
Abstract
In Escherichia coli hosts, hydrogen peroxide is one of the factors that may cause induction of λ prophage. Here, we demonstrate that H2O2-mediated λ prophage induction is significantly enhanced in the oxyR mutant host. The mRNA levels for cI gene expression were increased in a λ lysogen in the presence of H2O2. On the other hand, stimulation of the p M promoter by cI857 overproduced from a multicopy plasmid was decreased in the ΔoxyR mutant in the presence of H2O2 but not under normal growth conditions. The purified OxyR protein did bind specifically to the p M promoter region. This binding impaired efficiency of interaction of the cI protein with the OR3 site, while stimulating such a binding to OR2 and OR1 sites, in the regulatory region of the p M promoter. We propose that changes in cI gene expression, perhaps in combination with moderately induced SOS response, may be responsible for enhanced λ prophage induction by hydrogen peroxide in the oxyR mutant. Therefore, OxyR seems to be a factor stimulating λ prophage maintenance under conditions of oxidative stress. This proposal is discussed in the light of efficiency of induction of lambdoid prophages bearing genes coding for Shiga toxins.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Upon entering host cells, bacteriophage genomes can propagate either as extrachromosomal genetic elements or as DNA modules integrated into bacterial chromosomes. Bacteriophage λ is a temperate phage, which can enter one of two alternative developmental pathways upon infection of its host, Escherichia coli. These two alternatives are lytic or lysogenic developments (for reviews see Echols 1986; Taylor and Węgrzyn 1998; Węgrzyn et al. 2001; Węgrzyn and Węgrzyn 2002, 2005). When the lysogenic pathway takes place, phage DNA is incorporated into the E. coli chromosome forming a prophage that can be maintained in this state for many cell generations. Stable maintenance of the prophage is achieved through repression of the main lytic promoters, p L and p R, by binding of the phage cI repressor to their operators. Apart from its function as a repressor, cI also stimulates activity of its own promoter, p M. However, high levels of cI repress p M. This autoregulatory mechanism, involving both positive and negative regulation of transcription, sets a constant level of cI required for stable maintenance of the prophage (Dove et al. 1997; Węgrzyn and Węgrzyn 2005).
Studies on the control of λ prophage maintenance and induction are important not only in basic science (to understand basic regulatory processes operating in cells), but also in bio-medical applications. Namely, certain pathogenic E. coli strains (known as Shiga toxin-producing E. coli, STEC) bear prophages of the λ family (so called lambdoid phages) that have genes encoding Shiga toxins (Brussow et al. 2004; Herold et al. 2004). Moreover, phage regulatory circuits contribute significantly to expression of these genes. A lambdoid phage-encoded cI repressor ultimately controls Shiga toxin production and/or release in such a way that effective synthesis of this toxin is possible only after prophage induction (Waldor and Friedman 2005). Recent studies supported the prediction that bacteriophage λ may serve as a model in studies on biology of Shiga toxin-converting phages (Nejman et al. 2009).
Although λ prophage can be stably maintained in lysogenized E. coli host for a long time under normal growth condition, factors causing DNA damage and inducing the SOS response of a bacterial cell provoke prophage excision. This process is mediated by RecA-dependent cleavage of the cI repressor (Echols 1986; Taylor and Węgrzyn 1998; Węgrzyn and Węgrzyn 2002, 2005; Węgrzyn et al. 2001). Agents that induce oxidative stress, like hydrogen peroxide, may also cause DNA damage (Imlay and Linn 1987; Imlay et al. 1988), and recent studies indicated that H2O2-mediated prophage induction occurs in bacterial strains bearing various lambdoid prophages, including STEC lysogenized with Shiga toxin-converting phages (Łoś et al. 2009, 2010).
OxyR is a transcription factor that activates expression of genes coding for proteins involved in antioxidant defensive response. This transcription factor is sensitive to oxidation and only the oxidized form of the protein is capable of activating transcription (Aslund et al. 1999). It was also demonstrated that expression of some genes may be repressed by OxyR (Zheng et al. 2001a). OxyR is reduced, and thus inactive, during normal growth. However, even low concentrations of hydrogen peroxide lead to OxyR oxidation (formation of an intramolecular disulfide bond between two cysteine residues) (Zheng et al. 1998).
Since oxidative stress provides conditions potentially causing λ prophage induction, and OxyR is a crucial regulator of the antioxidant response, we investigated induction of the prophage in wild-type host and oxyR mutant and aimed to learn about a mechanism for H2O2- and OxyR-mediated control of the prophage induction/maintenance.
Materials and methods
Escherichia coli strains and bacteriophages
E. coli MG1655 (wild-type) (Jensen 1993) and MC1061 (hsdR mcrB araD139 Δ(araABC-leu)7679 ΔlacX74galU galK rpsL thi) (Meissner et al. 1987) strains were used as host in all experiments. The ΔoxyR::kan strain, GSO9 (Zheng et al. 1999) and recA56 zfi::Tn10 mutant (strain BM244) (Wegrzyn et al. 1995) were used as donors in P1 transduction. Bacteriophages λpapa (from our collection) and λcI857S7 (Goldberg and Howe 1969) were employed. E. coli lysogenic strains were obtained by infection of host bacteria with phage λ, titration of cells on LB agar plates (Sambrook et al. 1989) and screening of survivors for resistance to superinfection by λ, sensitivity to UV irradiation and presence of high titer of phage λ after induction of liquid cultures with UV or mitomycin C.
