
Abstract
Summary
Type V osteogenesis imperfecta (OI) is a form of OI characterized by radial head dislocation (RHD), calcification of interosseous membrane (CIM), and hyperplastic callus (HPC). In this study, we characterized the clinical features of 28 type V OI patients. We presented that dysfunctions of elbow, hip joint, and abnormal epiphyseal growth plate were associated with ectopic calcification and summarized the history of HPC progression and treatment.
Introduction
The current study aims to systematically characterize the skeletal phenotypes of patients with type V OI and suggested possible surgical solutions.
Methods
A total of 28 patients were admitted for inpatient care at The Hong Kong University-Shenzhen Hospital diagnosed with type V OI (either clinically diagnosed or genetically confirmed with the IFITM5 c.-14C > T mutation).
Results
Prevalence of type V radiological features was comparable to previous literatures (RHD, 100%; CIM, 100%; HPC, 44%; and scoliosis, 50%). Novel skeletal phenotypes were presented including extension of coronoid process, acetabular labrum, acetabular protrusion, spontaneous autofusion of the hip, bulbous epiphysis, and popcorn calcification. Significant increase in BMD was observed in patients with bisphosphonate treatment. Twenty-five percent (3/12) of patients with preoperative use of indomethacin developed HPC postoperatively, and HPCs were absorbed in 2 young patients 2 years later.
Conclusion
This retrospective study summarized the clinical features and highlighted the abnormalities in elbow, hip joint, and growth plate in type V OI patients. Our study contributed to a more comprehensive clinical spectrum of type V OI. We also characterized the natural progression of HPC formation and resorption in patients in different ages. The use of bisphosphonate treatment is effective in improving bone mineral density in type V OI patients, and whether indomethacin can reduce incidence of HPC formation deserves further investigation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Bone mass and homeostasis are maintained by the balance between bone forming osteoblasts and bone resorbing osteoclasts. Dysregulation of bone remodeling can lead to bone mass disorders such as osteoporosis and osteogenesis imperfecta (OI). OI is an inherited bone dysplasia and connective tissue disorder. Individuals with OI are characterized by low bone mass and recurrent fractures after incidents of low energy trauma, resulting in susceptibility to long bone deformity, fracture, and vertebral compression. Initially, typing of osteogenesis imperfecta was made on a clinical basis, ranging from occasional fractures to perinatal lethality (types I, IV, III, and II, from mild to severe). In addition, extra-skeletal manifestations such as blue sclera, dentinogenesis imperfecta, and hearing loss are commonly seen [1,2,3]. Most patients are heterozygous carrier of dominant mutations in COL1A1 or COL1A2, the main components of extracellular matrix in bone and skin. Genetic analysis has identified more than 19 OI genes involved in collagen post-translational modification and processing, bone mineralization, and osteoblast differentiation [1, 4,5,6]. With the increasing application and availability of genetic testing, classification based on gene mutations and the relevant molecular pathways have also been adapted.
In 2000, Glorieux et al. described a novel type of osteogenesis imperfecta (type V) that had a number of distinct characteristics [7]. While many aspects, such as skeletal fragility, high fracture rate, and bone deformity, were similar to type IV OI, type V OI presents a number of characteristic radiological features including hyperplastic callus (HPC), radial head dislocation (RHD), calcification of interosseous membrane (CIM), irregularly arranged lamellae, and mesh-like histology. Type V OI (OMIM 610,967) is a nonlethal skeletal dysplasia with various clinical presentations and severity [8, 9]. Subsequent studies have since ascertained that type V OI, instead of collagen defect, was caused by an autosomal dominant point mutation (c.-14C > T) in the 5′ UTR of IFITM5 (interferon induced transmembrane protein 5, also known as BRIL), resulting in five amino acids (Met-Ala-Leu-Glu-Pro) added to the N-terminus of the protein and alteration of its function [10, 11]. IFITM5 is an osteoblast-specific protein and plays a role in matrix mineralization, osteoblast maturation, and prenatal bone formation [12, 13].
