
Abstract
AIDS (Acquired immunodeficiency syndrome) is one of the chronic and potentially life-threatening epidemics across the world. Hitherto, the non-existence of definitive drugs that could completely cure the Human immunodeficiency virus (HIV) implies an urgent necessity for the discovery of novel anti-HIV agents. Since integration is the most crucial stage in retroviral replication, hindering it can inhibit overall viral transmission. The 5 FDA-approved integrase inhibitors were computationally investigated, especially owing to the rising multiple mutations against their susceptibility. This comparative study will open new possibilities to guide the rational design of novel lead compounds for antiretroviral therapies (ARTs), more specifically the structure-based design of novel Integrase strand transfer inhibitors (INSTIs) that may possess a better resistance profile than present drugs. Further, we have discussed potent anti-HIV natural compounds and their interactions as an alternative approach, recommending the urgent need to tap into the rich vein of indigenous knowledge for reverse pharmacology. Moreover, herein, we discuss existing evidence that might change in the near future.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
HIV (human immunodeficiency virus) infection remains a global public health concern and is predominantly serious in low- and middle-income countries (Notario-Pérez et al. 2017). As stated in the 2021 report of the United Nations Programme on HIV/AIDS (UNAIDS), about 37.7 million people were currently living with HIV/AIDS worldwide in 2021, among which 28.2 million people were accessing antiretroviral therapy (ART). An estimated 1.5 million people were newly infected, the majority being above the age of 15. Despite the scientific advances in this field, nearly 680,000 people have died due to AIDS-associated diseases, with a large population being from Eastern and Southern Africa (Siwe-Noundou et al. 2019; UNAIDS 2021).
Due to the inability to completely cure HIV, averting its spread is of paramount priority. To achieve the goal to end AIDS by 2030, UNAIDS has formulated the 90–90–90 target (Duwal et al. 2018; Mikasi et al. 2021). According to the National AIDS Control Organization 2020 Annual Report, the majority of people living with HIV in India projected Mizoram with the most cases of adult HIV prevalence (2.37%), followed by Nagaland and Meghalaya. Although in the past decade there has been a 33.3% decline, the same organization reported the national adult HIV prevalence to be about 0.22% with 0.23% being males and 0.20% being females, accounting for about 23.19 lakh people living with HIV (majority originating from Maharashtra). By March 2020 about 15 lakh people were enrolled for the ART programme (NACO 2019). Due to this, the Indian government has pledged 22 million USD to the Global Fund’s Sixth Replenishment for 2020–2022 to support the global response to HIV (Avert 2020). There is no published evidence as to what proportion of ART-eligible people living with HIV are lost before starting the therapy or the dropout rates of people exiting the programme.
HIV is an enveloped virus grouped in the genus Lentivirus within the family of Retroviridae and subfamily Orthoretrovirinae. It is 50 to 60 nm in radius and has a lipid envelope that is embedded with glycoproteins, beneath which lies the matrix protein (p17), the core proteins (p24 and p6), and the nucleocapsid protein (p7 bound to diploid, 9.8 kb positive-sense RNA), refer to Fig. 1 (Blut and Blood 2016; Rossi et al. 2021). The HIV genome encodes about 16 viral proteins that are involved in its life cycle and play crucial roles. The chief genes are gag (encodes structural proteins—matrix, capsid, nucleocapsid, and p6), pol (encodes viral enzymes—integrase (IN), protease, and reverse transcriptase), and env (encodes envelope proteins gp41 and gp120). The other genes proteins, such as tat and rev, code for regulatory, while vpr, vif, vpu (in HIV-1) or vpx (in HIV-2), and nef code for accessory proteins (Li and De Clercq 2016; Rossi et al. 2021).

Schematic representing the structure of HIV virion. This has been simplified for clarity. For instance, the gp120 and gp41 envelope glycoproteins are trimers in reality and not monomers as depicted here (Blut and Blood 2016; Rossi et al. 2021). This figure has been modified from Servier Medical Art (http://smart.servier.com/), licenced under a Creative Commons Attribution 3.0 Unported License
Both serotypes of the virus, HIV-1 and HIV-2 (comparatively less virulent), lead to acquired immunodeficiency syndrome (AIDS), which is the most advanced stage of HIV infection, in which an immunodeficient state is reached (Notario-Pérez et al. 2017). HIV uses the bloodstream as a means to spread (Evans et al. 2021); it destroys or impairs the functionality of the host’s cell-mediated immunity by specifically attacking the CD4+ cells from the monocyte–macrophage lineage, dendritic, microglial, and resting T-cells via gp120 (Wallet et al. 2019; Kruize and Kootstra 2019; Crisan and Bora 2021). Ultimately, it establishes a provirus reservoir causing a progressive deterioration of immunity and making the host’s body easily accessible to be attacked by a myriad of opportunistic infections, disorders, and cancers (Putim et al. 2018; Siwe-Noundou et al. 2019; Gong et al. 2020), further allowing them to persist in multiple organs, even while on ART (Evans et al. 2021).
Studies suggest that the expression of receptors CCR5, CXCR4, CCR3, and CD4 on these cells is directly proportional to susceptibility to HIV infection. Neurons usually lack CD4 receptors which reduce for them the probability of being infected directly by the HIV-1 virion, but the infected macrophages and microglia release lethal constituents, like inflammatory cytokines and chemokines along with viral proteins, which damage the neurons and cause HIV-1-associated neurocognitive disorders (Gong et al. 2020). The innate quiescent nature of the virus and its latency makes it difficult to identify compounds that could be employed as anti-HIV agents (Dahabieh et al. 2015).
Highly active antiretroviral therapy (HAART)
A personalized cocktail of diverse classes of drugs commonly referred to as the HAART exists to manage the disease. At present, about 26 antiretroviral drugs are FDA approved, which do not eradicate HIV infection but suppress its activity (Crisan and Bora 2021), as depicted in Fig. 2. Each HAART drug targets and suppresses different stages of HIV’s replication: (i) viral entry (co-receptor CCR5/CXCR4 antagonists; (ii) fusion of virion (fusion inhibitors); (iii) viral complementary DNA (vcDNA) synthesis (reverse transcriptase inhibitors); (iv) vcDNA-host DNA integration (INSTIs); and (v) virion release and maturation (protease inhibitors) (Notario-Pérez et al. 2017; Siwe-Noundou et al. 2019). HAART effectively reduces the viral load (viraemia) and assists the immune functions to recover in HIV/AIDS patients, prolonging death by more than 7 to 10 years or even longer when compared to single drug-treated patients (Park et al. 2021).

Schematic representation of diverse classes of HAART drugs targeting different stages of the HIV life cycle. CCR5 C–C chemokine receptor 5, CXCR4 C–X–C chemokine receptor type 4 (Fusin), NRTIs Nucleoside reverse transcriptase inhibitors, NNRTIs Non-nucleoside reverse transcriptase inhibitors, INSTIs Integrase strand transfer inhibitors (Li and De Clercq 2016; Rossi et al. 2021)
The World Health Organization and the National AIDS Control Organization (NACO) initially recommend a combination of ART for HIV patients, which includes reverse transcriptase inhibitors—2 nucleoside NRTIs and a non-nucleoside NNRTIs or INSTIs or a ritonavir-assisted protease (PR) inhibitor (Akanbi et al. 2012; NACO 2013; Huang et al. 2019; Raj et al. 2021; National Institutes of Health 2022). Despite the substantial success (decreased mortality and morbidity of virus) achieved with the aid of HAART, the continued emergence of cross-resistant viral strains and drug-related adverse effects on patients persist significant challenges to a sustained battle against HIV. Hidden virus in cellular reservoirs is the foremost barrier to functional therapy. This implies a pressing need for the identification of novel anti-HIV agents with enhanced pharmacokinetic profiles, potency, and least side effects and cross-resistance, to defeat HIV on a global scale (Wallet et al. 2019).
These strict requirements are addressed by employing several computational techniques, like molecular docking (Tewtrakul et al. 2015; Ercan et al. 2019; Arslan 2019; Dogan and Durdagi 2021; Esmaeili et al. 2021), pharmacophore modelling (Martin 2014; Bhatt et al. 2014; Xue et al. 2014; Islam and Pillay 2016), molecular dynamics (MD) simulation (Chen et al. 2014; Islam and Pillay 2016; Chitongo et al. 2019; Samorlu et al. 2019), quantitative structure–activity relationships (QSAR) (Gupta et al. 2013; Reddy et al. 2013; Zhou et al. 2021; Xuan et al. 2021), and many more. The human cells lack structural or functional integrase homologues, making IN a desirable drug target for HAART (Siwe-Noundou et al. 2019); further IN depicts greater tolerability, high efficacy, and fewer drug–drug interactions as compared to other HAART classes (Brooks et al. 2019).
Ross (2015) conducted a QikProp (developed by Schrödinger) analysis for the FDA-approved INSTIs to detect the compounds with unfavourable drug pharmacology and concluded that all of the IN inhibitors had a star score of 0, except for cabotegravir which had 1 depicting that one of its chemical properties (human serum albumin binding prediction) did not lie within the range of drug pharmacology. The more the star score, the more the compound is less drug-like owing to its predicted properties and outlying descriptors. Moreover, its potency is lower than the other two second-generation drugs and the development of Q148R/K mutation with its regimen can lead to a high-level cross-resistance to all INSTIs (Smith et al. 2018; Oliveira et al. 2018). Therefore, in this study, we have focused particularly on integrase inhibitors (raltegravir, elvitegravir, dolutegravir, and bictegravir) owing to their gaining popularity and aforementioned reasons.
Integrase inhibitors
IN is a crucial nucleotidyltransferase enzyme in the HIV’s life cycle that decides the virion’s fate, as its establishment leads to persistent HIV-1 infection (Brooks et al. 2019; Mbhele et al. 2021). It is present at the 3’-end of the pol gene and is produced during the cleavage of gag-pol polyprotein during early virus maturation (Mbhele et al. 2021). This enzyme catalyses the insertion of virally reverse-transcribed cDNA into the patient’s genome via dual stages (SN2 nucleophilic reaction) (Siwe-Noundou et al. 2019; Zhou et al. 2021). The first step is ‘3’-processing’ (3’P), wherein a dinucleotide (GT) pair is cleaved from both the cDNA’s 3’-ends, forming the pre-integration complex (PIC). This complex then integrates with the host’s cellular genome with the aid of lens epithelial-derived growth factor (LEDGF or p75) in the second step called strand transfer (ST), infecting the individual (Varadarajan et al. 2013; Park et al. 2021). Both these reactions are energetically independent as well as set apart, both spatially and temporally as the 3’P occurs in the cytoplasm of the infected cells, whereas the ST activity takes place in the nuclei (Arora et al. 2013). Once integrated, it is now referred to as a provirus, which acts as a post-integrative template for viral infection, allowing its self-transcription to release more infectious copies of itself (Thierry et al. 2017).
