pancreatic cancer have all witnessed a global surge over the last 25 years, and projections indicate a further alarming increase of approximately 1.97-fold by 2060 (Mario et al. 2018).
Anticancer drugs continue to be a vital element in the treatment protocol for pancreatic cancer, whether used as standalone therapies or in conjunction with other modalities such as surgery, radiotherapy, and immunotherapy (Pliarchopoulou and Pectasides 2009; Neoptolemos et al. 2018).
Several plant extracts and metabolites have demonstrated remarkable cytotoxic activity against Panc1 cells, either when used alone or in combination with chemotherapeutic drugs. Notably, extracts derived from Scutellaria barbata, bitter apricot, Moringa oleifera, Delonix regia, Mesua ferrea, and the lipophilic fraction of Senecio creuntus have been reported to exhibit potent cytotoxic activity against Panc1 (Berkovich et al. 2013; Rajendran et al. 2016; Prescott et al. 2018; Wang et al. 2019; Aamazadeh et al. 2020; Awad et al. 2020; Shameli Rajiri et al. 2021; Malak et al. 2023).
Moreover, a variety of plant metabolites have exhibited potent cytotoxic activity against Panc1 cancer cells through diverse mechanisms. For example, berberine, a natural anticancer agent, has been identified as an inhibitor of Panc1 proliferation and an inducer of apoptosis (Rauf et al. 2021). Damnacanthal, isolated from Garcinia huillensis, has demonstrated the ability to induce necrotic death in Panc1 cells (Dibwe et al. 2012). Furthermore, curcumin, rhein, ellagic acid, embelin, metformin, and eruberin A are additional plant metabolites derived from various plant sources that exhibit cytotoxic effects against Panc1, with their respective mechanisms of action confirmed (Ramakrishnan et al. 2020). These findings suggest that further exploration of plant extracts and metabolites may lead to the discovery of novel sources of anticancer agents against Panc1, making them an area of significant scientific interest.
(Z)-3-hexenyl-β-D-glucopyranoside is a natural metabolite that has been extracted from various plants, including Thymus vulgaris, Mallotus furetianus, Schizonepeta tenuifolia, Camellia sinensis, Xanthoxylum piperitum, Celosia argentea, and Epimedium glandiflorum (Jiang et al. 2001; Kishida et al. 2005; Lee et al. 2008; Cui et al. 2016; Huang et al. 2018). Despite its prevalence in nature, there have been relatively few studies conducted on the biological activity of (Z)-3-hexenyl-β-D-glucopyranoside. One notable example is a study that identified antisteatotic activity in (Z)-3-hexenyl-β-D-glucopyranoside isolated from Mallotus furetianus (Huang et al. 2018).
Further research is warranted to delve into the potential biological activities and therapeutic applications of this natural metabolite.
Experimental
Solvents and chemicals for extraction and isolation of compounds
Hexane, methanol, dichloromethane, and ethyl acetate (Alfa chemicals, Cairo, Egypt) were employed for the extraction and isolation of metabolites. TLC plates (Merk, Darmstadt, Germany), RP-TLC plates (Merk, Darmstadt, Germany), normal column silica gel plates (Merk, Darmstadt, Germany), reversed phase C18 silica gel plates (Merk, Darmstadt, Germany), and Sephadex LH20 (Merk, Darmstadt, Germany) were utilized for the compound isolation process.
Plant materials
The leaves of Calamus rotang L. (Fig. 1) were gathered from the Aswan Botanical Garden, located in Aswan, Egypt, in October 2018. Dr. Amr M. M. Mahmoud, the Director of Aswan Botanical Garden at the Horticultural Research Institute, Agriculture Center, Egypt, completed the taxonomical identification of the plant. A voucher specimen with the number A20220906 was deposited at the herbarium of the Department of Pharmacognosy, School of Pharmacy, Assiut University, Egypt.

Photo of C. rotang leaf
Isolation of compounds 1–3
The dried powdered leaves (2 kg) were subjected to maceration in 70% methanol (Alfa chemicals, Cairo, Egypt) (5 L × 3 times) at room temperature. The solvents were combined, and the resulting solution was concentrated under reduced pressure, yielding 300 g of residue. This residue was then suspended in 10% methanol (1 L) and fractionated using n-hexane (Alfa chemicals, Cairo, Egypt) (1 L × 6 times), dichloromethane (Alfa chemicals, Cairo, Egypt) (1 L × 6 times), and ethyl acetate (Alfa chemicals, Cairo, Egypt) (1 L × 6 times). Each solvent fraction was subsequently concentrated individually using a rotavapor (Buchi, Labortechnik AG, Flawil, Switzerland), resulting in the isolation of the n-hexane fraction (70 g), dichloromethane fraction (25 g), ethyl acetate fraction (32 g), and aqueous fraction (150 g). The dried ethyl acetate fraction was then subjected to chromatographic isolation of the bioactive compounds. In summary, the ethyl acetate fraction was subjected to sub-fractionation using chloroform: methanol solvent mixtures with varying percentages of methanol (0%, 5%, 10%, 15%, 20%, 30%, 50%, and 100%). Compound 1 (10 mg) was isolated from sub-fraction 4, which was eluted using 15% methanol in chloroform. Upon TLC analysis sprayed with 10% H2SO4, compound 1 exhibited a reddish-black spot. Purification of compound 1 was achieved through multiple open columns employing normal silica gel, RP-18, and Sephadex LH-20. Compounds 2 and 3 (100 mg and 200 mg, respectively) were isolated from sub-fraction 5, eluted using 20% methanol in chloroform. Both compounds 2 and 3 yielded brown spots on the TLC sprayed with 10% H2SO4 and were isolated using the same purification method as compound 1. The isolated compounds were subsequently dried and stored for NMR spectroscopic analyses (Bruker, Billerica, Massachusetts, USA) and biological assays.