Although a number of studies have revealed the various genotypic and phenotypic characteristics of type V OI, there is a lack of literature regarding elbow and hip dysfunction, epiphyseal abnormalities, and therapeutic strategies. We aimed to systematically describe the clinical and radiological characteristics of type V OI patients, focusing on the functional disability of elbow, hip, and knee joints. Our study contributed to a more comprehensive clinical spectrum of type V OI patients, and the observed outcome of indomethacin on HPC would deserve further case–control study with multicenter collaborations.
Materials and methods
Patients
This study was approved by the Institutional Review Broad of the University of Hong Kong – Shenzhen Hospital ([2016]08 and [2020]190). Patients who were admitted for inpatient care at HKU-Shenzhen Hospital between January 2016 and May 2022 diagnosed with type V OI [7] were recruited for this study. Informed consent was obtained from all patients and their family members. Detailed medical history and physical examination were assessed and collected by clinicians. Peripheral blood was obtained from available family members for genetic test. Excised skeletal tissues from deformity correction operation were collected for osteoblast isolation and histological analysis.
Targeted amplicon sequencing
Targeted amplicon sequencing was conducted as previously described [14]. DNA isolated from peripheral blood was subjected to targeted amplicon sequencing [15]. Nineteen OI causative genes (COL1A1, COL1A2, IFITM5, SERPINF1, CRTAP, P3H1, PPIB, SERPINH1, FKBP10, BMP1, SP7, TMEM38B, WNT1, CREB3L1, SPARC, TENT5A, MBTPS2, MESD, KDELR2) and 5 OI related genes (PLOD2, P4HB, SEC24D, PLS3, LRP5) were included in the targeted sequencing panel.
Bone histology
The specimens were fixed in 4% paraformaldehyde and decalcified with 0.5 M EDTA before embedding in paraffin. The 6-µm sections were rehydrated and stained with Goldner’s trichrome and visualized with Leica DM3000 microscope.
Results
Epidemiology and general clinical features of type V OI patients
In the study cohort, 257 individuals diagnosed with OI (family history, low bone mass, frequent bone fractures, bone deformity) were recruited for genetic test using targeted amplicon sequencing [14]. Twenty-three patients were confirmed with the IFITM5 c.-14C > T mutation. Five patients (HKU-SZH-247, 248, 260, 261, and 262) were clinically diagnosed with type V OI. The diagnostic criterion included recurrent fracture history, RHD, CIM, and HPC formation. Two patients (HKU-SZH-053 and 180) were half siblings sharing the same mother, and other 26 patients were from different families. Type V patients accounted for 10.6% of our OI cohort (28/262), comparable to the reported prevalence of type V OI in Chinese ethnicity [16]. Two patients also harbored mutations in COL1A1 (HKU-SZH-030: c.1856A > T, p.Gln619Leu; HKU-SZH-047: c.1298_1299insT, p.Gly434Argfs*2). Both mutations, which may result in qualitative and quantitative defects in type I collagen, were not found in the OI variant database (https://databases.lovd.nl/shared/genes/COL1A1). These two patients presented severe limb deformity, and Patient-030 also developed severe scoliosis. Whether combined effect of IFITM5 and COL1A1 mutations contribute to the severity needs to be proven.
Clinical and typical radiographic features are summarized in Table 1. BMD was measured in 23 patients by DXA scan (dual-energy X-ray absorptiometry). Z-score was measured by comparing average bone density of individuals of the same age and gender, ranged from − 5.6 to 1.0 at the lumbar spine (L1–L4) and − 7.1 to − 2.2 at the femoral neck. Ten out of 23 patients had Z-score below − 2.0, which indicated osteoporosis. The median Z-score was − 1.8 (interquartile range (IQR) − 4.70 to − 0.75). The age of first fracture ranged from newborn to 3 years. Nineteen out of 28 patients were prescribed with bisphosphonate (BP). To evaluate the effect of BP treatment, median Z scores of both groups (BP+ and BP−) were compared. Significant increase in BMD was observed in patients with BP treatment (BP+, median Z-score − 1.2, IQR − 1.40 to − 0.50; BP− median Z-score − 4.7, IQR − 5.05 to − 3.23; p = 0.000003), indicating the effects of BP treatment in improving bone mineral density in type V OI patients.