HIV-1 IN has a molecular weight of 32 kDa and comprises 288 amino residues, which can be distributed into three particular functional domains:
-
(i)
Residues 1–49: N-terminal domain (NTD)
The zinc finger motif HHCC (histidines 12 and 16, cysteines 40 and 43) present in NTD is responsible for the stability of the whole enzyme, favouring the multimerization of the protein.
-
(ii)
Residues 50–212: Catalytic core domain (CCD)
High enzymatic activity is maintained by proper chelation of the CCD catalytic triad DDE motif (i.e., Asp64-Asp116-Glu152) of the active site and the 2 divalent cofactor ions (Mn2+ or Mg2+). Mutation of either of the DDE residues inactivates the enzyme. In the apoprotein, one Mg2+ is coordinated between D64 and D116. Once the vcDNA joins, the other cation is coordinated between D64 and E152. This domain also encompasses a flexible 10 residues loop (140–149) that is adjacent to the catalytic site, involved in vcDNA substrate recognition.
-
(iii)
Residues 213–288: C-terminal domain (CTD)
This region serves as a non-specific binding site for the vcDNA and further contributes to stabilizing the IN–vcDNA complex (Arora et al. 2013; Ross 2015; Thierry et al. 2017; Nusrath Unissa et al. 2017; Zhou et al. 2021).
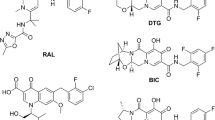
IN inhibitors (INSTIs) are a potentially attractive class of next-generation antiretroviral (ARV) drugs that have gained an audience in the past 15 years (Zhou et al. 2021). These comprise a polycyclic core with heteroatom triads and a halogenated benzyl group that interacts with vcDNA attached to the CCD by a flexible linker (Smith et al. 2021a). These disrupt the strand transfer step during viral infection by binding to the CCD of IN by chelating the catalytically essential divalent metal ions and blocking the enzyme’s active site, thereby displacing the reactive 3’-hydroxyl group of the terminal A17 away from the active site, obstructing the virion from hijacking the CD4+ cells (Chitongo et al. 2019; Park et al. 2021). The first INSTI to get approved was raltegravir (RAL) in 2007 as a therapeutic INSTI by the US Food and Drug Administration (FDA), followed by elvitegravir (EVG) in 2012, dolutegravir (DTG) in 2013 (Thierry et al. 2017), bictegravir (BIC) in 2018 and injectable formulation of cabotegravir (CAB) in 2021 (Phillips et al. 2021); their chemical structures have been depicted in Fig. 3. These are acclaimed for having a higher genetic barrier to resistance compared to other PR and RT inhibitors, especially DTG and BIC (Mbhele et al. 2021).

Chemical 2D structures of FDA-approved INSTIs along with a schematic of a common INSTI pharmacophore scaffold interacting with the DDE motif. Among mentioned INSTIs, RAL and EVG are first-generation inhibitors while others are second-generation inhibitors. The red circle represents a triad of coplanar oxygen atoms that chelate the pair of divalent metal ions, the green triangles represent a halogenated phenyl ring that invades the pocket natively occupied by the viral DNA extremity, and the purple boxes indicate linkers of variable length and flexibility which distinguish the earlier mentioned components (Thierry et al. 2017; Smith et al. 2018; Mbhele et al. 2021)
Although the mentioned INSTIs share various properties, including mechanism of action, antiviral potency, good tolerability, and safety profile, they have other significant traits that differ from each other, like pharmacokinetics, interactions, resistance profile, and pharmacodynamics (Blanco et al. 2015; Roulet et al. 2018), refer to Table 1. However, the advent of INSTI-resistant strains is an impending task (Xuan et al. 2021). Although numerous structurally diverse INSTIs have been reported, like coumarin derivatives, quinolone acids, diketo acids, salicylhydrazide derivatives, aryl cyclic compounds, sulphur nitrogen thiazepines, diazonaphthalene derivatives, pyrimidine ketones, benzenesulfonamides, and so on, the need to develop novel molecules still persists (Bhatt et al. 2014; Zhou et al. 2021).
Some of the IN inhibitor-based HAARTs have revealed antiviral potency against Middle East respiratory syndrome (MERS), severe acute respiratory syndrome (SARS), and viruses associated with COVID-19 (Rameshrad et al. 2020; Makoti and Fielding 2020). However, no clinical improvements beyond standard care were noticed, when administered to severe COVID-19 patients (Crisan and Bora 2021).
Computational studies
Computational approaches have created substantial interest by decreasing the time essential for preclinical assessments, clinical trials, as well as in terms of expenses and resources. These aid in predicting the significance of mutations involved in making the strain resistant, elucidating the molecular mechanisms of resistance associated to IN-ligand complex, and lastly, disclosing precise insights vital to designing new potent INSTIs, refer to Fig. 4 for major INSTI resistance-associated mutations (RAMs) (Islam and Pillay 2016; Han et al. 2016; Zhou et al. 2021; Crisan and Bora 2021). Consequently, the approved anti-HIV agents can be employed either as reference molecules to guide the design of novel ones or can be scrutinized for their re-application to other viruses or non-virus diseases using computational as well as experimental methods (like in the case of COVID-19) (Rameshrad et al. 2020; Indu et al. 2020; Alavian et al. 2021; Crisan and Bora 2021).

Schematic representation of RAMs in the IN gene susceptible to INSTIs. The pink bar indicates insertion mutation along with the specified loci (position). The row above it indicates a wild-type amino acid, while the row below indicates the substituted amino acid conferring resistance to INSTIs. Mutations in red depict the highest level of reduced susceptibility to the indicated INSTI, the ones in bold have a significant impact on susceptibility reduction, whereas the plain text indicates that they reduce the susceptibility but in combination with other INSTI resistance mutations (Stanford HIV-1 Drug Resistance Database 2019; Wensing et al. 2019)
Raltegravir (isentress, isentress HD)
The era of the therapeutic INSTIs for HIV treatment began with the approval of RAL in 2007 (Brooks et al. 2019); it predominantly suppresses the ST activity while inhibiting the 3’P activity only up to a certain limit (100-fold less specific than ST). The study of RAL’s binding to the HIV-1 IN and its conformational inclinations represent an essential task for identifying the molecular factors that contribute to its pharmacological action. This pyrimidinone analogue acts as a multipotent drug owing to the presence of 2 polydentate pharmacophores (1,3,4-oxadiazole-2-carboxamide and carbonylamino-1-N-alkyl-5-hydroxypyrimidinone) which are capable of hitting more than one target in HIV-1, the vcDNA, the unbound IN, or IN–vcDNA complex (Arora and Tchertanov 2013). The inhibition process initiates by recognition of RAL by the processed vcDNA bound to a transient intermediate IN, providing a potentially favourable route to the design of INSTIs with enhanced affinity and selectivity (Arora et al. 2013). Since no crystal structures of full-length HIV-1 IN are available, Tn5 transposase and prototype foamy virus (PFV) IN are considered surrogate models or scaffolds for studying the mechanism of action of INSTIs (Arora 2013; DeAnda et al. 2013) (Fig. 5).

The most impactful INSTIs inhibit both wild-type (WT) IN as well as a variety of familiar mutants. However, because there are mutations that reduce the potency of all available INSTIs, novel and enhanced compounds are needed (Nusrath Unissa et al. 2017; Smith et al. 2021a). With recurrent use, resistance patterns are likely to impact future INSTI-based regimen selection specifically concerning the suitability for use of long-acting injectables. The combinations of these mutations along with accessory mutations can lead to a greater reduction in drug susceptibility representing a threat to the ARV drug’s long-term efficacy (Brooks et al. 2019; Cook et al. 2020; Smith et al. 2021a). RAMs impend the enduring triumph of combination ART (cART) consequences for HIV-1 treatment (Chitongo et al. 2019). Of the available INSTIs, the first-generation RAL and EVG share an overlapping resistance profile (N155H, Q148K/R/H, and Y143R/C/H) and a low genetic barrier, resulting in a greater propensity for loss of virologic activity secondary to resistance. Due to this, patients failing one of the either containing regimens should not be transitioned to another (Nusrath Unissa et al. 2017; Brooks et al. 2019; Chitongo et al. 2019; Smith et al. 2021b, a). On the other hand, DTG and BIC have a comparatively lower possibility of developing resistance due to higher genetic barriers owing to their extended tricyclic scaffolds (Wainberg and Han 2015b; Smith et al. 2021a).
RAL further depicts E/Z configurational isomerism and high structural flexibility due to the presence of non-amide bonds joining the aromatic ring of the 8 aliphatic single bonds, characterizing the relative locus of the vicinal 1–4 and 1–5 oxygen atoms. This also determines the chelating properties of deprotonated or protonated drugs (Arora and Tchertanov 2013; Arora et al. 2013). These oxygen atoms of carbonylamino-1-N-alkyl-5-hydroxypyrimidinone and β-ketoenolate can chelate the pair of cations simultaneously by competing with the metal-binding sites (Arora 2013). Subsequently, when RAL is docked to the unbound IN, it adopts different configurations within a large binding cavity formed by the catalytic active site and the adjacent loop (Arora et al. 2013), further contributing to the recognition and binding of different substituents in topologically distinctive regions of IN by relating its molecular and conformational characteristics. It acts as a divalent metal cation chelating ligand and can bind to them by both pharmacophores in varying isomerisation states (Arora and Tchertanov 2013). All observed RAL poses show the absence of specific interactions with the targets lacking vcDNA intermediate (Arora et al. 2013). Moreover, the structural flexibility of RAL allows itself to fit into the relatively large binding pocket of IN–vcDNA PIC with low docking scores, thus yielding various RAL docked conformations/poses and indicating it to be a biologically relevant target of the INSTI. This also imposes that the DNA substrate is required to bind for the precise configuration of residues that are required for catalysis. (Arora and Tchertanov 2013; Arora et al. 2013).
The MD simulation studies have verified the significance of the catalytic site loop flexibility for catalysis. A major conformational change, concerning the E152 residue of the DDE motif, was observed upon vcDNA binding. This occurs due to a change in the position of the Mg2+ cation in the active site that induces a switch of E152 side-chain conformation towards the active site, without any changes in the configuration of the overall catalytic loop. This is crucial to decrease the flexibility induced, for instance, the G140A/G149A mutations result in lower levels of activity despite minimal effects on vcDNA binding. MD simulations of the influence of RAL-selected mutations on IN structure and, chiefly, on the structure of the catalytic loop, indicated strict conservation of IN CCD structure, including the Ω-shaped hairpin in the active site loop (Arora 2013).