Cell culture
The cell lines PANC-1 (pancreatic carcinoma), MCF7 (breast carcinoma), HepG2 (hepatocellular carcinoma), and WI-38 (normal lung fibroblast cells) were obtained from the American Type Culture Collection (ATCC) located in Manassas, Virginia, United States. These cell lines were cultured in DMEM (Invitrogen, Life Technologies, Rockville, MD, USA) medium supplemented with 10% FBS (Fetal Bovine Serum, Hyclone, Thermo Fisher Scientific, Waltham, MA, USA) and 1% penicillin (Sigma-Aldrich, Louis, MO, USA). The cells were maintained at 37°C in a 5% CO2 and 95% humidity environment for a maximum period of two weeks to ensure their viability for further experimentation. Cell detachment was achieved using a solution containing 0.25% (w/v) trypsin (Sigma-Aldrich, Louis, MO, USA) and 0.53 mM EDTA ((Sigma-Aldrich, Louis, MO, USA), followed by re-cultivation in fresh media.
MTT assay
The cultivated cells were seeded in 96-well plates at a concentration of 1 × 104 cells/100 μL per well (Naik et al. 2014). The cultured plates were then incubated for 24 hours at 37°C. Stock solutions of the isolated compounds (1-3) and staurosporine (reference compound) were prepared at a concentration of 1 mg/mL in 10% DMSO (Sigma-Aldrich, Louis, MO, USA) in ddH2O. The tested concentrations of the compounds (1, 10, 30, and 100 µg/mL) in 0.01 - 0.1% DMSO in ddH2O were prepared by diluting the stock solutions with double-distilled water (ddH2O). After a 24-hour incubation of cells in 96-well plates, the media were replaced, and the tested concentrations of each compound, along with staurosporine, were added in triplicates (Esharkawy et al. 2022). A negative control using 0.1% DMSO in ddH2O was also included. The treated cells were further incubated for 48 hours at 37 °C. The media of the treated and control plates were removed and replaced by fresh media containing MTT (Sigma, Louis, MO, USA) reagent in a concentration of 1 mg/mL (Ponnusamy et al. 2016), then incubated for 2 hours. The viability of the cancer cells was determined by measuring the amount of formazan formed by viable cells. The produced formazan was solubilized by adding 100 µL 10% DMSO in ddH2O per each treated and control well. The plates were gently shacked for 5 min. The intensity of the produced colour was measured using an ELISA plate reader (Bio-Tek EL 800, Agilent technology, Santa Clara, CA, USA) at wavelength of 570 nm. The percent of cells viability was calculated by the following equation:
- AA:
-
Average of triplicate absorbances for each sample concentration
Calibration curves of the tested and reference compounds were then prepared to calculate the IC50 (the concentration that inhibits 50% of cancer cells) (Esharkawy et al. 2022).
Flow cytometry analysis of the cell DNA contents in Panc1 cells treated with (Z)-3-hexenyl-β-D-glucopyranoside
All reagents and kits for this assay were obtained from Abcam (Abcam Technology, Boston, MA, USA). The detection of DNA cell contents and cell cycle status was conducted following the established method (Xu et al. 2001). To provide a brief overview, Panc1 pancreatic cancer cells (ATCC, Manassas, Virginia, USA) were cultured in a single-cell suspension in DMEM medium (Invitrogen, Life Technologies, Rockville, MD, USA). Subsequently, the cells were fixed in 66% ethanol (Sigma-Aldrich, Louis, MO, USA) and kept on ice for 2 hours. Afterward, the cancer cells were washed with a PBS (Sigma-Aldrich, Louis, MO, USA) solution (5 mL of 10X PBS + 45 mL water), re-cultured in fresh medium, and incubated at 37 ºC for 24 hours. A concentration of 10 µM of (Z)-3-hexenyl-β-D-glucopyranoside was added in triplicate, chosen based on previously reported data (Ponnusamy et al. 2016; Xu et al. 2001) for 24h. The second dose of 10 µM of (Z)-3-hexenyl-β-D-glucopyranoside was added to the Panc1 cells then incubated for 24 h. Following a 48-hour incubation period from the first dose, the cells were trypsinized, fixed, and stained with propidium iodide-RNase enzyme reagent (9.45 mL PBS + 500 μL 20X propidium iodide + 50 μL 200X RNase) from Abcam Technology, Boston, MA, USA. The intensity of propidium iodide fluorescence, and thereby the amount of cellular DNA in each stage of the Panc1 cell cycle, was quantified using a flow cytometer (Novocyte, Agilent technology, Santa Clara, CA, USA) with an excitation maximum of 493nm and an emission maximum of 636 nm. The incubation time for fluorescence quantification was 30 minutes. The experiment was repeated twice.