All patients with available radiographs presented RHD (Fig. 1a–b) and CIM between radioulnar (Fig. 1c) or tibiofibular areas (Fig. 1d–e), which limited the pronation-supination movement. Biopsy of calcified tibiofibular interosseous membrane from Patient249 was obtained after surgical correction for histological analyses by Goldner trichrome staining. No obvious Haversian structure was observed in transaxial sections, and collagen alignment was disorganized in coronal plane (Fig. 1f–g). Apart from RHD, 15 patients presented extension of coronoid process (Fig. 1h–l), resulting in elbow flexion and extension limitations, and eventually autofusion of humeroulnar joint (Fig. 1k–l). One patient (HKU-SZH-247) underwent radial head and partial ulnar resection due to severe elbow joint dysfunction. Eleven patients (44.0%) developed hyperplastic callus, the majority of which were located in the femur except one in humerus (HKU-SZH-087) (Fig. 1j). The percentage was generally lower than the previous cohort studies (65.3%) [17]. The prevalences of HPC formation in different locations along the femur, namely, proximal, mid shaft and distal femur, were 35%, 40%, and 25% respectively.

Interosseous ossification and radial head dislocation as typical features of type V OI patients. a–b Radial head dislocation. Arrow (black) indicated dislocated radial head. c–e Interosseous ossification between radius and ulna (c), tibia, and fibula (d–e). f–g Transaxial and coronal sections of calcified interosseous membrane between tibia and fibula stained with Goldner trichrome staining, Scale bar = 100 μm. h–l Extension of coronoid process. Red arrows indicated extended coronoid process. j Boxed region indicated HPC formation in the humerus. k–l Autofusion of humeroulnar joint
Scoliosis
Scoliosis was a major problem in type V OI patients, with a prevalence of around 52% in other type V OI cohorts (references including 157 individuals) [17]. Consistently, half of the patients in our center (14/28, 50.0%) developed scoliosis (defined by Cobb angle > 10°). Most of the curve (11 out of 14) were located in the thoracic region (Fig. 2a–e). Eight patients presented severe scoliosis with Cobb angle above 45° (Fig. 2c–e). Four patients (HKU-SZH-027, 142, 247, and 252) received surgical correction and spinal fusion due to compromised cardiopulmonary function. Two patients (HKU-SZH-056 and 188) were classified as early onset scoliosis with development of spinal curvature before the age of 10 [18]. Considering the relationship between age and severity of scoliosis, a trend was observed that Cobb angle increased with age. Patients with more severe scoliosis (Cobb angle > 60°) were all from the older age group (above 21 years old). Other spinal deformities such as compression fracture were observed in 23 patients (82.14%) (Table 1).

Spinal abnormalities of type V OI patients. a–e Representative radiological images of 5 patients with different degrees of scoliosis. Scoliosis, ranging from mild (a), progressive (b–c), and severe (d–e). Ages of the patients were labelled
Hip dysfunction
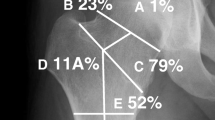
Hip abnormality was observed in 17 patients (60.71%) (Table 1). The hip joint space was generally reduced in these patients, and the hip range of motion (ROM) was significantly limited. Associated limitation in ROM related to the sustained presence of hyperplastic callus was found in 4 patients (Fig. 3a–b). Seven patients displayed concomitant acetabular protrusion (center–edge angle of Wiberg > 35° and migration of the femoral head medial to the acetabular ilioischial line) (Fig. 3b–d, Table 1). In some severe cases (HKU-SZH-053 and 180), fusion of hip joint and femoral head was noticed (Fig. 3c). Similar to what happened in the coronoid process, ectopic calcification and extension of acetabular labrum were observed in some patients (Fig. 3d–e). Three patients underwent hip surgeries, including hip soft tissue release, excision arthroplasty (girdlestone operation) (Fig. 3f–g), femur extension osteotomy with internal fixation, and callus resection. Hip ROM (including flexion/extension/internal rotation/external rotation) was improved after surgeries (Supplementary Table 1).