RAL is preferred over NRTIs and is employed clinically in first-line and second-line ART regimens because of its efficacy and better tolerance (Huang et al. 2019). Like other antiretroviral inhibitors, even RAL develops/induces resistance, which is linked with amino acid substitutions, specifically either N155H or Q148R/H/K or Y143R (in rare cases) primary mutations (Quashie et al. 2012; Arora and Tchertanov 2013; Smith et al. 2021b). RAL forms links with the catalytic triad and interacts with the five residues, T66, E92, Y143, Q148, and N155, involved in primary resistance (Arora 2013). RAL’s oxadiazole ring interacts via van der Waals contact with Y143 and P145 and makes a π-stacking connection with them (Ross 2015). G140S, a secondary mutation, further increases the RAL resistance by rescuing the replication defects from the Q148R/H/K mutation. This chiefly occurs by disturbing the position of the active site Mg2+ ions and not by affecting the affinity of the metals (Arora and Tchertanov 2013). RAL is independent of HIV-1 co-receptor tropism (CCR5 and CXCR4) owing to its mechanism of action and is also active against other classes of ARV (Arora 2013).
A study conducted by Nusrath Unissa et al. (2017), showed RAL forming 4 conventional H-bonds with Asp116 (2 bonds), Ile151 and Glu152 residues. Alike EVG, the halogen atom of RAL was also involved in interacting with Pro142 residue. The primary mutations Q148H/R/K decrease IN susceptibility to RAL by about 5 to 50 folds, whereas the double mutants E138K/A with Q148H or Q148H/R/K and G140S/A or Y143C/R with T97A (naturally occurring due to immune escape) reduces susceptibility by more than 100-fold (Ross 2015; Acosta et al. 2019). The role of these primary mutation pathways has been elaborated by Anstett et al. (2017), in dominating RAL selections in vivo and in vitro.
MK-2048 is a second-generation INSTI which is formulated as an intravaginal ring as pre-exposure prophylaxis for HIV prevention (Hoesley et al. 2019; Brooks et al. 2019), that has a duration of action fourfold longer than RAL and a dissociation half-life of 32 h on WT-IN (Arora 2013). It can specifically select a G118R resistance mutation, as demonstrated in its selection studies (Quashie et al. 2012). This ring is clinically proven safe and has depicted good tolerance along with detectable drug levels in the cervical tissue, cervicovaginal fluid, and plasma when continuously used for more than 28 days (Brooks et al. 2019). Another novel drug that advanced into phase 1 clinical trials is a non-catalytic site IN inhibitor, BI-224436 (BI2), which inhibits the virion replication via binding to a conserved allosteric pocket of IN, disrupting the interactions between IN and LEDGF protein, located in the CCD dimer interphase at residues A128, E170, A124, T125, and H171. Despite BI2’s promising results, it was put on hold (Ross 2015).
Elvitegravir (Vitekta, Stribild, Genvoya)
EVG is the next-generation INSTI therapy to garner FDA approval in 2012 for clinical use as monotherapy or in combination with other anti-HIV drugs (Brooks et al. 2019). It is a cobicistat-boosted quinolone-3-carboxylic INSTI that is administered in a quad pill formulation with the RT inhibitors as Stribild (Ross 2015). EVG inhibits the process of vcDNA strand transfer with a promising 50% maximal inhibitory concentration (IC50) of less than 0.06 μmol L−1 in vitro, thereby averting virion from being integrated into the patient’s chromosome and obstructing provirus formation and infection proliferation (Abdelhameed 2015). The emergence of certain mutational patterns in IN affects inhibitor binding, thereby modifying the degree of IN mutant susceptibility to EVG, and limiting their therapeutic efficiency (Masso et al. 2014). Due to multiple primary mutations at E92Q, T97A, R263K, S147G, E138K/A/T, T66I/A/K, G140A/S/C, Q148K/H/R, G118R, and N155H, it leads to resistance against EVG and hence virologic failure both in vitro and in vivo (Ross 2015; Unger et al. 2016; Stanford HIV-1 Drug Resistance Database 2019; Smith et al. 2021b). EVG substantially inhibits the RAL-resistant mutant Y143R owing to the absence of chemical moiety that stacks with the phenol group of Y143 (Smith et al. 2021b).
A study conducted by Khoury et al. (2019) showed that EVG binding to intasome has a slight preference over DTG and RAL, indicating that additional interactions of the diketo acid groups of the drugs with DNA could be essential to further enable preferential binding of DTG. EVG affects not only the residues in direct contact but also the outer-shell residues indirectly bound, particularly the double mutations, like Q148R/H and G140S/A, which has a synergetic effect on its efficacy. Its halobenzyl group targets the highly conserved 5′CpA 3′/5′TpG 3′ on the vcDNA ends with its centralized pharmacophore coordinating the pair of catalytic divalent cations. The X-ray structural analysis demonstrated that in the IN–vcDNA complex, the halobenzyl ring stacks over C16 (upstream), while the C–X bond points towards the centre of the G4 (downstream) of the second strand. Therefore, a surge in the drug–vcDNA complex stability draws a parallel with an increase in drug activity and a decline in viral resistance, indicating the vitality of vcDNA end recognition for the binding affinity of INSTIs to the intasome (Khoury et al. 2019).
Another study by Kang Birken et al. (2019) discussed 2 cases involving HIV rebound due to a possible drug–drug interaction between EVG, cobicistat, emtricitabine, tenofovir alafenamide, and calcium-containing products, which were the first reports of the rapid emergence of mutation. Further, a study involving ligand-EVG complexed with WT-IN protein demonstrated a conventional H-bond between EVG and the residue Asp116 along with two carbon H-bond formations with the same residue and an unfavourable negative–negative interaction between EVG and Asp60. Fascinatingly, two halogens (F and Cl) bonds were formed between EVG and with residues Pro142 and Asn144 of WT, respectively (Nusrath Unissa et al. 2017). Cross-resistance with RAL is most prominent in over 90% of cases as the RAL-resistant virus also retains resistance to EVG; however, despite viral mutations conferring resistance to RAL and EVG, 64% of the mentioned cases were susceptible to second-generation DTG (Unger et al. 2016).
Dolutegravir (Tivicay, Triumeq)
DTG was the earliest second-generation and first unboosted INSTI to get approval in 2013, owing to its favourable tolerability profile against RAL and EVG. This prominently distinctive resistance profile with a higher resistance barrier has decreased the pill burden and has further maintained activity against several INSTI resistance mutations as witnessed with first-line INSTI-based regimens. This high barrier is usually attributed to its long binding half-life to IN (Wainberg and Han 2015b; Malet et al. 2017; Brooks et al. 2019).
Metal ion chelation is a unifying feature mutual to all approved INSTIs. Structural and computational analyses reveal that the INSTI-resistant double mutation G140S/Q148H can affect all INSTI binding by disturbing the secondary coordination shells of the divalent ions in the IN active site (Nusrath Unissa et al. 2017; Smith et al. 2021a). However, variations to the INSTI scaffold can recompense for this loss of binding affinity, like in the case of DTG (presence of oxazine ring) and BIC (presence of oxazepine ring), which allow these compounds to have stabilizing engagements with backbone atoms of G118 and N117 in the β4-α2 loop of the IN, contributing to the enhanced activity of the second-generation INSTIs against the mutants (Cook et al. 2020). DTG chelates the Mg2+ ions in the active site, and the halo benzyl group forms a hydrophobic π–π stack with the base of the penultimate cytosine, interacting with the terminal adenine of the vcDNA (see Fig. 6), and the longer the linker, the better it allows a bound DTG to flexibly adjust to the alterations in the active site of IN mutants (Smith et al. 2021b).

(a) Binding orientation and polar bonds (in black) of DTG*3OYA (PDB ID) interactions. The green spheres represent magnesium ions, the orange sticks represent DTG, and the pink sticks are the amino acid residues involved in the ligand*receptor interaction. (b) Structural models of the Prototype foamy virus (PFV) 3OYA intasome with BIC as ligand. The coiling orange structure represents the viral complementary DNA (vcDNA) fragment, wherein the ligand can be seen interacting via the π-stacking bonding with the terminal 3′-deoxyadenosine 17 (DA17) of the vcDNA. Image created with the help of PyMol (Schrödinger and DeLano 2020)
Multiple reports on binding modes of all INSTIs to a surrogate for the HIV-1 intasome, the PFV intasomes, have revealed that an extended linker of DTG allows its halogen groups to enter beyond the pocket within the IN active site than other INSTIs. Further, DTG can regulate its structure and conformation in retort to structural changes within the active sites of ARV drug-resistant INs (Wainberg and Han 2015b, a). Using residue interaction network (RIN) analysis and MD simulation, Xue et al. (2013) disclosed that P145 in the 140S loop (Gly140–Gly149) of the intasome depicts strong hydrophobic interactions with INSTIs and is involved in a conformational rearrangement at the active site of the HIV-1 intasome which leads to a decrease in the chelating ability in INSTIs to Mg2+ in the active site of the resistant IN further causing cross-resistance.
As per the Stanford HIV-1 Drug Resistance Database—HIVDB (https://hivdb.stanford.edu/), residues T66K, E92Q, G118R, E138K/A/T, G140A/S/C, Q148H/R/K, N155H, and R263K are liable for IN resistance against DTG (Ross 2015; Stanford HIV-1 Drug Resistance Database 2019). Several non-polymorphic mutations (S153, R263, and G118) confer low-level in vitro DTG phenotype changes (Malet et al. 2017). A study conducted by Nusrath Unissa et al. (2017) demonstrated that DTG simultaneously formed two H-bonds between Gln62 and Asp116 residue of WT-IN and 3 conventional H-bonds amid DTG and Asp116, Gly149, and Glu152 residues. Remarkably, a halogen bond between the fluorine atom of the DTG’s pharmacophore and residue Asp116 was also formed. In addition, π–anion bonds were formed between DTG and residues Asp116 and Glu152, respectively, highlighting the role of residue Asp116 in multiple interactions. Further, alkyl bonds were also formed between DTG and residues Ile141 and Ile151, respectively. In the WT complex, the DTG formed a pair of stacking interactions, the first amid its 2, 4-difluorobenzyl ring and cytosine C16 of vcDNA and the next amid the metal-chelating pharmacophore and the viral 3′-deoxyadenosine (Malet et al. 2018).
This monocyclic carbamoyl pyridine derivative (Smith et al. 2021b) extends to the full width of the active site but only spans half of its height (DeAnda et al. 2013; Ross 2015). Few mutations among other accessory mutations involved in IN resistance to DTG both in vitro and in vivo include E138K, F121, G118R, S153Y, and R263K (Wainberg and Han 2015b; Seatla et al. 2021). The most typical mutation that arises in subtype B and recombinant A/G viruses when DTG is employed in vitro is R263K. These mutations not only confer low-level cross-resistance to DTG but either alone or in combination with other secondary mutations (like M50I, Q95K, T97A, V206I, G149A, H51Y/H, D67N, E157Q, A128T, and D232N) also diminish the viral fitness, and are rarely testified in patients (Wainberg and Han 2015b; Nusrath Unissa et al. 2017; Brooks et al. 2019; Seatla et al. 2021; Mikasi et al. 2021). Molecular modelling conducted by Quashie et al. (2012) proposes that R263K can initiate catalytic and structural changes within IN via partially impairing the IN–vcDNA binding, elucidating its selection by DTG.