Plasmids and gene fusion
Plasmid pACYC184 (Chang and Cohen 1978) and its derivatives, pGW857 and pACλCI, carrying the cI857(ts) and wild-type cI alleles, respectively, under control of the lac promoter (Dove et al. 1997; Szalewska-Palasz and Wegrzyn 1995) were employed. For measuring activity of the p M promoter, we constructed a p M-lacZ fusion plasmid (pAHA1). For this, the p M promoter region (248 bp) was amplified by PCR using the λ plasmid pKB2 (Kur et al. 1987) as a template, and the following primers: 5′-GCC GGA TCC CCA TCT TGT CTG C and 5′-TAT GCG TTG TTA GCT ATA GAC TCC TTA GTA C. The product of the amplification was digested with BamHI and cloned between the BamHI and SmaI sites of the plasmid vector pHG86 (Giladi et al. 1992), carrying the lacZ gene downstream of the cloning sites.
For overexpression of the oxyR gene, plasmid pBAD24oxyR was constructed as follows. The oxyR gene was amplified by PCR by using E. coli MG1655 chromosomal DNA and the primers OXYR1 (5′-GCG AAT TCA TGA ATC GTG ATC TTG AGT ACC TGG TGG C) and OXYR2 (5′-CGT CAA GCT TGC TAT CGG GTA GCT GCG TTA AAC G). The PCR product (a 917 bp DNA fragment) was cleaved with restriction endonucleases EcoRI and HindIII and inserted into corresponding sites of the pBAD24 vector (Guzman et al. 1995). DNA sequence of the plasmid insert was validated by sequencing.
Efficiency of λ prophage induction
Lysogenic bacterial cultures growing in LB medium (Sambrook et al. 1989) were treated with H2O2 or mitomycin C. Number of phage virions produced by one bacterial cell before and after induction was estimated by phage titration (using MG1655 strain as host); number of cells was also estimated by titration (plating).
One-step-growth experiment
Lytic development of bacteriophage λ in E. coli cells, either after phage infection or after mitomycin C-mediated prophage induction, was investigated by one-step growth experiments according to a procedure described previously (Szalewska et al. 1994).
Measurement of β-galactosidase activity
Activity of β-galactosidase in bacterial cells was measured according to Miller (1972). Since we used a multicopy lacZ fusion, the measured activities of β-galactosidase were calculated per plasmid copy number, estimated as described previously (Węgrzyn et al. 1996), to compensate for possible copy number variation between strains. In fact, we detected no appreciable differences in the plasmid copy number in all tested hosts.
Overproduction and purification of the OxyR protein
Overproduction of the OxyR protein was performed in E. coli BL21 cells (GE Healthcare Biosciences, Pittsburgh, PA, USA) harboring pBAD24oxyR plasmid. The culture was grown at 37°C to an A595 of 0.6, and L(+)arabinose was added to a final concentration of 0.1% to induce expression. The culture was incubated for another 2 h. Cells were harvested and suspended in 30 ml of buffer A (20 mM Tris–HCl, pH 7.5; 30 mM NaCl; 1 mM DTT), and then frozen in liquid nitrogen and stored at −70°C. Purification of the OxyR protein was performed as described previously (Tao et al. 1991), with some modifications. Namely, after sonication, streptomycin sulfate was added (0.3 vol of 5% solution) to the supernatant instead of Polymin- P. The OxyR protein was dialyzed against 2 l of the buffer STDG (500 mM NaCl; 20 mM Tris–HCl, pH 7.5; 1 mM DTT; 50% (v/v) glycerol); frozen in liquid nitrogen and stored at −70°C. OxyR purified as described above represents an oxidized form of this protein.
Purification of His-tagged cI protein
Plasmid pT7λcISa109His6 (Nickels et al. 2002) was used for overproduction of C-terminally His-tagged cI protein in the E. coli BL21(DE3) strain. Purification of the cI repressor was performed as described previously (Nickels et al. 2002).
OxyR binding to the pM promoter region
Electrophoretic mobility shift assay (EMSA) was performed using oligonucleotides bearing putative OxyR binding site present in the p M promoter region (Fig. 1). For control, oligonucleotides were used whose critical positions for OxyR binding were changed. One of the complementary oligonucleotides for each probe was biotinylated on its 5′ end. The sequences of these oligonucleotides were as follows: OXYR5, containing wild-type sequence: 5′-ATG GTT TCT TTT TTG TGC TCA TAC GTT AAA TCT ATC ACC GCA AGG GAT AAA TAT CTA ACA CCG TGC GTG TTG ACT ATT T), and OXYR6, containing mutated sequence in the putative OxyR binding site: 5′-ATG GTT TCT TTT TTG TGC TCA TAA GGT AAA TAT TAC ACC GAA TGG TAT AAA CAT CTA ACA CCG TGC GTG TTG ACT ATT T. The complementary oligonucleotides were called OXYR5′ and OXYR6′, respectively.

Schematic map of the p M-p R promoter region of bacteriophage λ genome. The OR3 sequence is underlined and the putative OxyR binding site is framed
To test activity of the isolated OxyR protein, EMSA was performed on a biotinylated DNA fragment, encompassing the oxyR promoter region (297 bp). This fragment was obtained by PCR with primers OXYR3 (5′-TGG TCC GCT CCA GCA ACA TC) and OXYR4 (5′-biot-ACG GGC AGT GAC TTC AAG GG).
All binding reactions were carried out in a total volume of 24 μl, in a buffer containing: 1 mM DTT, 25 mM Tris–Cl pH 7.9, 6.25 mM MgCl2, 5% glycerol, 0.5 mM EDTA, 0.05% Igepal CA-630 and 50 mM KCl. DNA fragments (0.2 nM) were incubated with increasing amounts of the OxyR protein for 15 min at 37°C, and subsequently, 1 μl of a loading buffer (40% sucrose, 0.025% xylenecyanol, 0.025% bromophenol blue) was added, and samples were separated electrophoretically in a 6% polyacrylamide gel, containing 2.5% glycerol, in 0.5 × TBE. The gel was run at 85 V in a cold-room. After electrophoresis, DNA was transferred on a nylon membrane and detected using Chemiluminescent Nucleic Acid Detection Module (Pierce), according to manufacturer’s instructions.