Annexin V-FITC cellular apoptotic assay
The reagents used in this assay included the Annexin V-FITC kit (Abcam Technology, Boston, MA, USA), 1X binding buffer (Abcam Technology, Boston, MA, USA), and propidium iodide (Bio Vision Research Products, Mountain View, CA, USA). Panc1 cells were seeded in 6-well plates and incubated at 37 ºC for 24 hours. Compound 1 ((Z)-3-hexenyl-β-D-glucopyranoside) was then added to the cultivated Panc1 6-well plates at a concentration of 10 µM and incubated for 48 hours. Subsequently, the cancer cells were trypsinized and centrifuged for 10 minutes at 300 rpm. The resulting precipitate was resuspended in 500 µL of 1X binding buffer. Annexin V-FITC (5 µL) and propidium iodide (5 µL) reagents were added to the treated cancer cell plates and incubated for 5 minutes in the dark at room temperature. A flow cytometer (Novocyte, Agilent technology, Santa Clara, CA, USA) with an excitation maximum of 488 nm and an emission maximum of 530 nm was employed to measure the intensity of annexin-binding phosphatidylserine (PS) and consequently determine the amount of apoptotic cells (Koopman et al. 1994).
RNA isolation and SYBR® Green RT-PCR assay
The effects of 10 µM of (Z)-3-hexenyl-β-D-glucopyranoside on the mRNA expression of caspase3, bax, and Bcl-2 genes were investigated using the reverse transcription-polymerase RT-RNA technique. After a 48-hour incubation of (Z)-3-hexenyl-β-D-glucopyranoside in Panc1 (pancreatic carcinoma) cells, mRNA isolation of the selected proteins was performed using the RN easy extraction kit (Qiagen, Hilden, Germany), following previously described methods (Janicke et al. 1998). For real-time quantitative PCR of the RNA templates, a one-step RT-PCR kit with SYBR® Green (Bio-Rad SYBR Green PCR MMX, Hercules, California, USA) was added to a 25 μL 2X SYBR® Green RT-PCR reaction mix (Bio-Rad, Hercules, California, USA). The reaction mix contained 2X reaction buffer with 0.4 mM of each dNTP, magnesium chloride, iTaq DNA polymerase, 20 nM fluorescein SYBR® Green I dye, stabilizers, 11 μL nuclease-free water, 1.5 μL reverse primer (10 μM), 1.5 μL forward primer (10 μM), 1 μL iScript reverse transcriptase, and RNA template (1 pg to 100 ng total RNA). The cDNA synthesis and PCR amplifications were performed in the same tube using Rotor-Gene RT-PCR (Qiagen, Hilden, Germany) software 1.7 (Abed et al. 2021). The primers used in the RT-PCR reaction were obtained from Corbett Life Science Division (Reg. No. QEC21313), Corbett Research, Mortlake, Australia. The primer sequences used in the RT-PCR reaction were as follows:
-
Caspase3: F 5’-GGAAGCGAATCAATGGACTCTGG-3’,
-
Caspase3: R 5'-GCATCGACATCTGTACCAGACC -3',
-
Bcl-2: F 5’-ATGTGTGTGGAGACCGTCAA -3’,
-
Bcl-2: F 5’-GCCGTACAGTTCCACAAAGG -3’,
-
Bax: F 5’-TCAGGATGCGTCCACCAAGAAG-3’,
-
Bax: R 5’-TGTGTCCACGGCGGCAATCATC-3’
The Rotor-Gene RT-PCR reaction protocol was as follows: cDNA synthesis for 10 minutes at 50 ºC, iScript reverse transcriptase inactivation for 5 minutes at 95 ºC, PCR cycling and detection (30 to 45 cycles) for 10 seconds at 95 ºC, then 30 seconds at 55°C to 60°C (data collection step), and melt curve analysis for 1 minute at 95°C, 1 minute at 55°C, then 10 seconds at 55°C (80 cycles, increasing each by 0.5°C each cycle).
Statistical analysis
The mean standard error of three measurements was used to calculate the IC50 of the tested reference compounds. ANOVA was used for statistical comparisons. P-values less than 0.05 were considered significant in the differences of treated cell lines by compounds 1-3 and reference stourosporine compared with the solvent control (0.1% DMSO).