However, treatment-emergent resistance mutations are of concern in patients, predominantly those with prior exposure to RAL or EVG (Cook et al. 2020). The second-generation INSTIs are effective against multiple RAMs that confer resistance to RAL and EVG; further, DTG considerably retains potency against the RAL-resistant mutants N155H, Y143R, and G140S/Q148H and the EVG-resistant mutants E92Q and T66I (Smith et al. 2021b). However, few standard mutations, mainly the ones involved in Q148 and N155 pathways, have been linked with the failure of DTG treatment, along with the selection of additional mutations, such as H51Y, L74M, T66A/I, E92Q, G118R, E138K/E/A, G140S/A, and S153Y/F, resulting in reduced susceptibility to DTG (Wainberg and Han 2015a; Brooks et al. 2019; Chitongo et al. 2019; Cook et al. 2020; Smith et al. 2021b, a). Furthermore, the effect of G140S mutation on subtype C IN has provided acumen into reduced drug affinity because of DTG resistance occurring due to reduced IN stability and greater flexibility around the 140 loop region in the mutant system (Chitongo et al. 2020).
A case study by Hardy et al. (2015) identified novel DTG mutational pathways in a RAL-resistant patient harbouring N155H when substituted with a DTG regimen for around 10 months. The mutations at T97A and E138K in IN led to a 37 FC resistance to DTG. After employing DTG for 10 further months, the sequential acquisition of mutations at L68FL, A49P, and L234V led to further resistance to DTG (~ 63 FC). Of these, except for L68FL, the other two are novel mutational pathways leading to emergent DTG resistance while on salvage therapy with the N155H mutation. In a similar case, an HIV-infected patient with high-level resistance to all PR and RT inhibitors and N155H, S119R, and E157Q mutations in IN after RAL failure was treated with DTG and RAL was subsequently removed from the regimen. A month later, the viral load was undetectable, and after about 8 months, a pair of novel mutations T97A and S147G in IN were detected (Wainberg and Han 2015b, a). These outcomes propose that DTG should be used with caution in INSTI salvage therapy for RAL failures or in patients with multiple RAMs (Alaoui et al. 2019).
Another study (Fulcher et al. 2018) showed that no INSTI is impervious to resistance, no matter how high its resistance barrier is. They showed virologic failure in treatment-naive individuals owing to the rapid evolution of IN mutations (T66I and M184V) in the presence of DTG, along with a simultaneous spike in plasma viraemia. In another study, the interesting association of polymorphic secondary mutation K156N (including other minor mutations leading to low susceptibility—K211R and E212T) with N155H was depicted, which was enough to confer impactful resistance to DTG; about fivefold change (FC, which is the decrease in potency of IN mutant against WT) was observed in IC50 as paralleled with N155H alone (Malet et al. 2018). DTG failure has been detected predominantly via the acquisition of manifold mutations at a given time.
DTG is susceptible to several other combinations of mutations, such as triple mutants M50I + S119R + R263K (FC ~ 28), G140S + Q148H + G149A (FC ~ 67), G140S + Q148H + T122N (FC ~ 37), E138K + G140S + Q148H (FC ~ 43), G140S + Q148H + N155H (FC ~ 49), E138K + Q148K + G140A (FC ~ 133), and V72I + E138K + Q148K (FC ~ 68) and the quadruple mutants L74M + V75A + G140S + Q148H (FC ~ 55) and L74M + G140S + S147G + Q148K (FC ~ 221) (Smith et al. 2021b).
Bictegravir (Biktarvy, GS-9883)
BIC was the next second-generation INSTI to be approved in 2018. It was designed and developed using structure–activity relationship data of DTG by modifying its oxazine ring with a series of oxaza-bridging and a 2,4,6-trifluorophenyl group, which led to an enhanced potency against G140S + Q148R double mutant. Being potent it has several advantages over first-generation agents, including a low potential for drug–drug interactions, a high genetic barrier to resistance like DTG, and administration without regard to meals or a boosting agent (Brooks et al. 2019). Although BIC has a high resistance barrier, the in vitro IN resistance selection tests over a long time frame verify the emergence of HIV-1 resistance to BIC comprising S153Y/F or R263K along with or without M50I (FC ~ 7) mutations. These substitutions have low-level reduced susceptibility to BIC and may occur in vivo after its wider use (Acosta et al. 2019; Smith et al. 2021b).
Similar to other INSTIs, even BIC coordinates the pair of chelating motifs, Mg2+ ions and stacks the halobenzyl moiety with the base of the penultimate cytosine. The favourable π-stacking interaction amid the halobenzyl tail with the 3’ cytosine of the vcDNA aids in its potency and resistance profile. The displaced terminal adenosine adapts to multiple rotameric conformations. Its oxazepine ring makes several interactions (van der Waals) with the β4α2 loop near the IN active site, resulting in a tighter binding. The methylene bridge in its ring imparts rigidness to its structure, allowing BIC to retain the ability to bind despite the changes in the geometry of the mutant IN active site (Smith et al. 2021b), refer to Fig. 7.

(a) Binding orientation and polar bonds (in black) of BIC*3OYA (PDB ID) interactions. The green spheres represent magnesium ions, the yellow sticks represent BIC, and the pink sticks are the amino acid residues involved in the ligand*receptor interaction. (b) Structural models of the PFV 3OYA intasome with BIC as ligand. The coiling orange structure represents the viral complementary DNA (vcDNA) fragment, wherein the ligand can be seen interacting via the π-stacking bonding with the terminal DA17 of the vcDNA. Image created with the help of PyMol (Schrödinger and DeLano 2020)
As per the HIVDB, residues T66K, E92Q, G118R, E138K/A/T, G140A/S/C, Q148H/R/K, N155H, and R263K are liable for IN resistance against BIC (Stanford HIV-1 Drug Resistance Database 2019). A study conducted by Anstett et al. (2017) demonstrated that the second-generation INSTIs faced the highest FCs in resistance due to the Q148 pathway when at least a pair of secondary mutations were present. However, these FCs are almost always below those that have been observed with first-generation INSTIs. In several phase III clinical studies, BIC has shown non-inferiority to DTG in treatment-naïve patients. However, it has led to weight gain in some patients (Smith et al. 2021b).
The in vitro susceptibility studies and animal models depict a 14 FC and 6 FC spike in the EC50 (i.e. 50% effective concentration) due to the G118R and R263K mutations, respectively, whereas a simultaneous 4.8 FC decrease in susceptibility to BIC is observed due to G140S + Q148H combination of mutations. If E138K joins this latter combination, it further reduces the BIC susceptibility 10 FC. To avoid these susceptible mutations and those observed during virologic failure of DTG monotherapy, BIC is co-formulated with tenofovir alafenamide or emtricitabine (Acosta et al. 2019; Wensing et al. 2019). Isaacs et al. (2020) conducted docking and EC50 experiments and derived a linear correlation between the inhibition constant values and experimental values for INSTIs, proposing that DTG and BIC are the most powerful IN inhibitors among all FDA-approved INSTIs.
An experiment conducted by Ghasabi et al. (2022) showed that the mutational combination of a naturally occurring polymorphism, M50I with a primary mutation, R263K and a minor accessory mutation L74I, reduces the susceptibility to BIC potency. Another study (Smith et al. 2020) showed that the IN with triple or quadruple mutations having RAL-resistant G140S + Q148H substitutions along with an accessory mutation at G140S/Q148H/V75A, G140S/Q148H/T122N, and G140S/Q148H/L74M/V75A reduced the BIC potency by more than 2.5 FC. The IN mutants developed by the team (C56S, G149A, and G140S + G149A) were all susceptible to BIC. Moreover, only BIC exhibited the potency to inhibit the RAL-resistant double mutant G140S/Q148H and the DTG-resistant IN mutants R263K, G118R, and H51Y/R263K with EC50 values less than 5 nM (Smith et al. 2018). Therefore, they concluded that RAMs with single substitutions that are located about 5 Å from the divalent ions bound at the IN active site generally do not cause an extensive reduction in susceptibility to BIC potency (Smith et al. 2020).
While BIC has not yet been employed in patients with prior INSTI failure, in vitro data propose that BIC and DTG have an extensively overlapping resistance profile (Scutari et al. 2020). Although BIC retains potency (FC ~ 5) against the DTG mutants (T66I + L74M + E138K + S147G + Q148R + S230N = FC ~ 33 towards DTG) by inhibiting the replication of a broad spectrum of IN mutants (Smith et al. 2020), it was susceptible to several other combinations of mutations, such as triple mutants G140S + Q148H + G149A (FC ~ 13), G140S + Q148H + N155H (FC ~ 30), E138K + G140A + Q148K (FC ~ 117), and V72I + E138K + Q148K (FC ~ 36) and the quadruple mutant L74M + G140S + S147G + Q148K (FC ~ 147). Moreover, these INSTI-resistant triple mutants when tested against CAB depicted a substantial decrease in their susceptibility (Smith et al. 2021b, a). These remarks emphasize the earlier mentioned tenacious need to develop novel INSTIs that can maintain potency against evolving IN mutations, particularly those that preserve potency against the variants capable of causing multiple mutations.
The mutation scores (MtS) refer to the summation of individual RAMs penalty score for a drug and determines its estimated resistance level.
-
0 ≤ MtS < 10 → no evidence of reduced ARV susceptibility (against WT),
-
10 < MtS < 14 → potential low-level resistance (might contain mutations either due to prior contact with ARV or might express when they occur with other mutations),
-
15 < MtS < 29 → low-level resistance (these depict reduced in vitro ARV susceptibility),
-
30 < MtS < 59 → intermediate resistance (high probability of reduced susceptibility to ARV),
-
MtS ≥ 60 → high-level resistance (not much virological response to treatment is observed in vitro) (Stanford HIV-1 Drug Resistance Database 2019).
To summarize, the primary substitution mutations in IN conferring INSTI resistance are Q148R/K/H, S147G, N155H/S, T66A/I/K, E92G/Q, T97A, Y143C/H/R, R263K, and F121Y, whereas the secondary IN mutations include L74M, Q95K/R, M50I, H51Y, L68I/V, V72A/N/T, G118R, G163K/R, S119P/R/T, F121C, P145S, A128T, E138A/K, G140A/C/S, Q146I/K/L/P/R, V151A/L, E170A, S153A/F/Y, and E157K/Q (Acosta et al. 2019); the majority of these mutational pathways and patterns are discussed in Table 2.