Primer extension experiments
Overnight bacterial cultures were diluted 1:100 in fresh LB medium and incubated without aeration at 37°C untill OD575 = 0.1. Next, the cultures were divided in half, and one of them was supplemented with 1 mM H2O2. Samples were taken at indicated times, and total RNA was isolated using Total RNA kit (A&A Biotechnology). RNA from each sample was subjected to primer extension reaction with the use of a 32P-labeled primer (5′-TTT AAG GCG ACG TGC GTC CTC AAG). In the first stage, a mixture (4 μl) of RNA template (20 μg) and a primer (0.75 pmol) was supplemented with 270 mM KCl and heated to 94°C for 3 min, subsequently, primer was allowed to hybridize at 51°C for 15 min. Primer extension reaction was initiated by addition of 12 μl of a primer extension mix containing: 20 mM Tris–Cl (pH 8.4), 10 mM MgCl2, 1.6 mM of each of four dNTPs, 10 mM DTT, 2 U of RNase inhibitor (Fermentas) and 24 U of RevertAid H minus Reverse Transcriptase (Fermentas). Following 1-h incubation at 42°C, reactions were stopped by addition of 1/2 volume of a loading buffer (95% deionized formamide, 20 mM EDTA, 0.05% xylenecyanol, 0.05% bromophenol blue) and subjected to polyacrylamide gel electrophoresis in 8% sequencing gel (19:1 acrylamide:bis, 7 M urea in 1 × TBE) and autoradiography. Bands’ intensities were analyzed by densitometry.
DNase I footprinting
A DNA fragment was obtained by PCR using a primer pair (5′-32P- TTA AGG CGA CGT GCG TCC TC and 5′-ATA TAC GCC GAG ATC TTT AGC TGT C), encompassing the putative OxyR binding site and cI operators (λ phage genome coordinates 37,881–38,118). DNA (20–40 ng) was incubated at 37°C for 20 min with indicated amounts of OxyR protein in the buffer containing: 1 mM DTT, 25 mM Tris–Cl pH 7.9, 6.25 mM MgCl2, 5% glycerol, 0.5 mM EDTA, 0.05% Igepal CA-630 and 50 mM KCl. Total reaction volume was 50 μl. Where indicated, the cI protein was added and incubation was prolonged for another 10 min. Subsequently, 50 μl of DNase I cofactor solution was added (10 mM MgCl2, 5 mM CaCl2) followed by addition of 5 μl of appropriate DNase I (Fermentas) dilution (0.005 U for OxyR-bound DNA and 0.0025 U for other reactions). After 2-min digestion at 37°C, reaction was stopped by addition of 100 μl of a solution containing: 1% SDS, 200 mM NaCl, 20 mM EDTA, extracted by eqal volume of phenol:chloroform (1:1) mixture and precipitated with 2 volumes of 96% ethanol in the presence of glycogen. DNA pellet was dissolved in 4 μl of TE. Equal volume of the loading buffer (95% deionized formamide, 20 mM EDTA, 0.05% xylenecyanol, 0.05% bromophenol blue) was added, and samples were subjected to polyacrylamide gel electrophoresis as described for the primer extension assay.
Analysis of the regulatory modules of lambdoid bacteriophages
We have searched for the putative OxyR binding sites in the p M-p R region of the regulatory modules of lambdoid bacteriophages infecting Escherichia coli using the positional weight matrix approach with two tools: Target Explorer (http://trantor.bioc.columbia.edu/Target_Explorer/); (Sosinsky et al. 2003) and MatInspector (http://www.genomatix.de/) (Cartharius et al. 2005). The weight matrices were constructed using the alignment of 16 OxyR binding sites identified experimentally (http://www.lecb.ncifcrf.gov/~toms/paper/zheng.storz2001/supplement/) (Zheng et al. 2001a). Since the OxyR binding site is symmetrical, the complementary strand was included when building the matrices. The regulatory regions of lambdoid phages were retrieved from GenBank. The database was browsed for sequences of lambda-like viruses with the Taxonomy Browser tool (http://www.ncbi.nlm.nih.gov/Taxonomy/taxonomyhome.html/), and the NonRedundant database was searched for sequences similar to lambdoid repressor proteins with tBLASTn (http://www.ncbi.nlm.nih.gov/blast/). Thus, retrieved sequences were analyzed for the presence of another helix-turn-helix protein in the vicinity but in the opposite orientation, but the sequences very similar at the nucleotide level to those already in the data set were not considered further. In total, we have collected eight sequences of lambdoid coliphages with a structure similar to that of bacteriophage λ: two helix-turn-helix proteins (putative cI and Cro homologs) separated by a region containing two promoters with a position similar to that of pM and pR of bacteriophage lambda (see Fig. 1). The GenBank accession numbers for phage DNA sequences are AF034975 (H19B), AF125520 (933 W), AJ413274 (Nil2), J02459 (λ), NC_001901 (N15), NC_002167 (HK97), X13065 (ϕ80), X16093 (HK022). The position of the promoters was investigated using a Neural Network Promoter Prediction tool (http://www.fruitfly.org/seq_tools/promoter.html) (Reese 2000).