Results
Identification of compounds 1–3
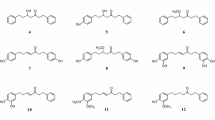
The isolated compounds (Fig. 2) were identified by using NMR spectroscopy (Bruker 400 MHz, USA) and confirmed by a comparison with the previously published data.

Chemical structure of compounds 1–3
-
(Z)-3-hexenyl-β-D-glucopyranoside (1) (10 mg), colorless needle. 1H-NMR (Methanol-d4, 400 MHz) (Fig. S1): δ 5.50-5.36 (2H, m, H3, H4), δ 4.29 (1H, d, J = 7.76, H1'), 3.89-3.86 (2H, m, H6', H1), δ 3.70-3.66 (1H, m, H6'), δ 3.58-3.52 (1H, m, H1), δ 3.38-3.34 (3H, m, H2', H4', H5'), δ 3.21-3.16 (1H, dd, J = 7.96, 8.8 H3'), δ 2.40 (2H, dd, J = 6.96, 7.24, H2), δ 2.10 (2H, dq, J = 7.44, 7.36, H5), δ 0.98 (3H, t, J = 7.56). 13C-NMR (Methanol-d4, 100 MHz) (Fig. S2): δ 134.50 (C4), δ 125.85 (C3), δ 104.34 (C1'), δ 78.12 (C4'), δ 77.88 (C5'), δ 75.11 (C3'), δ 71.64 (C2'), δ 70.46 (C1), δ 62.75 (C6'), δ 28.78 (C2), δ 21.65 (C5), δ 14.62 (C6). The identification of compound 1 was confirmed by 2 D NMR (HSQC & HMBC) as shown in Figs. S3 and S4, in addition to a comparison the 1H and 13C-NMR data with those previous reported (Kishida et al. 2005).
-
Gallic acid (2) (100 mg), white powder. 1H-NMR (DMSO-d6, 400 MHz) (Fig. S5): δ H 6.93 (2H, s, H2, H6). 13C-NMR (DMSO-d6, 400 MHz) (Fig. S6): δ 167.9 (C7), 138.51 (C4), 145.89 (C3, C5), 109.13 (C2, C6), 120.87 (C1). The compound was confirmed by a comparison the NMR data with those preciously reported (Nawwar et al. 1982).
-
Pyrogallol (3) (200 mg), white amorphous powder.1H-NMR (400 MHz, DMSO-d6) (Fig. S7): δ 6.42 (1H, dd, J = 7.6, 7.6 Hz, H4), δ 6.27 (2H, d, J = 8 Hz, H5, H6). 13CNMR (100 MHz, DMSO- d6) (Fig. S8): δ 146.44 (C2, C6), 133.21 (C1), 118.72 (C4), 107.33 (C3, C5) (Liu et al. 1999).
Cytotoxic activity of compounds 1–3
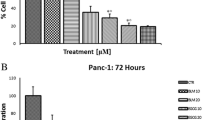
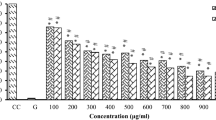
The cytotoxic activities of the isolated compounds 1-3 are depicted in Figs. 3 and S2 and Table 1. In this study, we present the novel finding of the cytotoxic activity of (Z)-3-hexenyl-β-D-glucopyranoside (1) (Figs. 3A, B, and 4) against pancreatic (Panc1), hepatic (HepG2), and breast (MCF7) cancer cells, as well as WI-38 normal cells. It should be noted that compounds 2 and 3 have previously been reported as cytotoxic natural agents (Faried et al. 2007; Jiang et al. 2022; Maurya et al. 2010; Revathi et al.). In this study, we describe their cytotoxic activity against Panc1 cells, as shown in Table 2 and Fig. 3C.

A Cytotoxic activity of (Z)-3-hexenyl-β-D-glucopyranoside (E-W-4) against HepG2, MCF7 and Panc1 cancer cells. B Cytotoxic activity of (Z)-3-hexenyl-β-D-glucopyranoside (E-W-4) against W138. C Cytotoxic activity of gallic acid (G) and pyrogallol (P) against Panc1 cancer cells. D Cell cycle profiling of treated panc1 cells with (Z)-3-hexenyl-β-D-glucopyranoside (E-W-4) and control panc1 cells. Statistically significant (P < 0.05) differences in treated cell lines by compounds 1–3 and reference stourosporine compared with the solvent control (0.1% DMSO) were indicated by asterisk symbol (*)

Representative images (40X) of Panc1 cells treated by different concentrations of (Z)-3-hexenyl-β-D-glucopyranoside compared to control (0.1% DMSO). A: Control Panc1 cells, B: 1 µg/mL treated Panc1 cells, C: 3 µg/mL treated Panc1 cells, D: 10 µg/mL treated Panc1 cells, E: 30 µg/mL treated Panc1 cells, F: 100 µg/mL treated Panc1 cells
Panc1 cell cycle DNA arrest and apoptotic effects of (Z)-3-hexenyl-β-D-glucopyranoside
Figures 3D and 5, along with Table S1, demonstrate that treatment of Panc1 cancer cells with (Z)-3-hexenyl-β-D-glucopyranoside (10 µM) resulted in cell growth arrest at the S phase and a significant decrease in cell DNA at the G2/M phase. Additionally, (Z)-3-hexenyl-β-D-glucopyranoside (10 µM) induced 26.15% early and 9.85% late apoptosis in Panc1 cells, as well as 5.52% necrosis (Table 3 and Fig. 5D).