Hip abnormalities in type V OI patients. a–b Hyperplastic callus formation (white arrows) in the femurs, affecting the ROM of the hip joint. b–d Acetabular protrusion with center–edge angle of Wiberg > 35° (blue line) and migration of the femoral head medial to the acetabular ilioischial line (red dotted line). c Autofusion of the hip. d–e Ectopic calcification of extended acetabular labrum (red arrows). f–g Patient images before and after girdlestone operation
Bulbous epiphysis and popcorn calcification
Longitudinal bone growth occurs at the epiphyseal growth plate through the process of endochondral bone formation. The chondro-osseous progression from the epiphysis to the mid-shaft of bones includes chondrocyte proliferation, hypertrophy and trans-differentiation into osteoblasts. Abnormalities in the epiphyseal growth plate have been observed in severe OI patients [19, 20]. Metaphyseal radiodense lines were presented in 13 patients (Fig. 4a). Radiological examination further revealed popcorn calcification in 3 patients and bulbous epiphysis in 4 patients (Fig. 4b–e). Most of the epiphyseal abnormalities were located in the distal femur and proximal tibia. Presence of popcorn calcification was independent of bisphosphonate and intramedullary implant, as popcorn calcification was seen in patients that had not received bisphosphonate treatment or rod implant. Bilateral popcorn formation demonstrated an effect in growth retardation and bone deformity, while unilateral femoral popcorn calcification resulted in leg length discrepancy (Fig. 4b–c). Figure 4d–e illustrated the bulbous enlargement of both distal femurs. Intramedullary rodding did not seem to be the cause of bulbous epiphysis formation, as Patient 053 did not implant any rodding, and Patient 180 developed bilateral bulbous epiphysis but with rodding only in the right femur. Popcorn calcification and bulbous epiphysis may represent a cartilaginous dysplasia at the developing growth plate associated with primary bone defects. Independent occurrence of these abnormalities suggested that the mechanisms underlying the development and progression of each lesion might be different.

Radiodense line, popcorn calcification, and bulbous epiphysis in type V OI patients. Radiological images showing a radiodense metaphyseal lines, b–c popcorn calcification, and d–e bulbous epiphysis
Pre-operative treatment of Indomethacin and post-operative hyperplastic callus formation
Presence of HPC is a characteristic feature of type V OI patients, though its etiology is still unclear at the present. Eleven patients in our cohort developed hyperplastic callus. We tracked the fracture and callus formation history in patients with available clinical images. Noticeably, no HPC formation was observed in the spine and tibia, even after surgical interventions. We summarized the callus resorption into three categories: able to resorb in good shape (HKU-SZH-048, Fig. 5a–b), able to resorb but in irregular shape (HKU-SZH-137, Fig. 5c–d), and unable to resorb (HKU-SZH-142, Fig. 5e–f).