The similarity relations between the regulatory regions of lambdoid phages were analyzed assuming that they correspond to the relations between the cI repressor proteins. This allowed the analysis of distantly related regions, since the distances were calculated at the amino acid level. Six additional protein sequences of repressor proteins of lambdoid viruses infecting enterobacteria and carrying a regulatory region with the structure described above were added to the dataset. The sequences were aligned using ClustalW (Thompson et al. 1994); the columns with gaps were removed. The distanced were calculated using the BLOSUM62 matrix with program TREE-PUZZLE (Schmidt et al. 2002). Programs in the PHYLIP package (Felsenstein 2005) were used to construct the neighbor-joining trees (NEIGHBOR), to obtain 1,000 boostrap pseudomatrices (SEQBOOT), and to calculate the bootstrap values (CONSENSE). Program WeightLESS (Sanjuán and Wróbel 2005) was used to test the interior branches in the neighbor-joining tree with weighed least squares likelihood ratio test.
Results
Induction of λ prophage in hydrogen peroxide-treated cultures of oxyR+ and ΔoxyR lysogens
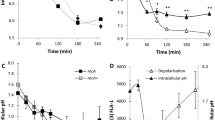
As demonstrated previously, hydrogen peroxide, an agent causing oxidative stress, may induce λ prophage to some extent (Łoś et al. 2009, 2010). We found, however, that this induction is significantly more effective in the ΔoxyR::kan lysogenic strain than in a wild-type host (Fig. 2). The most efficient prophage induction occurred in exponentially growing lysogenic bacteria at final H2O2 concentration of 1 mM (Fig. 2, and data not shown). The highest difference between oxyR + and ΔoxyR::kan strains was observed when bacterial cultures were grown with agitation of flask cultures (which enables aerobic growth) before addition of H2O2 and without agitation (under conditions resembling anaerobic growth) after induction (Fig. 2); however, significant differences were also detected under all other combinations of aeration conditions tested, e.g. cultures in agitated test tubes (data not shown).

λpapa prophage induction in MG1655 wild-type lysogenic strain (open symbols) and its ΔoxyR::kan derivative (closed symbols) after treatment with 1 mM (final concentration) hydrogen peroxide at time 0 (circles) or without such a treatment (squares). Cultures were grown with agitation of flask cultures (at 200 rpm) before addition of H2O2 and without agitation after the induction. Number of plaque forming units was estimated in each sample, and the results are expressed as number of phages per cell
Since efficiency of prophage induction was measured as an increase in the number of mature phages (plaque forming units) per cell, higher number of phage particles in the ΔoxyR::kan strain relative to wild-type bacteria could result from either higher frequency of prophage induction in the mutant host or more efficient lytic development in this strain. The latter hypothesis was tested by inducing the phage with mitomycin C. Subsequent treatment with hydrogen peroxide did not stimulate lytic development, and it was in fact even less efficient in the ΔoxyR::kan mutant than in wild-type bacteria (Table 1). Therefore, we conclude that treatment with H2O2 results in enhanced frequency of induction of λ prophage.
Nature of hydrogen peroxide-mediated λ prophage induction
We found that under normal growth conditions, frequency of spontaneous λ prophage induction was similar in both ΔoxyR::kan and wild-type hosts (Fig. 2 time 0, and Table 2). Similarly, no significant difference between these two lysogenic strains was observed when efficiency of mitomycin C-mediated prophage induction was investigated (Table 1). The hydrogen peroxide-mediated prophage induction was found to proceed normally as it was dependent on the function of the recA gene. Namely, only negligible prophage induction (with frequency decreased two to three orders of magnitude relative to recA + hosts) was observed in both recA56 oxyR + and recA56 ΔoxyR::kan strains treated with H2O2 (data not shown).
H2O2-mediated prophage induction and the SOS response
One could speculate that the most straight-forward explanation of the enhanced λ prophage induction in the ΔoxyR::kan mutant by hydrogen peroxide might be based on potentially higher levels of DNA lesions in the absence of OxyR-dependent antioxidant defensive response. This would lead to more effective induction of the SOS response, and thus more effective production and activation of the RecA protein, causing rapid degradation of the cI repressor and subsequent prophage induction. However, it was demonstrated experimentally that treatment with H2O2 results, in both wild-type and ΔoxyR bacteria, in very similar levels of activation of recA and other genes from the SOS regulon (Zheng et al. 2001b). Although these measurements were performed 10 min after addition of H2O2 (Zheng et al. 2001b) and it was demonstrated that the SOS response typically takes longer than this (Friedman et al. 2005), the period of 10 min is definitely enough to induce the SOS response and to stimulate the expression of SOS genes by several fold (Friedman et al. 2005). Although in our experiments, the cells were incubated with H2O2 for 4 h to allow production of phage progeny (Tables 1 and 2), we assume that the results of Zheng et al. (2001b) can be considered in analysis of data presented in here. Therefore, enhanced λ prophage induction in the hydrogen peroxide-treated ΔoxyR::kan host is unlikely to be due to more efficient induction of the SOS response relative to wild-type cells.
OxyR binds specifically to the pM promoter region and influences cI binding
The OxyR protein is known to bind DNA and to regulate transcription from various promoters, including its own promoter (Aslund et al. 1999; Zheng et al. 2001a). The positional weight matrix approach allowed us to identify a potential binding site in the p M region of λ DNA (Fig. 1). We assumed that if OxyR binds to this DNA fragment, it might significantly influence binding and action of the cI protein at the p M promoter. Therefore, we tested whether OxyR is able to bind this sequence specifically.