Effect of (Z)-3-hexenyl-β-D-glucopyranoside on gene expression of caspase3, Bax and Bcl-2:
(Z)-3-hexenyl-β-D-glucopyranoside (10 µM) upregulated both caspase-3 and bax proapoptotic genes in Panc1 cells by 4.064 and 3.173-fold, respectively, while downregulating the Bcl-2 antiapoptotic gene by 0.231-fold (Table 4 and Fig. 6).
Discussion
The leaf ethyl acetate extract of Calamus rotang L. was subjected to isolation and characterization, resulting in the identification of compounds 1-3. In this study, we report the cytotoxicity of (Z)-3-hexenyl-β-D-glucopyranoside (1) for the first time, along with its molecular pathway. Additionally, two well-known natural anticancer compounds, gallic acid (2) and pyrogallol (3), were also isolated and characterized. (Faried et al. 2007; Tang et al. 2007; Maurya et al. 2010; Subramanian et al. 2015; Revathi et al. 2018, 2019; Jiang et al. 2022). These compounds have been previously reported and are commercially available, having been obtained from various medicinal plants (Khan et al. 2002; Verma et al. 2013). The compounds were identified based on NMR data in comparison with previously reported data (Nawwar et al. 1982; Liu et al. 1999; Kishida et al. 2005). (Z)-3-hexenyl-β-D-glucopyranoside (1) exhibited the highest cytotoxicity against Panc1 cells (IC50 = 7.6 µM) in a dose response manner. It was found to be twice as active as staurosporine (IC50 = 15.5 µM) and three times more active than gallic acid (IC50 = 21.8 µM) (Figs. 3 and 4, Table 1). However, it showed weak or no cytotoxicity against HepG2 and MCF7 cancer cells (IC50 = 45.8 and 108.7 µM, respectively) (Fig. 3A, Table 1). The cytotoxic activity of (Z)-3-hexenyl-β-D-glucopyranoside (1) on normal cells (WI-38) was also evaluated, and it exhibited weak to no cytotoxic activity (IC50 = 194.6 µM) compared to staurosporine (IC50 = 41 µM) (Fig. 3B, Table 1). Gallic acid (compound 2) demonstrated strong cytotoxic activity against Panc1 cells (IC50 = 21.8 µM), which is consistent with the previous reported data (Haddad and Rowland-Goldsmith 2014; Sharma et al. 2019). On the other hand, pyrogallol (compound 3) showed weak cytotoxicity against Panc1 cells (IC50 = 198.2 µM), although it has been reported to exhibit cytotoxic activity against other cancer cells (Mitsuhashi et al. 2008; Yang et al. 2009). Further investigation demonstrated that treatment with two doses of (Z)-3-hexenyl-β-D-glucopyranoside (10 µM) resulted in the induction of apoptosis in Panc1 cancer cells at the S1 stage of the cell cycle, as compared to the control Panc1 cells (Fig. 3D). This apoptotic effect was clearly observed in Fig. 5, where it is evident that (Z)-3-hexenyl-β-D-glucopyranoside caused a significant 42% increase in apoptosis in Panc1 cancer cells, with 26.15% early apoptosis and 9.85% late apoptosis. These findings are also summarized in Table 3.

Apoptotic effects of (Z)-3-hexenyl-β-D-glucopyranoside (E-W-4) on Panc1 cells. A Cell count at various stages of controlled Panc1 cell cycle. B Annexin V/PI (Controlled Panc1). C Cell count at various stages of treated Panc1 cell cycle with 10 µM (Z)-3-hexenyl-β-D-glucopyranoside (E-W-4). D Annexin V/PI treated Panc1 with 10 µM (Z)-3-hexenyl-β-D-glucopyranoside (E-W-4)
Furthermore, the molecular analysis of the treated Panc1 cells with (Z)-3-hexenyl-β-D-glucopyranoside (10 µM) revealed notable changes in gene expression. Specifically, there was a significant increase in caspase-3 and Bax expression by 4.064 and 3.173-fold, respectively, indicating their involvement in the apoptotic pathway. Conversely, the expression of the antiapoptotic gene Bcl-2 decreased by 0.231-fold. These results are depicted in Figs. 3D, 5D, and 6, and further details can be found in Tables 3 and 4.