Natural anti-HIV molecules
Recently, to treat HIV/AIDS there has been a surge in isolating the natural substances as active leads from plants and other natural sources. The chief obstacle is the affordability of the treatment that does not involve any risk of drug resistance and serious side effects, thereby creating a strong demand to assess drugs derived from plants as well as their derivatives (Kaur et al. 2020). Some plant compounds are known for modulating multiple cellular factors involved in HIV replication, which makes them potential anti-HIV candidates. Therefore, it is vital to search for new antiretroviral agents which can be combined with or can substitute with the present antiretroviral drugs. Although several studies on multiple natural compounds have depicted enormous potential to become drug leads owing to their anti-HIV activity that may yield effective and affordable therapeutic agents, it has not been well documented or its literature is vague and scanty (Kurapati et al. 2015).
Multiple bioactive compounds can inhibit HIV, these are broadly categorized as phenolics, flavonoids, triterpenes, alkaloids, polyphenolics, lectins, coumarins, saponins, lignans, tannins, lactones, iridoids, O-caffeoyl derivatives, xanthones, sulphated polysaccharides, polyketides, quinones, and phospholipids (Salehi et al. 2018). There are also some ribosome-inactivating plant proteins, like MAP30 and MRK29, from Momordica charantia fruits, Trichosanthin from Trichosanthes kirilowii roots, and GAP31 from Gelonium multiflorum seeds that inhibit the HIV-1 infection in T-cells and monocytes via IN inhibition, except for MRK29 which inhibits RT. Moreover, these have an N-glycosidase activity on 28S ribosomal RNA and a topological activity on long terminal repeats of HIV (Mandal et al. 2020; Kaur et al. 2020). More such plants comprising proteins employed in HIV/AIDS have been discussed by Kaur and their team (2020). Further, there are proofs where plant extracts depicted anti-HIV activity considerably, but need supplementary studies to isolate the major active compounds responsible for the same. Several such prominent compounds and plants have been summarized in Online Resource Table S1.
Mandal et al. (2020) and Kurapati et al. (2015) have summarized the clinical trials of HIV/AIDS employing natural compounds. As reported in 2018, out of 717 species, HIV-RT, HIV-PR, and HIV IN inhibitory activities have been tallied to about 206, 254, and 43 species, respectively. Apart from these studies, researchers have also estimated nearly 390 species for other enzyme inhibition studies which are categorized under the umbrella of anti-HIV activities (Salehi et al. 2018). Since the main focus of this review paper are INSTIs, we will be elaborating on the docking interactions for a few bioactive compounds/plants exhibiting IN inhibition.
Online Resource Table S1 indicates that the potential IN enzyme inhibition is due to the presence of the catechol, galloyl, and sugar moieties. In the computational docking of the bioactive compounds composing the crude extract of Dioscorea bulbifera L, Chaniad et al. (2016) found myricetin to be a potent INSTI as it comprised the galloyl moiety which binds strongly with the IN amino acids. It interacts with several amino acid residues via hydrogen bonding—Thr66, His67, Asp116, Glu152, Asn155, and Lys159. Another compound, quercetin-3-O-β-d-glucopyranoside comprised both the catechol and the sugar moieties; the former was involved with the IN inhibition, whereas the latter enhanced this activity by increasing its solubility. It forms hydrogen bonds with the enzyme’s Thr66, Glu92, Asp116, Gln148, and Lys159 residues, exhibiting an IC50 of 21.80 μM (Kaur et al. 2020).
The computational docking of crude extract of Betula alnoides showed potential interactions of its compounds with IN active sites. The results directed that betulin interacted with Thr66, His67, and Lys159 residues involved in the ST reaction and with Asp64, a catalytic triad residue involved in 3′P of the integration process. The binding energy of betulinic acid was lower than that of botulin as it interacted with the same residues except for His67. Its oleanolic and ursolic acids showed weak interactions with Thr66 and Gln148, and Gln148 and Lys159, respectively (Chaniad et al. 2019). Similarly, catechin interacts with Thr66, Gly148, and Glu152 in the CCD, whereas protocatechuic acid was reported to interact with Thr66, His67, Glu152, Asn155, and Lys159. Both these compounds interact with Glu152 which is a catalytic triad residue; further catechin possesses higher activity and lower binding energy than protocatechuic acid, making it a better INSTI (Panthong et al. 2015; Kaur et al. 2020).
Another docking study of chicoric acid shows that it binds to several amino acid residues depending on its orientation, at times with hydrophobic interactions between its two phenyl arms and E152, I141, and Q148 and at times with E152, K156, C65, and E92 while pointing towards H67 and T66. Its phenyl arms are entirely anchored via hydrophobic interactions, one arm coordinated by the catalytic triad residue D116 and Q148 and the other arm by C65 (Nobela et al. 2018). Another study conducted by Siwe-Noundou et al. (2019) docked IN with L-chicoric acid (LCA) and Alchornea cordifolia’s methylgallate (MG). This protein–ligand interaction analysis indicated that one of the vicinal OH-group in MG formed an H-bond with the thiol group, Cys65. Unique to LCA was its ability to interact with Gln148 (which is found in the 140 s loop) facing the active site, for which its aromatic rings have to be perpendicular for optimum interaction.
Apart from anti-HIV natural substances extracted from plants, INSTIs can be of other origins too, like microbial (fungal, algal, cyanobacterial), marine vertebrates and invertebrates, etc. Despite marine invertebrates producing a variety of antivirals, limited products have reached clinical trials or have been approved for use. One such example is tris-phenethyl urea, molleurea A, and cyclopeptides, mollamides E (IC50 = 39 μM) and F, extracted from tunicate Didemnum molle’s methanol extract, which demonstrated the ability to inhibit HIV-1 replication as well as behaved like an INSTI. There have also been studies on Lamellarin alkaloids produced by the marine ascidian (mollusc), Lamellaria sp. (Riccio et al. 2020).
A study by Shiomi et al. (2005) identified a trio of phenalenones from the culture broth of Penicillium sp. FKI-1463 showed IN inhibiting properties. The compounds are atrovenetinone methyl acetal, erabulenol B, and funalenone with IC50 values of 19 μM, 7.9 μM, and 10 μM respectively. Another study (Ding et al. 2017) included the isolation of alternariol 5-O-methyl ether from the endophytic fungus Colletotrichum sp., which was found to be an HIV-1 PIC inhibitor and an INSTI (Raimi and Adeleke 2021). Similarly, a carboxybiphenyl INSTI, altenusin (IC50 = 25 μM), was isolated from the soil broth of the fungus Talaromyces pinophilus BioMCC-f.T.3979, epi-ophiobolin K (IC50 > 120 μM) and epi-ophiobolin C (IC50 > 120 μM) from fungus Neosartorya sp., nalanthalide (IC50 = 25 μM), and coprophilin (IC50 = 25 μM) from fungus Nalanthamala sp. MF 5638 (Singh et al. 2003; Waluyo et al. 2021), Integrasone (a bicyclic dihydroxy epoxide lactone with IC50 value of 41 μM) from an unidentified fungus MF6836 (Herath et al. 2004), and Aspochalasin L from Aspergillus flavipes (IC50 = 71.7 μM) (Rochfort et al. 2005). More such compounds of fungal origin with anti-HIV and INSTI properties have been elaborated by Linnakoski et al. (2018) and Biswajit Roy (2017).
A study demonstrated phlorotannins isolated from the alga Ecklonia cava Kjellman could effectively inhibit IN, similarly, a carmalol derivative, diphlorethohydroxycarmalol isolated from a marine brown alga, Ishige okamurae, with both their IC50 values being 25.2 μM (Vo and Kim 2010; Besednova et al. 2019). Another such INSTI isolated from cyanobacteria, Lyngbya majuscula is dolastatin 3. More such compounds of algal origin with anti-HIV properties have been elaborated by Liu et al. (2021) and Reynolds et al. (2021).
If one carefully observes Online Resource Table S1, it is evident that some bioactive molecules act as dual inhibitors (DIs); these comprise a pharmacophore that enables it to bind to two varying target enzymes to produce synergistic or additive effects. These are usually either IN and LEDGF-dependent IN or IN and RT-associated ribonuclease H inhibitors. The latter is the most common type of DIs, sharing a common catalytic site residue and geometry, like quinolinonyl β-diketo acid derivatives, 2-hydroxyisoquinoline-1,3(2H,4H)-diones, madurahydroxylactone, and pyrrolyl derivatives. Some examples of the former category include CHI-1043, CHIBA-3002, CHIBA-3003, CHIBA-3053, GSK1264, Tert-butoxy(4-phenyl-quinolin-3-yl)-acetic acid, and CX04328 (Di Santo 2014; Choi et al. 2018; Ercan et al. 2019). A universal drawback of ribonuclease H is that it has a human homolog; this obstructs the designing of DI that specifically inhibits the viral enzyme and keeps the human homolog intact. Consequently, it is better to find biological targets other than IN that can be inhibited simultaneously with IN when formulated as a DI, tackling the resistance problems faced earlier (Mahboubi-Rabbani et al. 2021).
Conclusion
-
Hitherto, INSTIs arguably are the most effective and well-tolerated ARV drugs to be discovered and developed. The introduction of INSTIs to the HIV therapeutic landscape provides safe, effective, and viable HIV treatment alternatives.
-
Selecting which regimen to use in initial HIV treatment acts as a crucial decision. It is predicted that the older first-generation INSTIs, RAL and EVG, will soon be phased out in near future owing to the potential for novel formulations to revolutionize patient care and advantages offered by second-generation INSTIs in terms of drug interactions, tolerability, resistance barrier, safety, cross-resistance, dosing strategy, and efficacy in both treatment-experienced and treatment-naïve patients.
-
Although INSTIs are highly potent drugs, as the frequency of use of INSTIs continues to rise, inevitable mutations will develop that confer resistance against all currently available ARVs. Therefore, more computational studies (in silico) and clinical experiments (in vitro and in vivo) are required for the characterization of the resistance profile of all FDA-approved INSTIs.
-
Promising natural or synthetic compounds that retain potency against the initial set of mutants can be tested against more complex IN mutants and for their ability to select additional mutants.
-
More sensitive next-generation techniques, assays, and equipment are vital for identifying low-level viraemia and minority low-level resistance variants.
-
To infer based on the available data, BIC is the most suitable and promising option. This and its structurally related analogues can be further studied and employed accordingly in a novel INSTI structure-based design having a better resistance profile than present drugs.
-
In addition to that, INSTIs designed to interact more strongly with the 3’-adenosine nucleotide could potentially adapt to the modified geometry of the IN active site that is responsible for some of the most problematic RAMs. With this, there is a high possibility that the HIV-1 intasome structures addressing numerous popular IN mutants will be solved soon.
-
Bioactive compounds alone or as DI or in combination with HAART can be prescribed for accepted toxicity and fewer RAMs. This optimistic outcome is bound to inspire researchers for a positive breakthrough in the near future.
Data Availability
All data generated or analyzed during this study are included in this published article [and its supplementary information files.