Hyperplastic callus formation and resorption in type V OI patients. a–d HPC resorption after fracture in young patients. e–f HPC in adult patient. No sign of HPC resorption was observed after 3 years. g–v Progression of HPC formation and resorption after operations
The initial phase of HPC formation resembles an inflammatory reaction associated with soft tissue swelling [21]. Glucocorticoids, indomethacin, and anti-inflammatory drugs have been proven effective in reducing inflammation and improving bone remodeling [17]. To assess the effects of indomethacin in reducing HPC formation, we monitored the postoperative HPC formation in 45 operations of 19 patients within the recording period. Sites of operations, usage of indomethacin, and HPC formation for each individual were summarized in Supplementary Table 2. Oral indomethacin was prescribed according to body weight in over 80% of the operations (38 out of 45), 4 postoperative HPC were formed in the femurs of 3 patients (HKU-SZH-056, 179 and 249) (Fig. 5g–v). There was no obvious pattern found regarding HPC formation after osteotomy. Patient-056 (7 years old, female) had operations on bilateral femurs, but HPC only developed on the right femur and was resorbed after one and a half years (Fig. 5g–l). Patient-179 (5 years old, male) had operations twice on the right femur, but HPC only developed in the first operation. Noticeably, we observed a callus formed in the proximal femur without obvious lesion sites (Fig. 5k). Resorption of HPC was only observed in younger patients below the age of 10 (HKU-SZH-048, 056, 137 and 179, Fig. 5a–d,g–l), but not in adult patients (HKU-SZH-142 and 249, Fig. 5e–f,s–v).
Discussion
The prevalence of type V OI (10.6%) caused by IFITM5 (c.-14C > T) mutation in our center was comparable with other cohort studies [1, 16, 22]. Clinical features including HPC, RHD, CIM, and scoliosis were also in the average range quoted by previous literature [8, 9, 11, 17, 23,24,25,26]. RHD and CIM were the most characteristic features of type V OI cohort. Similarly, femur was the most susceptible site for HPC formation. No callus was observed in vertebrae and tibia in any patients even after spine operations in our study. When HPC formed in proximal femur, it may associate with potential complications and severe limitations of hip joint functions and ROM. The severity of scoliosis varied from 17° to 112° in our patients. Compromised cardiopulmonary status was found in patients with severe scoliosis. It deserves attention from orthopedic surgeons in terms of regular surveillance and early surgical intervention such as spinal fusion, jeopardizing cardiopulmonary function, and increasing risks of surgeries. In general, type V patients do not present blue sclera or dentinogenesis imperfecta, which is mainly seen in patients with type I collagen mutations. However, one girl with IFITM5 mutation was reported to show dentinogenesis imperfecta in a cohort study [17]. In our genetic test, we detected compound mutations of IFITM5 and COL1A1 in two patients. Whether combined effect of mutations leads to more severe phenotypes should be considered.
Meanwhile, novel clinical features were observed in our cohort including extension of coronoid process and acetabular labrum due to ectopic calcification (limiting the ROM of elbow and hip joints), spontaneous hip autofusion, acetabular protrusion, popcorn calcification, and bulbous epiphysis. The underlying mechanisms of these particular features are still not clear. Type V OI phenotype is a combination of osteoporosis and exuberant bone formation. Cho et al. have postulated that IFITM5 mutation leads to impaired osteoblast function in the bone, ectopic ossification in interosseous membrane, and HPC at bone healing site [10]. Changes in bone mineralization arose from ectopic calcification may lead to an osteoporotic phenotype [8]. Whether the ectopic overgrowth of coronoid process and acetabular labrum, hip joint autofusion, and epiphyseal popcorn calcification develop in a similar mechanism has yet to be proven.