The binding of OxyR to DNA was assessed by using EMSA. Comparison of the efficiency of OxyR-mediated retardation of mobility of short DNA fragments in the gel during electrophoresis between the fragments bearing the p M promoter region and the oxyR promoter region (known to bind OxyR efficiently) indicated that OxyR binds to p M less efficiently than to its own promoter (Fig. 3a). To assess whether the binding to p M is specific, an analogous DNA template but containing several mismatch mutations, potentially impairing OxyR binding, was used. Contrary to the wild-type template, gel mobility of such a mutant DNA fragment was not affected in the presence of OxyR (Fig. 3b). Therefore, we conclude that binding of the OxyR protein to the λ p M promoter region is specific.

Binding of OxyR to the p M promoter. a Comparison of the efficiency of OxyR binding to p M and oxyR promoter regions. Gel mobility shift assays were performed as described in “Materials and methods” using the 258-bp PCR fragment encompassing pM and the 297-bp fragment of oxyR promoter region. Biding reactions were run on a 5% polyacrylamide gel. b EMSA analysis of OxyR binding to pM promoter region. Gel mobility shift assays were performed as described in “Materials and methods” using 79-bp double-stranded oligos, containing wild-type (lanes 6–10) or mutated sequence (lanes 1–5) of a putative oxyR box. Reactions were run on a 6% polyacrylamide gel
To learn more about OxyR binding to the p M promoter region, footprinting experiments were performed. We confirmed that OxyR binds specifically to this promoter (Fig. 4), in the region predicted as a putative OxyR binding site (compare Figs. 1 and 4). This binding also induced an appearance of a DNase hypersensitive site (Fig. 4), suggesting a DNA-bend caused by the OxyR protein.

DNase I footprinting of OxyR binding to the p M promoter region. OxyR protein, at indicated concentrations, was bound to a labeled DNA fragment, and DNase I digestion was performed as described in “Materials and methods”. Putative OxyR binding site was depicted. A site of enhanced DNA cleavage by DNase I, observed in the presence of OxyR, was marked by an arrow
Interestingly, we found that binding of the cI protein to the OR3 operator region (see Fig. 1) is less efficient in the presence of OxyR, while interaction of cI with OR2 and OR1 is stimulated by OxyR (Fig. 5). This was evident in both types of experiments, namely when binding of increased amounts of cI was tested in the presence of constant levels of OxyR, or when increased amounts of OxyR were used in experiments with constant levels of cI (Fig. 5a, b).

DNase I footprinting analysis of the cI repressor interaction with operator sites of p M-p R in the presence of the OxyR protein. OxyR, at indicated concentrations, was bound to a labeled DNA fragment. After 20 min incubation at 37°C, the cI repressor was added (as indicated), and incubation was continued for 10 min. DNase I footprinting was performed as described in “Materials and methods”. Operator sites for cI as well as OxyR binding site are marked. a and b represent different variants of the same experiment
Influence of H2O2 and OxyR on cI mRNA levels
Since we found that OxyR binds to the p M promoter region, and impairs binding of cI to the OR3 sequence while stimulating its binding to OR2 and OR1 at this promoter, we asked what are effects of H2O2 and OxyR functions on the cI gene expression. Therefore, primer extension experiments were performed to estimate levels of cI mRNA in wild-type and oxyR lysogenic bacteria treated or not with H2O2. An increase in the level of cI mRNA was detected after treatment of the culture of λ lysogenic E. coli with H2O2, and this effect was pronounced somewhat more strongly in the oxyR mutant (Fig. 6).

Primer extension analysis of the abundance of cI mRNA in oxyR + (open symbols) or ΔoxyR::kan (closed symbols) bacteria lysogenic for λ, either untreated (squares) or treated with 1 mM H2O2 at time 0 (circles). The analysis was performed as described in “Materials and methods”. Results shown are mean values ± SD
Since binding of cI to OR1 and OR2 repress transcription from p R and stimulates the activity of the p M promoter, whereas cI-OR3 interactions lead to repression of p M (for a review see Węgrzyn and Węgrzyn 2005), we assumed that the results presented in Fig. 6 may reflect the effects of the oxidative stress and subsequent SOS response, which might cause a decrease in cI level (kept high in lysogenic bacteria for the most of the cell cycle, thus, preventing further increase in the repressor concentration due to OR3 occupancy by cI), and subsequent activation of transcription from p M. If so, the presence of active OxyR protein would enhance binding of cI to OR1 and OR2 and prevent transcription from p R, while in the oxyR mutant the p R repression could be leaky, leading to an increased frequency of prophage induction. Moreover, one might assume that in the absence of active OxyR, a H2O2-caused damage of a fraction of cI molecules might cause a lower level of active λ repressor in the oxyR mutant than in wild-type cells, thus, leading to initial increase in transcription of the cI gene (as depicted in Fig. 6), followed by subsequent less effective activation of p M.
The above presented hypothesis could not be tested in the experimental system described above (primer extension assays using RNA isolated from lysogenic bacteria treated or not with hydrogen peroxide) due to unsynchronized prophage induction in population of lysogenic cells and simultaneously ongoing processes of lytic development (with subsequent cell lysis) in some cells and prophage maintenance in other cells, which would make the results of experiments impossible to interpret. Therefore, we decided to employ two other experimental systems. First, if enhanced λ prophage induction in the H2O2-teated oxyR mutant arise from impaired stimulation of expression of the cI gene and weakened repression of the p R-initiated transcription due to lower level and/or activity of the λ repressor, overexpression of this gene should abolish the oxyR-dependent defect of prophage maintenance in these cells. We found that in such cells, expression of the cI gene from a multicopy plasmid resulted in efficient stabilization of the prophage (Table 2), which supports the hypothesis.