References
Abdelhameed AS (2015) Insight into the interaction between the HIV-1 integrase inhibitor elvitegravir and bovine serum albumin: a spectroscopic study. J Spectrosc. https://doi.org/10.1155/2015/435674
Acosta RK, Willkom M, Martin R et al (2019) Resistance analysis of bictegravir-emtricitabine-tenofovir alafenamide in HIV-1 treatment-naive patients through 48 weeks. Antimicrob Agents Chemother. https://doi.org/10.1128/AAC.02533-18
Akanbi MO, Scarci K, Taiwo B, Murphy RL (2012) Combination nucleoside/nucleotide reverse transcriptase inhibitors. Exp Opin Pharmacother 13:65. https://doi.org/10.1517/14656566.2012.642865
Alaoui N, El Alaoui MA, El Annaz H et al (2019) HIV-1 integrase resistance among highly antiretroviral experienced patients from Morocco. Intervirology 62:65–71. https://doi.org/10.1159/000501016
Alavian G, Kolahdouzan K, Mortezazadeh M, Torabi ZS (2021) Antiretrovirals for prophylaxis against COVID-19: a comprehensive literature review. J Clin Pharmacol 61:581–590. https://doi.org/10.1002/JCPH.1788
Anstett K, Brenner B, Mesplede T, Wainberg MA (2017) HIV drug resistance against strand transfer integrase inhibitors. Retrovirology 14:36. https://doi.org/10.1186/S12977-017-0360-7
Arora R, Tchertanov L (2013) The HIV-1 integrase: modeling and beyond. Integr View Mol Recognit Toxinol. https://doi.org/10.5772/52344
Arora R, De Beauchene IC, Polanski J et al (2013) Raltegravir flexibility and its impact on recognition by the HIV-1 IN targets. J Mol Recognit 26:383–401. https://doi.org/10.1002/JMR.2277
Arora R (2013) Molecular mechanism of HIV-1 integrase inhibition by Raltegravir proposed by using of molecular modeling approaches
Arslan N (2019) Molecular docking study of four chromene derivatives as novel HIV-1 integrase inhibitors. J Turk Chem Soc Sect A Chem 6:133–142. https://doi.org/10.18596/JOTCSA.478772
Atta MG, De Seigneux S, Lucas GM (2019) Clinical pharmacology in HIV therapy. Clin J Am Soc Nephrol 14:435. https://doi.org/10.2215/CJN.02240218
Avert (2020) HIV and AIDS in India. In: Avert. https://www.avert.org/professionals/hiv-around-world/asia-pacific/india. Accessed 27 Feb 2022
Bar-Magen T, Sloan RD, Donahue DA et al (2010) Identification of novel mutations responsible for resistance to MK-2048, a second-generation HIV-1 integrase inhibitor. J Virol 84:9210. https://doi.org/10.1128/JVI.01164-10
Besednova NN, Zvyagintseva TN, Kuznetsova TA et al (2019) Marine algae metabolites as promising therapeutics for the prevention and treatment of HIV/AIDS. Metabolites. https://doi.org/10.3390/METABO9050087
Bhatt H, Patel P, Pannecouque C (2014) Discovery of HIV-1 integrase inhibitors: pharmacophore mapping, virtual screening, molecular docking, synthesis, and biological evaluation. Chem Biol Drug Des 83:154–166. https://doi.org/10.1111/CBDD.12207
Blanco JL, Whitlock G, Milinkovic A, Moyle G (2015) HIV integrase inhibitors: a new era in the treatment of HIV. Exp Opin Pharmacother 16:1313–1324. https://doi.org/10.1517/14656566.2015.1044436
Blut A, Blood S (2016) Assessment of PT by human immunodeficiency virus (HIV). Transfus Med Hemother 43:203. https://doi.org/10.1159/000445852
Brooks KM, Sherman EM, Egelund EF et al (2019) Integrase inhibitors: after 10 years of experience, is the best yet to come? Pharmacother J Hum Pharmacol Drug Ther 39:576–598. https://doi.org/10.1002/PHAR.2246
Cattaneo D, Gervasoni C (2019) Pharmacokinetics and Pharmacodynamics of cabotegravir, a long-acting HIV integrase strand transfer inhibitor. Eur J Drug Metab Pharmacokinet 44:319–327. https://doi.org/10.1007/S13318-018-0526-2/TABLES/4
Chaniad P, Wattanapiromsakul C, Pianwanit S, Tewtrakul S (2016) Anti-HIV-1 integrase compounds from Dioscorea bulbifera and molecular docking study. Pharm Biol 54:1077–1085. https://doi.org/10.3109/13880209.2015.1103272
Chaniad P, Sudsai T, Septama AW et al (2019) Evaluation of Anti-HIV-1 integrase and anti-inflammatory activities of compounds from betula alnoides buch-ham. Adv Pharmacol Sci. https://doi.org/10.1155/2019/2573965
Chen Q, Cheng X, Wei D, Xu Q (2014) Molecular dynamics simulation studies of the wild type and E92Q/N155H mutant of Elvitegravir-resistance HIV-1 integrase. Interdiscip Sci Comput Life Sci 7:36–42. https://doi.org/10.1007/S12539-014-0235-8
Chitongo R, Obasa AE, Mikasi SG et al (2019) Molecular dynamic simulations to investigate the structural impact of known drug resistance mutations on HIV-1C integrase-dolutegravir binding. BioRxiv. https://doi.org/10.1101/781120
Chitongo R, Obasa AE, Mikasi SG et al (2020) Molecular dynamic simulations to investigate the structural impact of known drug resistance mutations on HIV-1C Integrase-Dolutegravir binding. PLoS ONE. https://doi.org/10.1371/JOURNAL.PONE.0223464
Choi E, Mallareddy JR, Lu D, Kolluru S (2018) Recent advances in the discovery of small-molecule inhibitors of HIV-1 integrase. Futur Sci. https://doi.org/10.4155/FSOA-2018-0060
Cook NJ, Li W, Berta D et al (2020) Structural basis of second-generation HIV integrase inhibitor action and viral resistance. Science 367:806–810. https://doi.org/10.1126/SCIENCE.AAY4919/SUPPL_FILE/PAPV2.PDF
Crisan L, Bora A (2021) Small molecules of natural origin as potential anti-HIV agents: a computational approach. Life 11:722. https://doi.org/10.3390/LIFE11070722
Dahabieh MS, Battivelli E, Verdin E (2015) Understanding HIV latency: the road to an HIV cure. Annu Rev Med 66:407. https://doi.org/10.1146/ANNUREV-MED-092112-152941
DeAnda F, Hightower KE, Nolte RT et al (2013) Dolutegravir interactions with HIV-1 integrase-DNA: structural rationale for drug resistance and dissociation kinetics. PLoS ONE 8:e77448. https://doi.org/10.1371/JOURNAL.PONE.0077448
Deeks ED (2018) Bictegravir/emtricitabine/tenofovir alafenamide: a review in HIV-1 infection. Drugs 78:1817. https://doi.org/10.1007/S40265-018-1010-7
Di Santo R (2014) Inhibiting the HIV integration process: past, present, and the future. J Med Chem 57:539–566. https://doi.org/10.1021/JM400674A/ASSET/IMAGES/LARGE/JM-2013-00674A_0014.JPEG
Ding J, Zhao J, Yang Z et al (2017) Microbial natural product alternariol 5-O-methyl ether inhibits HIV-1 integration by blocking nuclear import of the pre-integration complex. Viruses 9:105. https://doi.org/10.3390/V9050105
Dogan B, Durdagi S (2021) Drug re-positioning studies for novel HIV-1 inhibitors using binary QSAR models and multi-target-driven in silico studies. Mol Inform 40:2000012. https://doi.org/10.1002/MINF.202000012
Duwal S, Dickinson L, Khoo S, von Kleist M (2018) Hybrid stochastic framework predicts efficacy of prophylaxis against HIV: an example with different dolutegravir prophylaxis schemes. PLOS Comput Biol 14:e1006155. https://doi.org/10.1371/JOURNAL.PCBI.1006155
El Khoury L, El HK, Piquemal J-P et al (2019) Spectrometric and computational studies of the binding of HIV-1 integrase inhibitors to viral DNA extremities. PeerJ Phys Chem 1:e6. https://doi.org/10.7717/PEERJ-PCHEM.6
Ercan S, Şenyiğit B, Şenses Y (2019) Dual inhibitor design for HIV-1 reverse transcriptase and integrase enzymes: a molecular docking study. J Biomol Struct Dyn 38:573–580. https://doi.org/10.1080/0739110220191700166
Esmaeili S, Mosaddeghi H, Ravari F (2021) Molecular docking studies of HIV-1 protease-, integrase- and reverse-transcriptase with delta-9-tetrahydrocannabinol and curcumin as two herbal ligands. J Evol Biochem Physiol 57:281–288. https://doi.org/10.1134/S0022093021020101
Evans N, Martinez E, Petrosillo N et al (2021) SARS-CoV-2 and human immunodeficiency virus: pathogen pincer attack. HIV/AIDS Res Palliat Care 13:361–375. https://doi.org/10.2147/HIV.S300055
Fenwick C, Amad M, Bailey MD et al (2014) Preclinical profile of BI 224436, a novel HIV-1 non-catalytic-site integrase inhibitor. Antimicrob Agents Chemother 58:3233. https://doi.org/10.1128/AAC.02719-13
Fernandez C, van Halsema CL (2019) Evaluating cabotegravir/rilpivirine long-acting, injectable in the treatment of HIV infection: emerging data and therapeutic potential. HIV AIDS (auckl) 11:179. https://doi.org/10.2147/HIV.S184642
Fulcher JA, Du Y, Zhang TH et al (2018) Emergence of integrase resistance mutations during initial therapy containing dolutegravir. Clin Infect Dis 67:791–794. https://doi.org/10.1093/CID/CIY228
Ghasabi F, Hashempour A, Khodadad N et al (2022) First report of computational protein–ligand docking to evaluate susceptibility to HIV integrase inhibitors in HIV-infected Iranian patients. Biochem Biophys Rep 30:101254. https://doi.org/10.1016/J.BBREP.2022.101254
Gong Y, Zhi K, Nagesh PKB et al (2020) An elvitegravir nanoformulation crosses the blood–brain barrier and suppresses HIV-1 replication in microglia. Viruses 12:564. https://doi.org/10.3390/V12050564
Gupta P, Sharma A, Garg P, Roy N (2013) QSAR study of curcumine derivatives as HIV-1 integrase inhibitors. Curr Comput Aided Drug Des 9:141–150. https://doi.org/10.2174/157340913804998793
Han D, Su M, Tan J et al (2016) Structure–activity relationship and binding mode studies for a series of diketo-acids as HIV integrase inhibitors by 3D-QSAR, molecular docking and molecular dynamics simulations. RSC Adv 6:27594–27606. https://doi.org/10.1039/C6RA00713A