Caspase3, Bax and Bcl-2 gene expression levels in controlled Panc1 (untreated), positive controlled (Staurosporine treated) and tested Panc1 with 10 µM (Z)-3-hexenyl-β-D-glucopyranoside (E-W-4)
Conclusion
The findings of this study strongly suggest that (Z)-3-hexenyl-β-D-glucopyranoside (1) holds great potential as an effective cytotoxic compound against Panc1 cells. Its mechanism of action involves inhibiting the proliferation of Panc1 cancer cells specifically during the S phase of the cell cycle through the induction of apoptosis. The observed upregulation of proapoptotic markers, such as caspase-3 and bax, along with the simultaneous downregulation of the antiapoptotic marker Bcl-2, provides further evidence of the cytotoxic activity of (Z)-3-hexenyl-β-D-glucopyranoside (1).
These findings shed light on the promising cellular activity exhibited by (Z)-3-hexenyl-β-D-glucopyranoside (1) in inducing cytotoxicity, particularly in Panc1 cells. Consequently, it is essential to conduct further investigations to explore its potential in vivo anticancer activity against pancreatic carcinoma. Future studies should focus on evaluating the efficacy and safety of (Z)-3-hexenyl-β-D-glucopyranoside (1) as a potential therapeutic agent for pancreatic cancer.
Data availability
Correspondence and requests for materials should be addressed to A.M.Z.
Abbreviations
- IC50 :
-
The half maximal inhibitory concentration
- Bcl-2:
-
B-cell lymphoma 2
- Bax:
-
Bcl-2-associated X protein
- Casp3:
-
Caspase 3
- °C:
-
Celsius temperature degree
- µg/mL:
-
Microgram/milliliter
- Kg:
-
Kilogram
- Panc1:
-
Pancreatic carcinoma
- MCF7:
-
Breast carcinoma
- HepG2:
-
Hepatocellular carcinoma
- WI-38:
-
Normal lung fibroblast cells
- NMR:
-
Nuclear Magnetic Resonance
- 1H-NMR:
-
Proton-Nuclear Magnetic Resonance
- 13C-NMR:
-
Carbon-Nuclear Magnetic Resonance
- HMBC:
-
Heteronuclear Multiple Bond Correlation
- HSQC:
-
Heteronuclear Single Quantum Coherence
- PCR:
-
Polymerase Chain Reaction
- RT-PCR:
-
Reverse Transcription Polymerase Chain Reaction
- L:
-
Liter
- G:
-
Gram
- MHz:
-
Megahertz
- DMSO-d 6 :
-
Deuterated dimethyl sulfoxide.
References
Aamazadeh F, Ostadrahimi A, RahbarSaadat Y, Barar J (2020) Bitter apricot ethanolic extract induces apoptosis through increasing expression of Bax/Bcl-2 ratio and caspase-3 in Panc-1 pancreatic cancer cells. Mol Biol Rep 47:1895–1904
Abed N, El-hallouty S, El-Zayat E, El-Sherief A (2021) In vitro anticancer activity of Nanazoxid drug against colorectal cancer cell line and its molecular pathways. EAJAST 1:29–37
Awad MG, Ali RA, El-Monem A, Dalia D, El-Magd MA (2020) Graviola leaves extract enhances the anticancer effect of cisplatin on various cancer cell lines. MCT 16:385–399
Berkovich L, Earon G, Ron I, Rimmon A, Vexler A, Lev-Ari S (2013) Moringa Oleifera aqueous leaf extract down-regulates nuclear factor-kappaB and increases cytotoxic effect of chemotherapy in pancreatic cancer cells. BMC Complement Altern Med 13:1–7
Cui J, Katsuno T, Totsuka K, Ohnishi T, Takemoto H, Mase N, Toda M, Narumi T, Sato K, Matsuo T (2016) Characteristic fluctuations in glycosidically bound volatiles during tea processing and identification of their unstable derivatives. J Agr Food Chem 64:1151–1157
Dibwe DF, Awale S, Kadota S, Tezuka Y (2012) Damnacanthal from the Congolese Medicinal Plant Garcinia huillensis has a potent preferential cytotoxicity against human pancreatic cancer Panc-1 cells. Phytother Res 26:1920–1926
Esharkawy ER, Almalki F, Hadda TB (2022) In vitro potential antiviral SARS-CoV-19- activity of natural product thymohydroquinone and dithymoquinone from Nigella sativa. Bioorg Chem 120:105587
Faried A, Kurnia D, Faried LS, Usman N, Miyazaki T, Kato H, Kuwano H (2007) Anticancer effects of gallic acid isolated from Indonesian herbal medicine, Phaleria macrocarpa (Scheff.) Boerl, on human cancer cell lines. Int J Oncol 30:605–613