Second, if the hypothesis were true, stimulation of the p M promoter by overproduced cI protein should be impaired in the oxyR mutant treated with H2O2 relative to the oxyR + cells, in which OxyR negatively influences binding of cI to the OR3 site while stimulating its binding to OR2 and OR1. To test this, we employed a p M-lacZ fusion and a plasmid bearing the cI857 allele (pGW857), coding for a temperature-sensitive cI variant. Bacteria were cultured at 43°C (to keep the cI857 protein inactive), and then shifted to 30°C at the time of H2O2 addition (control cultures were not treated with hydrogen peroxide). Thus, only residual activity of p M was detected at 43°C and stimulation of this promoter by cI857 protein started after the shift to 30°C. We found no significant differences in cI-mediated stimulation of the p M promoter between oxyR + and ΔoxyR::kan strains growing under normal conditions (Fig. 7). Similarly, little differences in the residual activity of p M (without activation by cI) were found between these two strains (200–300 Miller units in all cases; Fig. 7). However, the activity of the p M-lacZ fusion was higher in wild-type bacteria treated with H2O2 than in untreated cells, whereas opposite results were obtained for the ΔoxyR::kan mutant (Fig. 7). Therefore, these results support the above described hypothesis.

Stimulation of the p M promoter activity by the cI protein in the oxyR + host (open columns) and its ΔoxyR::kan derivative (closed columns) bearing the p M-lacZ fusion and pGW857 plasmid. Bacteria were grown at 43°C and shifted to 30°C at time 0. At this time, hydrogen peroxide (HP) was added to half of each culture to final concentration of 1 mM. β-galactosidase activity was measured (in Miller units, MU) 30 min after the temperature shift. From obtained values, corresponding activities of β-galactosidase measured at time 0 (about 200–300 Miller units in all cases) were subtracted. Results presented are mean values from three experiments with SD indicated
Potential OxyR binding sites near the pM promoter in lambdoid prophages
As presented in Fig. 1, the putative OxyR binding site at the p M region of λ DNA encompasses OR3 (Fig. 1). The position of the residues with which the OxyR and cI proteins interact with DNA, according to the computational analysis (Papp et al. 1993; Schneider 1996; Zheng et al. 2001a), suggests that both proteins interact with the residues in the major groove on the same side of the DNA double helix (Fig. 1), which indicates that they could interfere with the binding of each other.
The results indicating that OxyR binds to the p M region of λ DNA corroborated our suggestion that this protein may regulate prophage maintenance by influencing transcription of the cI gene. On the other hand, prophage induction is crucial for expression of toxin genes in STEC. This is why we have also analyzed the regulatory regions of other lambdoid phages. The comparative analysis of the lambdoid regulatory regions, some very distantly related to the bacteriophage λ region (Fig. 8), suggests that six out of eight analyzed bacteriophages might carry a potential OxyR binding sites at a site very close to the start of cI gene transcription. These sites were found in genomes of phages H19B, 933 W, N15, HK97, λ and HK022 (though in HK022 and H19B they were evident when using only one of the two searching methods used) but not in genomes of ϕ80 and Nil2. This suggests a conservation of the site for functional reasons. The presence of the OxyR binding sequence near the p M transcription start site in phages 933 W and H19B is especially intriguing as these phages are carriers of Shiga toxin genes (Herold et al. 2004; Waldor and Friedman 2005), and the 933 W cI repressor is functionally very similar to that of λ (Koudelka et al. 2004).

Neighbor-joining tree of repressor proteins of eight lambdoid coliphages: λ (GenBank protein identification number: AAA96581, H19B (AAD04644), 933 W (AAD25430), ϕ80 (CAA31471), HK022 (CAA34222), Nil2 (CAC95084), HK97 (NP_037735) and N15 (NP_046933), and five related bacteriophages infecting enterobacteria: L (a Salmonella typhimurium phage; CAA63999), ST104 (a Salmonella typhimurium phage; YP_006379), Sf6 (a Shigella flexneri phage; AAQ12228), PY54 (a Yersinia enterocolitica phage; CAD91799) and APSE-2 (a Candidatus Hamiltonella defense phage; ABA29387). The phages in which a potential OxyR binding site was identified were underlined, twice if the results of both methods coincided, once if a site was identified using only MatInspector, but not Target Explorer. A letter W marks the interior branches judged significant by weighted least squares likelihood ratio test (Sanjuan andWróbel 2005); the bootstrap values were also marked over the corresponding branches
Discussion
In this report, we demonstrated that hydrogen peroxide-mediated induction of λ prophage is significantly enhanced in the oxyR mutant and that the mechanisms of this phenomenon may be based on OxyR-mediated regulation of the cI gene expression by influencing activity of the p M promoter, and perhaps also the p R promoter. In fact, OxyR was found to be able to interact with the region we predicted in silico. Relatively low efficiency of in vitro binding of OxyR to the p M promoter region may either reflect the in vivo process or result from a lack of putative additional factors required for effective interaction with this region. It remains to be elucidated whether this weak interaction of OxyR with p M is sufficient to affect cI-mediated regulation of activity of this promoter in vitro.
The cI protein binds strongly and cooperatively to OR1 and OR2 sequences in the p M-p R region, and this binding leads to repression of p R and activation of p M. Binding to OR3 requires higher concentrations of cI and results in repression, rather than activation, of p M (for a review see Węgrzyn and Węgrzyn 2005). Thus, on the basis of results presented in this report, we propose the following hypothesis, which may explain the OxyR-mediated regulation of prophage maintenance in bacteria subjected to oxidative stress, and subsequent weak SOS response. These conditions may cause a decrease in the cI level—which is high in lysogenic bacteria but prevents further increase in the repressor concentration—and subsequent activation of transcription from p M, supported by OxyR-mediated impairment of binding of cI to OR3. Furthermore, the presence of the active OxyR protein can enhance binding of cI to OR1 and OR2 and prevent transcription from p R more effectively.