Hardy I, Brenner B, Quashie P et al (2015) Evolution of a novel pathway leading to dolutegravir resistance in a patient harbouring N155H and multiclass drug resistance. J Antimicrob Chemother 70:405–411. https://doi.org/10.1093/JAC/DKU387
Herath KB, Jayasuriya H, Bills GF et al (2004) Isolation, structure, absolute stereochemistry, and HIV-1 integrase inhibitory activity of integrasone, a novel fungal polyketide. J Nat Prod 67:872–874. https://doi.org/10.1021/NP0340504/SUPPL_FILE/NP0340504SI20040209_030413.PDF
Hill L, Smith SR, Karris MY (2018) Profile of bictegravir/emtricitabine/tenofovir alafenamide fixed dose combination and its potential in the treatment of HIV-1 infection: evidence to date. HIV AIDS (auckl) 10:203. https://doi.org/10.2147/HIV.S145529
Hodge D, Back DJ, Gibbons S et al (2021) Pharmacokinetics and drug–drug interactions of long-acting intramuscular cabotegravir and rilpivirine. Clin Pharmacokinet 60:835–853. https://doi.org/10.1007/S40262-021-01005-1/FIGURES/4
Hoesley CJ, Chen BA, Anderson PL et al (2019) Phase 1 safety and pharmacokinetics study of MK-2048/Vicriviroc (MK-4176)/MK-2048A intravaginal rings. Clin Infect Dis 68:1136–1143. https://doi.org/10.1093/CID/CIY653
Huang Y, Huang X, Chen H et al (2019) Efficacy and safety of raltegravir-based dual therapy in AIDS patients: a meta-analysis of randomized controlled trials. Front Pharmacol 10:1225. https://doi.org/10.3389/FPHAR.2019.01225/BIBTEX
Indu P, Rameshkumar MR, Arunagirinathan N et al (2020) Raltegravir, indinavir, tipranavir, dolutegravir, and etravirine against main protease and RNA-dependent RNA polymerase of SARS-CoV-2: A molecular docking and drug repurposing approach. J Infect Public Health 13:1856–1861. https://doi.org/10.1016/J.JIPH.2020.10.015
Isaacs D, Mikasi SG, Obasa AE et al (2020) Structural comparison of diverse HIV-1 subtypes using molecular modelling and docking analyses of integrase inhibitors. Viruses 12:936. https://doi.org/10.3390/V12090936
Islam MA, Pillay TS (2016) Structural requirements for potential HIV-integrase inhibitors identified using pharmacophore-based virtual screening and molecular dynamics studies. Mol Biosyst 12:982–993. https://doi.org/10.1039/C5MB00767D
Kang-Birken SL, El-sayed D, Prichard J (2019) HIV viral rebound due to a possible drug–drug interaction between elvitegravir/cobicistat/emtricitabine/tenofovir alafenamide and calcium-containing products: report of 2 cases. J Int Assoc Provid AIDS Care. https://doi.org/10.1177/2325958218821653
Kaur R, Sharma P, Gupta GK et al (2020) Structure-activity-relationship and mechanistic insights for anti-HIV natural products. Mol 25:2070. https://doi.org/10.3390/MOLECULES25092070
Kolakowska A, Maresca AF, Collins IJ (2019) Update on adverse effects of HIV integrase inhibitors. Curr Treat Options Infect Dis 114(11):372–387. https://doi.org/10.1007/S40506-019-00203-7
Kruize Z, Kootstra NA (2019) The role of macrophages in HIV-1 persistence and pathogenesis. Front Microbiol 10:2828. https://doi.org/10.3389/FMICB.2019.02828/BIBTEX
Kubin CJ, Hammer SM (2010) Antiretroviral agents. Infect Dis Third Ed 2:1434–1453. https://doi.org/10.1016/B978-0-323-04579-7.00145-3
Kurapati KRV, Atluri VS, Samikkannu T et al (2015) Natural products as anti-HIV agents and role in HIV-associated neurocognitive disorders (HAND): a brief overview. Front Microbiol. https://doi.org/10.3389/FMICB.2015.01444
Li G, De Clercq E (2016) HIV genome-wide protein associations: a review of 30 years of research. Microbiol Mol Biol Rev 80:679–731. https://doi.org/10.1128/MMBR.00065-15/SUPPL_FILE/ZMR003162428SO1.PDF
Linnakoski R, Reshamwala D, Veteli P et al (2018) Antiviral agents from fungi: diversity, mechanisms and potential applications. Front Microbiol 9:2325. https://doi.org/10.3389/FMICB.2018.02325/BIBTEX
Liu J, Obaidi I, Nagar S et al (2021) The antiviral potential of algal-derived macromolecules. Curr Res Biotechnol 3:120–134. https://doi.org/10.1016/J.CRBIOT.2021.04.003
Mahboubi-Rabbani M, Abbasi M, Hajimahdi Z, Zarghi A (2021) HIV-1 reverse transcriptase/integrase dual inhibitors: a review of recent advances and structure-activity relationship studies. Iran J Pharm Res IJPR 20:333. https://doi.org/10.22037/IJPR.2021.115446.15370
Makoti P, Fielding BC (2020) HIV and human coronavirus coinfections: a historical perspective. Viruses 12:937. https://doi.org/10.3390/V12090937
Malet I, Subra F, Charpentier C et al (2017) Mutations located outside the integrase gene can confer resistance to HIV-1 integrase strand transfer inhibitors. Mbio. https://doi.org/10.1128/MBIO.00922-17/ASSET/AAECBCCA-7EAF-4566-AC85-49E1B03887ED/ASSETS/GRAPHIC/MBO0051735020005.JPEG
Malet I, Ambrosio FA, Subra F et al (2018) Pathway involving the N155H mutation in HIV-1 integrase leads to dolutegravir resistance. J Antimicrob Chemother 73:1158–1166. https://doi.org/10.1093/JAC/DKX529
Mandal A, Biswas D, Hazra B (2020) Natural products from plants with prospective anti-HIV activity and relevant mechanisms of action. Stud Nat Prod Chem 66:225–271. https://doi.org/10.1016/B978-0-12-817907-9.00009-X
Martin YC (2014) Applications of pharmacophore mapping. Ref Modul Chem Mol Sci Chem Eng. https://doi.org/10.1016/B978-0-12-409547-2.11305-8
Masso M, Chuang G, Hao K et al (2014) Structure-based predictors of resistance to the HIV-1 integrase inhibitor Elvitegravir. Antiviral Res 106:5–12. https://doi.org/10.1016/J.ANTIVIRAL.2014.03.006
Mbhele N, Chimukangara B, Gordon M (2021) HIV-1 integrase strand transfer inhibitors: a review of current drugs, recent advances and drug resistance. Int J Antimicrob Agents 57:106343. https://doi.org/10.1016/J.IJANTIMICAG.2021.106343
Mikasi SG, Isaacs D, Chitongo R et al (2021) Interaction analysis of statistically enriched mutations identified in Cameroon recombinant subtype CRF02_AG that can influence the development of Dolutegravir drug resistance mutations. BMC Infect Dis 21:1–12. https://doi.org/10.1186/S12879-021-06059-X/FIGURES/4
NACO (2013) National guidelines on second-line and alternative first-line ART for adults and adolescents
NACO (2019) HIV Facts & Figures | National AIDS Control Organization | MoHFW | GoI. In: NACO. http://naco.gov.in/hiv-facts-figures. Accessed 4 Mar 2022
National Institutes of Health (2022) What’s New in the Guidelines? | NIH. In: Clin. INFO.HIV.GOV, NIH. https://clinicalinfo.hiv.gov/en/guidelines/adult-and-adolescent-arv/whats-new-guidelines. Accessed 25 Mar 2022
Nobela O, Renslow RS, Thomas DG et al (2018) Efficient discrimination of natural stereoisomers of chicoric acid, an HIV-1 integrase inhibitor. J Photochem Photobiol B Biol 189:258–266. https://doi.org/10.1016/J.JPHOTOBIOL.2018.10.025
Notario-Pérez F, Ruiz-Caro R, Veiga-Ochoa MD (2017) Historical development of vaginal microbicides to prevent sexual transmission of HIV in women: from past failures to future hopes. Drug Des Devel Ther 11:1767–1787. https://doi.org/10.2147/DDDT.S133170
Nusrath Unissa A, Swathi S, Ramya Lakshmi A, Elizabeth Hanna L (2017) Insights into Integrase resistance to dolutegravir, elvitegravir and raltegravir-strand transfer inhibitors of HIV-1: a computational approach. Orig Res Artic Saudi J Pathol Microbiol 2:167–175
Oliveira M, Ibanescu RI, Anstett K et al (2018) Selective resistance profiles emerging in patient-derived clinical isolates with cabotegravir, bictegravir, dolutegravir, and elvitegravir. Retrovirology 15:56. https://doi.org/10.1186/S12977-018-0440-3
Panthong P, Bunluepuech K, Boonnak N et al (2015) Anti-HIV-1 integrase activity and molecular docking of compounds from Albizia procera bark. Pharm Biol 53:1861–1866. https://doi.org/10.3109/13880209.2015.1014568
Park KH, Kim M, Bae SE et al (2021) Study on suitable analysis method for HIV-1 non-catalytic integrase inhibitor. Virol J 18:1–9. https://doi.org/10.1186/S12985-020-01476-X/FIGURES/4
Phillips AN, Bansi-Matharu L, Cambiano V et al (2021) The potential role of long-acting injectable cabotegravir–rilpivirine in the treatment of HIV in sub-Saharan Africa: a modelling analysis. Lancet Glob Heal 9:e620–e627. https://doi.org/10.1016/S2214-109X(21)00025-5/ATTACHMENT/092D2DBF-6FBA-4226-8B75-0924D14C906A/MMC1.PDF
Podany AT, Scarsi KK, Fletcher CV (2017) Comparative clinical pharmacokinetics and pharmacodynamics of HIV-1 integrase strand transfer inhibitors. Clin Pharmacokinet 56:25. https://doi.org/10.1007/S40262-016-0424-1
Putim C, Phaonakrop N, Jaresitthikunchai J et al (2018) Secretome profile analysis of multidrug-resistant, monodrug-resistant and drug-susceptible Mycobacterium tuberculosis. Arch Microbiol 200:299–309. https://doi.org/10.1007/S00203-017-1448-0/FIGURES/5
Quashie PK, Mesplède T, Han Y-S et al (2012) Characterization of the R263K mutation in HIV-1 integrase that confers low-level resistance to the second-generation integrase strand transfer inhibitor dolutegravir. J Virol 86:2696–2705. https://doi.org/10.1128/JVI.06591-11/ASSET/C94B8E9C-DC59-486E-AEC2-9C3D1E3BD09A/ASSETS/GRAPHIC/ZJV9990957020005.JPEG
Raimi A (2021) Adeleke R (2021) Bioprospecting of endophytic microorganisms for bioactive compounds of therapeutic importance. Arch Microbiol 2035(203):1917–1942. https://doi.org/10.1007/S00203-021-02256-Z
Raj CTD, Kandaswamy DK, Danduga RCSR et al (2021) COVID-19: molecular pathophysiology, genetic evolution and prospective therapeutics—a review. Arch Microbiol 203:2043–2057. https://doi.org/10.1007/S00203-021-02183-Z/TABLES/1