Haddad L, Rowland-Goldsmith M (2014) Assessment of the effects of caffeine, gallic acid, and epigallocatechin-3-gallate on cell inhibition, PIM-3 and E. cadherin protein levels in two lines of pancreatic cancer cells. In: Student scholar symbosium abstracts and posters. Chapman University. https://digitalcommons.chapman.edu/cusrd_abstracts/
Huang X, Xu M, Shirahata T, Li W, Koike K, Kojima-Yuasa A, Yuasa I, Kobayashi Y (2018) Anti-steatosis compounds from leaves of Mallotus furetianus. Nat Prod Res 32:1459–1462
Janicke RU, Sprengart ML, Wati MR, Porter AG (1998) Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J Biol Chem 273:9357–9360
Jiang L, Kojima H, Yamada K, Kobayashi A, Kubota K (2001) Isolation of some glycosides as aroma precursors in young leaves of Japanese pepper (Xanthoxylum piperitum DC.). J Agr Food Chem 49:5888–5894
Jiang Y, Pei J, Zheng Y, Miao Y-j, Duan B-z, Huang L-F (2022) Gallic Acid: A potential anti-cancer agent. Chin J Integr Med 28:661–671
Khan MTH, Lampronti I, Martello D, Bianchi N, Jabbar S, Choudhuri MSK, Datta BK, Gambari R (2002) Identification of pyrogallol as an antiproliferative compound present in extracts from the medicinal plant Emblica officinalis: effects on in vitro cell growth of human tumor cell lines. Int J Oncol 21:187–192
Kishida M, Fujii M, Ida Y (2005) Chemoenzymatic synthesis of naturally occurring (Z)-3-hexenyl 6-O-glycosyl-β-D-glucopyranosides. Heterocycles 65:2127–2137
Koopman G, Reutelingsperger CP, Kuijten GA, Keehnen RM, Pals ST, Van Oers MH (1994) Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood 84:1415–1420
Lee I-K, Kim M-A, Lee S-Y, Hong J-K, Lee J-H, Lee K-R (2008) Phytochemical constituents of Schizonepeta tenuifolia Briquet. Nat Prod Sci 14:100–106
Liu SL, Zhao BL, Shi DY, Xin WJ (1999) Auto-oxidation mechanism of pyrogallol and its free radical effects studied by means of ESR, UV and NMR. Chem Res Chin Univ 15:146–151
Malak YA, Mohamed KM, Abd El-Mawla AM, Zaher AM (2023) Cytotoxic and antimicrobial effects of selected Egyptian Asteraceae species as well as GC-MS metabolite profiling of Senecio cruentus lipophilic fraction. Bull Pharm Sci Ass Unive 46(1):39–49
Mario C, Marilisa F, Kryssia IR-C, Pellegrino C, Ginevra C, Chiara M, Alberto B, Antonio N, Gioacchino L, Tiziana M (2018) Epidemiology and risk factors of pancreatic cancer. Acta BioMed 89:141
Maurya DK, Nandakumar N, Devasagayam TPA (2010) Anticancer property of gallic acid in A549, a human lung adenocarcinoma cell line, and possible mechanisms. J Clin Biochem Nut 48:85–90
Mitsuhashi S, Saito A, Nakajima N, Shima H, Ubukata M (2008) Pyrogallol structure in polyphenols is involved in apoptosis-induction on HEK293T and K562 cells. Molecules 13:2998–3006
Naik SK, Mohanty S, Padhi A, Pati R, Sonawane A (2014) Evaluation of antibacterial and cytotoxic activity of Artemisia nilagirica and Murraya koenigii leaf extracts against mycobacteria and macrophages. BMC Complement Altern Med 14:1–10
Nawwar MAM, Buddrus J, Bauer H (1982) Dimeric phenolic constituents from the roots of Tamarix nilotica. Phytochemistry 21:1755–1758
Neoptolemos JP, Kleeff J, Michl P, Costello E, Greenhalf W, Palmer DH (2018) Therapeutic developments in pancreatic cancer: current and future perspectives. Nat Rev Gastroenterol Hepatol 15:333–348
Pliarchopoulou K, Pectasides D (2009) Pancreatic cancer: current and future treatment strategies. Cancer Treat Rev 35:431–436
Ponnusamy L, Mahalingaiah PK, Singh KP (2016) Chronic oxidative stress increases resistance to doxorubicin-induced cytotoxicity in renal carcinoma cells potentially through epigenetic mechanism. Mol Pharmacol 89:27–41
Prescott M, Mitchell J, Totti S, Lee J, Velliou E, Bussemaker M (2018) Sonodynamic therapy combined with novel anti-cancer agents, sanguinarine and ginger root extract: Synergistic increase in toxicity in the presence of Panc-1 cells in vitro. Ultrason Sonochem 40:72–80
Rajendran K, Reddy EV, Khanna A (2016) Anticancer effect of Mesua ferrea extracts on human pancreatic cancer cell line. Int J Life Sci Scienti Res 2:198–205