It is worth noting that the above hypothesis is only one of possible explanations of the effects of the oxidative stress and functions of OxyR on p M activity and λ prophage induction. For example, treatment with hydrogen peroxide leads to a transient decrease in DNA negative supercoiling (Weinstein-Fischer et al. 2000), and many promoters are sensitive to such changes in DNA topology. OxyR protein is known to induce DNA bending (Sun and Hattman 1996), and in fact, an enhancement of the intensity of some bands in the footprinting experiments with the p M region and the OxyR protein suggests such DNA topology changes at this promoter. One might speculate that this activity of the oxyR gene product could compensate for decreased DNA negative supercoiling at the p M promoter region and normalize activity of the promoter. Nevertheless, although oxyR deletion causes pleiotropic effects after exposition of host cells to H2O2 (Zheng et al. 2001b), and thus, there is a possibility that it may influence λ prophage induction indirectly, demonstration of the presence of the functional OxyR binding site at the p M region makes it more probable that the involvement of this protein in phage maintaince is direct, at least during oxidative stress.
As mentioned earlier, our results indicate that OxyR enhances maintenance of λ prophage in a host endangered by oxidative stress. One might consider such a mechanism surprising, as a general strategy for phage should be to escape from endangered lysogenic cells through prophage induction and lytic development. However, agents provoking oxidative stress, apart from causing some DNA lesions, also induce damage of other molecules, including proteins. Therefore, phage lytic development, which should lead to production of fully active infective virions, could be inefficient under such conditions. Since OxyR stimulates the expression of antioxidant defensive activities, it could be a better strategy for the phage to be maintained as a prophage when abundance of reactive oxygen species is low or moderate, i.e. when there is a high probability that OxyR-induced defensive response restores normal cellular functions. If levels of such destructive agents were sufficiently high to cause serious DNA damage, indicating that life of the cell is seriously endangered, the SOS response should be efficiently induced leading to degradation of the cI repressor, prophage excision and initiation of λ lytic development.
Since the OxyR binding site in the p M-p R promoter region is conserved among various lambdoid phages, including Shiga toxin-encoding phages 933 W and H19B, it is tempting to speculate that regulations similar to that described in this report for λ occur also in these viruses. If so, the cases of 933 W and H19B may be of special importance as stx genes (coding for Shiga toxins) are expressed only during phage lytic growth, which follows prophage induction (Herold et al. 2004, Waldor and Friedman 2005). Thus, understanding conditions and mechanisms of induction of 933 W, H19B and other prophages bearing stx genes may be crucial for development of procedures allowing protection against Shiga toxin-mediated diseases of humans.
References
Aslund F, Zheng M, Beckwith J, Storz G (1999) Regulation of the OxyR transcription factor by hydrogen peroxide at the cellular thiol-disulfide status. Proc Natl Acad Sci USA 96:6161–6165
Brussow H, Canchaya C, Hardt WD (2004) Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol Mol Biol Rev 68:560–602
Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T (2005) MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics 21:2933–2942
Chang AC, Cohen SN (1978) Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J Bacteriol 134:1141–1156
Dove SL, Joung JK, Hochschild A (1997) Activation of prokaryotic transcription through arbitrary protein-protein contacts. Nature 386:627–630
Echols H (1986) Bacteriophage λ development: temporal switches and the choice of lysis or lysogeny. Trends Genet 2:26–30
Felsenstein J (2005) PHYLIP: phylogenetic inference package [3.6]. Department of Genome Sciences, University of Washington, Seattle
Friedman N, Vardi S, Ronen M, Alon U, Stavans J (2005) Precise temporal modulation in the response of the SOS DNA repair network in individual bacteria. PLoS Biol 3:e238
Giladi H, Koby S, Gottesman ME, Oppenheim AB (1992) Supercoiling, integration host factor, and a dual promoter system participate in the control of the bacteriophage λ pL promoter. J Mol Biol 224:937–948
Goldberg AR, Howe M (1969) New mutations in the S cistron of bacteriophage lambda affecting host cell lysis. Virology 38:200–202
Guzman LM, Belin D, Carson MJ, Beckwith J (1995) Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130
Herold S, Karch H, Schmidt H (2004) Shiga toxin-encoding bacteriophages—genomes in motion. Int J Med Microbiol 294:115–121
Imlay JA, Linn S (1987) Mutagenesis and stress responses induced in Escherichia coli by hydrogen peroxide. J Bacteriol 169:2967–2976
Imlay JA, Chin SM, Linn S (1988) Toxic DNA damage by hydorgen peroxide through the Fenton reaction in vivo and in vitro. Science 240:640–642
Jensen KF (1993) The Escherichia coli “wild types” W3110 and MG1655 have rph frame shift mutation that leads to pyrimidine starvation due to low pyrE expression levels. J Bacteriol 175:3401–3407
Koudelka AP, Hufnagel LA, Koudelka GB (2004) Purification and characterization of the repressor of the shiga toxin-encoding bacteriophage 933 W: DNA binding, gene regulation, and autocleavage. J Bacteriol 186:7659–7669
Kur J, Górska I, Taylor K (1987) Escherichia coli dnaA initiation function is required for replication of plasmids derived from coliphage lambda. J Mol Biol 198:203–210