RHD is generally considered an outcome of CIM that leads to the growth discrepancy of proximal radius and ulna, and it is often thought that reduced ROM is associated with RHD. However, in this study, we observed that extension of coronoid process might also cause reduction of ROM of elbows. Therefore, in addition to radial head excision for patients with severe elbow dysfunction, excision of ectopic coronoid process might improve elbow ROM. Hip dysfunction was generally seen in severe OI patients. Previously, the presence of acetabular protrusion (AP) was described in a retrospective study including OI patients of different types (I, III, IV, and V) [27]. The prevalence of AP was relatively high in type III (69%) and type V (54%) OI patients. The lower percentage of AP (40.74%) in our cohort may due to difference in ethnicity, age distribution, and larger sample size. In addition to AP, hip autofusion and hyperplastic callus formation in proximal hip joint should be recognized as major complications yet infrequently mentioned. Hip autofusion influences patients in different aspects, including marked limitation of hip joints, ambulatory status, and compromised cardiopulmonary function. In case of asymmetrical fusion, patients may be unable to maintain an upright position when sitting. Surgeries including hip soft tissue release, excision arthroplasty (girdlestone operation), and callus resection could improve the hip ROM (Supplementary Table 1) and patients’ quality of life (Fig. 3). We also noticed that hip autofusion was mainly detected during or after puberty, while other ectopic calcification such as CIM and HPC were observed at younger age.
Indomethacin is known as a potent nonsteroidal anti-inflammatory drug inhibiting the production of prostanoids. It was proposed in previous studies that the use of indomethacin can limit the size of HPC, as the initial phase of HPC resembles the inflammatory reaction. However, no information or statistical analysis was provided [21]. We used indomethacin preoperatively, hoping to reduce HPC formation at the osteotomy site. Similarly, our study was a retrospective review in which no randomized control was set. Therefore, conclusions regarding the effectiveness of indomethacin on prevention of HPC formation could not be drawn. To date, the effectiveness of indomethacin in lowering the incidence or reduce the size of HPC has not been conclusively evaluated [17, 21]. The efficacy of indomethacin on HPC prevention and the underlying mechanisms deserve further investigation in case–control study with a bigger cohort size.
Conclusion
In summary, this study summarized the clinical and radiological features of 28 type V OI patients. The prevalences of IFITM5 (c.-14C > T) mutation, RHD, CIM, and scoliosis were compatible with previous literature. Significant increase in BMD was observed in patients with bisphosphonate treatment, indicating that BP treatment is effective in improving bone mineral density in type V OI patients. We highlighted the dysfunction of elbow and hip joint and its possible association with ectopic calcification. We also characterized the natural progression of HPC formation and resorption in patients at different ages. Whether indomethacin is effective in reducing HPC formation deserves further investigation with randomized controlled studies.
References
Marini JC et al (2017) Osteogenesis imperfecta. Nat Rev Dis Primers 3:17052
Sillence DO, Senn A, Danks DM (1979) Genetic heterogeneity in osteogenesis imperfecta. J Med Genet 16(2):101–116
Forlino A, Marini JC (2016) Osteogenesis imperfecta. Lancet 387(10028):1657–1671
Doyard M et al (2018) FAM46A mutations are responsible for autosomal recessive osteogenesis imperfecta. J Med Genet 55(4):278–284
Dubail J et al (2020) Homozygous loss-of-function mutations in CCDC134 are responsible for a severe form of osteogenesis imperfecta. J Bone Miner Res 35(8):1470–1480
Moosa S et al (2019) Autosomal-recessive mutations in MESD cause osteogenesis imperfecta. Am J Hum Genet 105(4):836–843
Glorieux FH et al (2000) Type V osteogenesis imperfecta: a new form of brittle bone disease. J Bone Miner Res 15(9):1650–1658