Ramakrishnan P, Loh WM, Gopinath SCB, Bonam SR, Fareez IM, Mac Guad R, Sim MS, Wu YS (2020) Selective phytochemicals targeting pancreatic stellate cells as new anti-fibrotic agents for chronic pancreatitis and pancreatic cancer. Acta Pharm Sin B 10:399–413
Rauf A, Abu-Izneid T, Khalil AA, Imran M, Shah ZA, Emran TB, Mitra S, Khan Z, Alhumaydhi FA, Aljohani ASM (2021) Berberine as a potential anticancer agent: A comprehensive review. Molecules 26:7368
Revathi S, Hakkim FL, Kumar NR, Bakshi HA, Rashan L, Al-Buloshi M, Hasson SSAA, Krishnan M, Javid F, Nagarajan K (2018) Induction of HT-29 colon cancer cells apoptosis by Pyrogallol with growth inhibiting efficacy against drug-resistant Helicobacter pylori. Anti-Cancer Agents in Med Chem 18:1875–1884
Revathi S, Hakkim FL, Kumar NR, Bakshi HA, Sangilimuthu AY, Tambuwala MM, Changez M, Nasef MM, Krishnan M, Kayalvizhi N (2019) In vivo anti cancer potential of pyrogallol in murine model of colon cancer. APJCP 20:2645
ShameliRajiri M, Aminsalehi M, Shahbandeh M, Maleki A, Jonoubi P, Rad AC (2021) Anticancer and therapeutic potential of Delonix regia extract and silver nanoparticles (AgNPs) against pancreatic (Panc-1) and breast (MCF-7) cancer cell. Toxicol Environ Health Sci 13:45–56
Sharma S, Mittal D, Verma AK, Roy I (2019) Copper-gallic acid nanoscale metal–organic framework for combined drug delivery and photodynamic therapy. ACS Appl Bio Mater 2:2092–2101
Subramanian AP, John AA, Vellayappan MV, Balaji A, Jaganathan SK, Supriyanto E, Yusof M (2015) Gallic acid: prospects and molecular mechanisms of its anticancer activity. RSC Adv 5:35608–35621
Tang G, Yang C-Y, Nikolovska-Coleska Z, Guo J, Qiu S, Wang R, Gao W, Wang G, Stuckey J, Krajewski K (2007) Pyrogallol-based molecules as potent inhibitors of the antiapoptotic Bcl-2 proteins. J Med Chem 50:1723–1726
Verma S, Singh A, Mishra A (2013) Gallic acid: molecular rival of cancer. Environ Toxicol Pharmcol 35:473–485
Wang L, Xu J, Yan Y, Liu H, Karunakaran T, Li F (2019) Green synthesis of gold nanoparticles from Scutellaria barbata and its anticancer activity in pancreatic cancer cell (Panc-1). Artif Cells Nanomed Biotechnol 47:1617–1627
Xu X, Hamhouyia F, Thomas SD, Burke TJ, Girvan AC, McGregor WG, Trent JO, Miller DM, Bates PJ (2001) Inhibition of DNA replication and induction of S phase cell cycle arrest by G-rich oligonucleotides. J Biol Chem 276:43221–43230
Yang C-J, Wang C-S, Hung J-Y, Huang H-W, Chia Y-C, Wang P-H, Weng C-F, Huang M-S (2009) Pyrogallol induces G2-M arrest in human lung cancer cells and inhibits tumor growth in an animal model. Lung Cancer 66:162–168
Zhang L, Sanagapalli S, Stoita A (2018) Challenges in diagnosis of pancreatic cancer. WJG 24:2047
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
A.M.Z.: Responsible for the aim of the current study, selection of the plant, interpretation of the NMR and biological data, writing the manuscript.
W.S.A: Extraction, fractionation and isolation of compounds 1–3 and participated in the data analysis.
I.A.M.A & M.A.M: Participate in the data analysis, and preparation of figures.
The authors declare that all data were generated in-house and that no paper mill was used.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Plant ethics
The plant was kindly identified by Dr. Amr M. M. Mahmoud, Director of Aswan Botanical Garden, Hort. Res. Institute, Agriculture Centre, Egypt. Voucher specimen of the plant leaves was placed in the herbarium of the Pharmacognosy Department, Faculty of Pharmacy, Assiut University, Egypt (Voucher no. A20220906). This experiment was carried out in accordance with relevant guidelines and regulations. Permission to collect plant material was obtained from Aswan Botanical Garden, Hort. Res. Institute, Agriculture Centre, Aswan, Egypt.
Consent to publish
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zaher, A.M., Anwar, W.S., Makboul, M.A. et al. Potent anticancer activity of (Z)-3-hexenyl-β-D-glucopyranoside in pancreatic cancer cells. Naunyn-Schmiedeberg's Arch Pharmacol 397, 2311–2320 (2024). https://doi.org/10.1007/s00210-023-02755-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-023-02755-4