Rameshrad M, Ghafoori M, Mohammadpour AH et al (2020) A comprehensive review on drug repositioning against coronavirus disease 2019 (COVID19). Naunyn Schmiedebergs Arch Pharmacol 393:1137. https://doi.org/10.1007/S00210-020-01901-6
Reddy KK, Singh SK, Tripathi SK, Selvaraj C (2013) Identification of potential HIV-1 integrase strand transfer inhibitors: In silico virtual screening and QM/MM docking studies. SAR QSAR Environ Res 24:581–595. https://doi.org/10.1080/1062936X.2013.772919
Reynolds D, Huesemann M, Edmundson S et al (2021) Viral inhibitors derived from macroalgae, microalgae, and cyanobacteria: a review of antiviral potential throughout pathogenesis. Algal Res 57:102331. https://doi.org/10.1016/J.ALGAL.2021.102331
Rhee SY, Grant PM, Tzou PL et al (2019) A systematic review of the genetic mechanisms of dolutegravir resistance. J Antimicrob Chemother 74:3135–3149. https://doi.org/10.1093/JAC/DKZ256
Riccio G, Ruocco N, Mutalipassi M et al (2020) Ten-year research update review: antiviral activities from marine organisms. Biomol 10:1007. https://doi.org/10.3390/BIOM10071007
Rochfort S, Ford J, Ovenden S et al (2005) A novel aspochalasin with HIV-1 integrase inhibitory activity from Aspergillus flavipes. J Antibiot (tokyo) 58:279–283. https://doi.org/10.1038/JA.2005.34
Ross K (2015) Hiv integrase mechanisms of resistance to Raltegravir, Elvitegravir. Graduate School of Wayne State University, Dolutegravir
Rossi E, Meuser ME, Cunanan CJ, Cocklin S (2021) Structure, function, and interactions of the HIV-1 capsid protein. Life 11:100. https://doi.org/10.3390/LIFE11020100
Roulet J, Taton A, Golden JW et al (2018) Development of a cyanobacterial heterologous polyketide production platform. Metab Eng 49:94–104. https://doi.org/10.1016/j.ymben.2018.07.013
Roy BG (2017) Potential of small-molecule fungal metabolites in antiviral chemotherapy. Antivir Chem Chemother 25:20–52. https://doi.org/10.1177/2040206617705500
Salehi B, Anil Kumar NV, Şener B et al (2018) Medicinal plants used in the treatment of human immunodeficiency virus. Int J Mol Sci 19:1459. https://doi.org/10.3390/IJMS19051459
Samorlu AS, Yelekçi K, Ibrahim Uba A (2019) The design of potent HIV-1 integrase inhibitors by a combined approach of structure-based virtual screening and molecular dynamics. SIMULATION 37:4644–4650. https://doi.org/10.1080/07391102.2018.1557559
Schrödinger L, DeLano W (2020) PyMOL. http://www.pymol.org/pymol. Accessed 18 Feb 2023
Scutari R, Alteri C, Vicenti I et al (2020) Evaluation of HIV-1 integrase resistance emergence and evolution in patients treated with integrase inhibitors. J Glob Antimicrob Resist 20:163–169. https://doi.org/10.1016/J.JGAR.2019.07.015
Seatla KK, Maruapula D, Choga WT et al (2021) HIV-1 subtype C drug resistance mutations in heavily treated patients failing integrase strand transfer inhibitor-based regimens in Botswana. Viruses 13:594. https://doi.org/10.3390/V13040594
Shiomi K, Matsui R, Isozaki M et al (2005) Fungal phenalenones inhibit HIV-1 integrase. J Antibiot 581(58):65–68. https://doi.org/10.1038/ja.2005.8
Singh SB, Jayasuriya H, Dewey R et al (2003) Isolation, structure, and HIV-1-integrase inhibitory activity of structurally diverse fungal metabolites. J Ind Microbiol Biotechnol 30:721–731. https://doi.org/10.1007/S10295-003-0101-X
Siwe-Noundou X, Musyoka TM, Moses V et al (2019) Anti-HIV-1 integrase potency of methylgallate from Alchornea cordifolia using in vitro and in silico approaches. Sci Rep 91(9):1–9. https://doi.org/10.1038/s41598-019-41403-x
Smith SJ, Zhao XZ, Burke TR, Hughes SH (2018) Efficacies of cabotegravir and bictegravir against drug-resistant HIV-1 integrase mutants. Retrovirology 15:1–18. https://doi.org/10.1186/S12977-018-0420-7/FIGURES/11
Smith SJ, Zhao XZ, Passos DO et al (2020) HIV-1 integrase inhibitors that are active against drug-resistant integrase mutants. Antimicrob Agents Chemother. https://doi.org/10.1128/AAC.00611-20/SUPPL_FILE/AAC.00611-20-S0001.PDF
Smith SJ, Zhao XZ, Passos DO et al (2021a) HIV-1 integrase inhibitors with modifications that affect their potencies against drug resistant integrase mutants. ACS Infect Dis 7:1469–1482. https://doi.org/10.1021/ACSINFECDIS.0C00819/SUPPL_FILE/ID0C00819_LIVESLIDES.MP4
Smith SJ, Zhao XZ, Passos DO et al (2021b) Integrase Strand transfer inhibitors are effective anti-HIV drugs. Viruses 13:205. https://doi.org/10.3390/V13020205
Stader F, Battegay M, Marzolini C (2021) Physiologically-based pharmacokinetic modeling to support the clinical management of drug-drug interactions with Bictegravir. Clin Pharmacol Ther 110:1231–1239. https://doi.org/10.1002/CPT.2221
Stanford HIV-1 Drug Resistance Database (2019) INSTI resistance notes—HIV drug resistance database. In: Stanford Univ. https://hivdb.stanford.edu/dr-summary/resistance-notes/INSTI/. Accessed 1 Apr 2022
Tewtrakul S, Chaniad P, Pianwanit S et al (2015) Anti-HIV-1 integrase activity and molecular docking study of compounds from Caesalpinia sappan L. Phytother Res 29:724–729. https://doi.org/10.1002/PTR.5307
Thierry E, Deprez E, Delelis O (2017) Different pathways leading to integrase inhibitors resistance. Front Microbiol 7:2165. https://doi.org/10.3389/FMICB.2016.02165/BIBTEX
UNAIDS (2021) Global HIV & AIDS statistics—fact sheet. In: UNAIDS. https://www.unaids.org/en/resources/fact-sheet. Accessed 27 Feb 2022
Unger NR, Worley MV, Kisgen JJ et al (2016) Elvitegravir for the treatment of HIV. Expert Opin Pharmacother 17:2359–2370. https://doi.org/10.1080/14656566.2016.1250885
van der Galiën R, ter Heine R, Greupink R et al (2019) Pharmacokinetics of HIV-integrase inhibitors during pregnancy: mechanisms, clinical implications and knowledge gaps. Clin Pharmacokinet 58:309–323. https://doi.org/10.1007/S40262-018-0684-Z/TABLES/4
Varadarajan J, McWilliams MJ, Hughes SH (2013) Treatment with suboptimal doses of raltegravir leads to aberrant HIV-1 integrations. Proc Natl Acad Sci U S A 110:14747–14752. https://doi.org/10.1073/PNAS.1305066110/SUPPL_FILE/PNAS.201305066SI.PDF
Vo TS, Kim SK (2010) Potential anti-HIV agents from marine resources: an overview. Mar Drugs 8:2871. https://doi.org/10.3390/MD8122871
Völker P, Haza L von, Wöntz C, Zakharchuk D (2020) Raltegravir
Wainberg MA, Han Y-S (2015a) HIV–1 resistance to dolutegravir: update and new insights. J Virus Erad 1:13–16. https://doi.org/10.1016/S2055-6640(20)31150-X
Wainberg MA, Han YS (2015b) Will drug resistance against dolutegravir in initial therapy ever occur? Front Pharmacol 6:90. https://doi.org/10.3389/FPHAR.2015.00090/BIBTEX
Wallet C, De Rovere M, Van Assche J et al (2019) Microglial cells: the main HIV-1 reservoir in the brain. Front Cell Infect Microbiol 9:362. https://doi.org/10.3389/FCIMB.2019.00362/BIBTEX
Waluyo D, Prabandari EE, Pramisandi A et al (2021) Exploring natural microbial resources for the discovery of anti-malarial compounds. Parasitol Int 85:102432. https://doi.org/10.1016/J.PARINT.2021.102432
Wensing AM, Calvez V, Ceccherini-Silberstein F et al (2019) 2019 update of the drug resistance mutations in HIV-1. Top Antivir Med 27:111
White KL, Osman N, Cuadra-Foy E et al (2021) Long dissociation of bictegravir from HIV-1 integrase-DNA complexes. Antimicrob Agents Chemother. https://doi.org/10.1128/AAC.02406-20
Xuan CHH, Kion LN, Rahman T et al (2021) Regression QSAR models for predicting HIV-1 integrase inhibitors. BioRxiv. https://doi.org/10.1101/2021.02.23.432583
Xue W, Jin X, Ning L et al (2013) Exploring the molecular mechanism of cross-resistance to HIV-1 integrase strand transfer inhibitors by molecular dynamics simulation and residue interaction network analysis. J Chem Inf Model 53:210–222. https://doi.org/10.1021/CI300541C/SUPPL_FILE/CI300541C_SI_001.PDF
Xue W, Liu H, Yao X (2014) Molecular modeling study on the allosteric inhibition mechanism of HIV-1 integrase by LEDGF/p75 binding site inhibitors. PLoS ONE 9:e90799. https://doi.org/10.1371/JOURNAL.PONE.0090799
Zeuli J, Rizza S, Bhatia R, Temesgen Z (2019) Bictegravir, a novel integrase inhibitor for the treatment of HIV infection. Drugs Today 55:669–682. https://doi.org/10.1358/DOT.2019.55.11.3068796
Zhou J, Hao J, Peng L et al (2021) Classification and design of HIV-1 integrase inhibitors based on machine learning. Comput Math Methods Med. https://doi.org/10.1155/2021/5559338
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
All the authors have contributed to the study's conception and design. Material preparation, data collection, and data analysis were performed by SKS and TS. The first draft of the manuscript was written by MQ and SKS and all the other authors are involved in critical revision, project administration, and supervision. All the authors have read and agreed to the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Communicated by Yusuf Akhter.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Sayyed, S.K., Quraishi, M., Jobby, R. et al. A computational overview of integrase strand transfer inhibitors (INSTIs) against emerging and evolving drug-resistant HIV-1 integrase mutants. Arch Microbiol 205, 142 (2023). https://doi.org/10.1007/s00203-023-03461-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00203-023-03461-8