Shapiro JR et al (2013) Phenotypic variability of osteogenesis imperfecta type V caused by an IFITM5 mutation. J Bone Miner Res 28(7):1523–1530
Brizola E et al (2015) Clinical and molecular characterization of osteogenesis imperfecta Type V. Mol Syndromol 6(4):164–172
Cho TJ et al (2012) A single recurrent mutation in the 5’-UTR of IFITM5 causes osteogenesis imperfecta type V. Am J Hum Genet 91(2):343–348
Semler O et al (2012) A mutation in the 5’-UTR of IFITM5 creates an in-frame start codon and causes autosomal-dominant osteogenesis imperfecta type V with hyperplastic callus. Am J Hum Genet 91(2):349–357
Moffatt P et al (2008) Bril: a novel bone-specific modulator of mineralization. J Bone Miner Res 23(9):1497–1508
Hanagata N et al (2011) Characterization of the osteoblast-specific transmembrane protein IFITM5 and analysis of IFITM5-deficient mice. J Bone Miner Metab 29(3):279–290
Chen P et al (2022) Phenotypic spectrum and molecular basis in a Chinese cohort of osteogenesis imperfecta with mutations in type I collagen. Front Genet 13:816078
Bybee SM et al (2011) Targeted amplicon sequencing (TAS): a scalable next-gen approach to multilocus, multitaxa phylogenetics. Genome Biol Evol 3:1312–1323
Liu Y et al (2017) Gene mutation spectrum and genotype-phenotype correlation in a cohort of Chinese osteogenesis imperfecta patients revealed by targeted next generation sequencing. Osteoporos Int 28(10):2985–2995
Cao YJ et al (2019) Expanding the clinical spectrum of osteogenesis imperfecta Type V: 13 Additional Patients and Review. Front Endocrinol (Lausanne) 10:375
El-Hawary R, Akbarnia BA (2015) Early onset scoliosis - time for consensus. Spine Deform 3(2):105–106
Obafemi AA et al (2008) Popcorn calcification in osteogenesis imperfecta: incidence, progression, and molecular correlation. Am J Med Genet A 146A(21):2725–2732
Brizola E, McCarthy E, Shapiro JR (2015) Bulbous epiphysis and popcorn calcification as related to growth plate differentiation in osteogenesis imperfecta. Clin Cases Miner Bone Metab 12(2):202–206
Cheung MS, Glorieux FH, Rauch F (2007) Natural history of hyperplastic callus formation in osteogenesis imperfecta type V. J Bone Miner Res 22(8):1181–1186
Bardai G et al (2016) DNA sequence analysis in 598 individuals with a clinical diagnosis of osteogenesis imperfecta: diagnostic yield and mutation spectrum. Osteoporos Int 27(12):3607–3613
Fitzgerald J et al (2013) Phenotypic variability in individuals with type V osteogenesis imperfecta with identical Ifitm5 mutations. J Rare Disord 1(2):37–42
Kim OH et al (2013) Osteogenesis imperfecta type V: clinical and radiographic manifestations in mutation confirmed patients. Am J Med Genet A 161A(8):1972–1979
Rauch F et al (2013) Osteogenesis imperfecta type V: marked phenotypic variability despite the presence of the IFITM5 c-14C > T mutation in all patients. J Med Genet 50(1):21–4
Zhang Z et al (2013) Phenotype and genotype analysis of Chinese patients with osteogenesis imperfecta type V. PLoS ONE 8(8):e72337
Violas P et al (2002) Acetabular protrusion in osteogenesis imperfecta. J Pediatr Orthop 22(5):622–625
Acknowledgements
We thank the Fu Tak Iam foundation (Hong Kong) and Chow Tai Fook charity foundation (Hong Kong) for covering part of the medical costs. ZT and PKC are supported by the Shenzhen Peacock Plan. We thank all our patients and their families for their participation in this study.
Funding
This work was supported by Shenzhen Key Medical Discipline Construction Fund (SZXK077), Guangdong Basic and Applied Basic Research Fund (2022A1515010987), and Hong Kong Health and Medical Research Fund (07181676).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Ethics approval
This study was approved by the Institutional Review Broad of the University of Hong Kong – Shenzhen Hospital ([2016]08 and [2020]190).
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Tan, Z., Shek, H.T., Dong, Z. et al. Retrospective analyses of clinical features in 28 Chinese patients with type V osteogenesis imperfecta: new perspectives in an old issue. Osteoporos Int 34, 369–377 (2023). https://doi.org/10.1007/s00198-022-06581-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-022-06581-x