Łoś JM, Łoś M, Węgrzyn G, Węgrzyn A (2009) Differential efficiency of induction of various lambdoid prophages responsible for production of Shiga toxins in response to different induction agents. Microb Pathog 47:289–298
Łoś JM, Łoś M, Węgrzyn A, Węgrzyn G (2010) Hydrogen peroxide-mediated induction of the Shiga toxin-converting lambdoid prophage ST2–8624 in Escherichia coli O157:H7. FEMS Immunol Med Microbiol 58:322–329
Meissner PS, Sisk WP, Berman ML (1987) Bacteriophage λ cloning system for the construction of directional cDNA libraries. Proc Natl Acad Sci USA 84:4171–4175
Miller JH (1972) Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Nejman B, Łoś JM, Łoś M, Wegrzyn G, Wegrzyn A (2009) Plasmids derived from lambdoid bacteriophages as models for studying replication of mobile genetic elements responsible for the production of Shiga toxins by pathogenic Escherichia coli strains. J Mol Microbiol Biotechnol 17:211–220
Nickels BE, Dove SL, Murakami KS, Darst SA, Hochschild A (2002) Protein-protein and protein-DNA interactions of σ70 region 4 involved in transcription activation by λcI. J Mol Biol 324:17–34
Papp PP, Chattoraj DK, Schneider TD (1993) Information analysis of sequences that bind the replication initiator RepA. J Mol Biol 233:219–230
Reese MG (2000) Computational prediction of gene structure and regulation in the genome of Drosophila melanogaster. Dissertation, University of Hohenheim
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory, Cold Spring Harbor
Sanjuán R, Wróbel B (2005) Weighted least-squares likelihood ratio test for branch testing in phylogenies reconstructed from distance measures. System Biol 54:218–229
Schmidt HA, Strimmer K, Vingron M, von Haeseler A (2002) TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 18:502–504
Schneider TD (1996) Reading of DNA sequence logos: prediction of major groove binding by information theory. Methods Enzymol 274:445–455
Sosinsky JA, Bonin CP, Mann RS, Honig B (2003) Target Explorer: an automated tool for the identification of new target genes for a specified set of transcription factors. Nucleic Acids Res 31:3589–3592
Sun W, Hattman S (1996) Escherichia coli OxyR protein represses the unmethylated bacteriophage Mu mom operon without blocking binding of the transcriptional activator C. Nucleic Acids Res 24:4042–4049
Szalewska A, Węgrzyn G, Taylor K (1994) Neither absence nor excess of λ O initiator-digesting ClpXP protease affects λ plasmid or phage replication in Escherichia coli. Mol Microbiol 13:469–474
Szalewska-Pałasz A, Węgrzyn G (1995) Inhibition of transcription starting from bacteriophage λ p R promoter during the stringent response in Escherichia coli: implications for λ DNA replication. Acta Biochim Pol 42:233–240
Tao K, Makino K, Yonei S, Nakata A, Shinagawa H (1991) Purification and characterization of the Escherichia coli OxyR protein, the positive regulator for a hydrogen peroxide-inducible regulon. J Biochem 109:262–266
Taylor K, Węgrzyn G (1998) Regulation of bacteriophage λ replication. In: Busby SJW, Thomas CM, Brown NL (eds) Molecular microbiology. Springer Verlag, Berlin, pp 81–97
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Waldor MK, Friedman DI (2005) Phage regulatory circuits and virulence gene expression. Curr Opin Microbiol 8:459–465
Węgrzyn G, Węgrzyn A (2002) Regulation of bacteriophage λ DNA replication. Curr Topics Virol 2:187–194
Węgrzyn G, Węgrzyn A (2005) Genetic switches during bacteriophage lambda development. Prog Nucleic Acid Res Mol Biol 79:1–48
Węgrzyn G, Węgrzyn A, Konieczny I, Bielawski K, Konopa G, Obuchowski M, Helinski DR, Taylor K (1995) Involvement of the host initiator function dnaA in the replication of coliphage λ. Genetics 139:1469–1481
Węgrzyn G, Węgrzyn A, Pankiewicz A, Taylor K (1996) Allele specificity of the Escherichia coli dnaA gene function in the replication of plasmids derived from phage λ. Mol Gen Genet 252:580–586
Węgrzyn G, Węgrzyn A, Barańska S, Czyż A (2001) Regulation of bacteriophage lambda development. Recent Res Dev Virol 3:375–386
Weinstein-Fischer D, Elgrably-Weiss M, Altuvia S (2000) Escherichia coli response to hydrogen peroxide: a role for DNA supercoiling, topoisomerase I and Fis. Mol Microbiol 35:1413–1420
Zheng M, Aslund F, Storz G (1998) Activation of the OxyR transcription factor by reversible disulfide bond formation. Science 279:1718–1721
Zheng M, Doan B, Schneider TD, Storz G (1999) OxyR and SoxRS regulation of fur. J Bacteriol 181:4639–4643
Zheng M, Wang X, Doan B, Lewis KA, Schneider TD, Storz G (2001a) Computation-directed identification of OxyR DNA binding sites in Escherichia coli. J Bacteriol 183:4571–4579
Zheng M, Wang X, Templeton LJ, Smulski DR, LaRossa RA, Storz G (2001b) DNA microarray-mediated transcriptional profiling of the Escherichia coli response to hydrogen peroxide. J Bacteriol 183:4562–4570
Acknowledgments
This work was supported by the Ministry of Science and Higher Education (Poland) (project grants No. N301 122 31/3747 to A. W. and N303 291234 to B.W.) and was partially supported by the European Union within European Regional Development Fund, through grant Innovative Economy (POIG.01.01.02-00-008/08).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by David Kelly.
Monika Glinkowska and Joanna M. Łoś contributed equally to this work.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Glinkowska, M., Łoś, J.M., Szambowska, A. et al. Influence of the Escherichia coli oxyR gene function on λ prophage maintenance. Arch Microbiol 192, 673–683 (2010). https://doi.org/10.1007/s00203-010-0596-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-010-0596-2