
Abstract
Summary
We analyzed polymorphism of the ALPL gene in patients with low serum levels of tissue-nonspecific alkaline phosphatase (TNAP). The presence of three or more of the less frequent alleles of ALPL polymorphisms was associated with significantly lower TNAP serum level and higher frequencies of metatarsal fractures, which may help confirm a clinical suspicion of adult hypophosphatasia.
Introduction
Alkaline phosphatases (ALPs) are membrane-bound enzymes that hydrolyze monophosphate esters at a high pH (pH 8–10). Inorganic pyrophosphate, pyridoxal 5-phosphate, the activated form of vitamin B6 (PLP), and phosphoethanolamine (PEA), are natural substrates of ALPs. Hypophosphatasia (HPP, OMIM 146300, 241500, 241510) is a heterogeneous rare metabolic bone disease caused by loss-of-function mutations in the tissue-nonspecific alkaline phosphatase gene (ALPL; MIM 171760) with a deficiency of TNAP. Clinical presentation of HPP in adults demonstrated a wide range of manifestations, many of which are nonspecific. In the present study, we screened the polymorphic genetic variants of ALPL in 56 subjects presenting low serum levels of TNAP and/or other clinical signs of adult HPP in order to evaluate a possible role of polymorphic variants in the diagnosis and management of HPP in adults.
Methods
Genomic DNA was extracted from peripheral blood and ALPL gene was sequenced by PCR-based Sanger technique.
Results
Fourteen different polymorphic variants were found in the study population. A lower serum level of TNAP and higher frequencies of metatarsal fractures were observed in patients bearing three or more of the minor frequency alleles (MFAs) of the ALPL polymorphic variants. The presence of some MFAs, mostly as a contemporary presence of three or more of them, was found to be mainly represented in patients having both a significantly lower level of TNAP and a higher level of vitamin B6.
Conclusion
The genetic analysis and presence of some polymorphic variants may be an instrument to confirm clinical and biochemical data, consider adult HPP, and help clinicians be cautious in the administration of anti-reabsorption drugs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Biomineralization, consisting in the deposition of hydroxyapatite crystals in the extracellular matrix of hard tissues [1], is a complex osteoblast-driven process, which requires appropriate concentrations of extracellular calcium and phosphate. Alkaline phosphatases (ALPs) are membrane-bound enzymes that hydrolyze monophosphate esters at a high pH (pH 8–10), releasing phosphate [2]. Human ALPs are classified into 4 types: tissue-nonspecific, intestinal, placenta, and germ cell. The tissue-nonspecific type (TNAP) is the most commonly expressed, accounting for approximately 95% of total serum ALP activity [3]. It is expressed in the cell membrane of many tissues, including liver, kidney, and hypertrophic chondrocytes, osteoblasts, and odontoblasts [2,3,4]. It hydrolyzes inorganic pyrophosphate (PPi) and provides inorganic phosphate (Pi) to promote mineralization. PPi, pyridoxal 5-phosphate, the activated form of vitamin B6 (PLP), and phosphoethanolamine (PEA) are all physiologic substrates of TNAP [1].
Hypophosphatasia (HPP, OMIM 146300, 241500, 241510) is a heterogeneous rare metabolic bone disease, with a prevalence between 1/100,000 and 1/300,000 of severe forms [5, 6], and a prevalence of moderate HPP, within the European the population, estimated at 1/6370, suggesting that the mild forms of the disease likely occur more frequently [6, 7].
HPP is caused by loss-of-function mutations in the tissue-nonspecific alkaline phosphatase gene (ALPL: MIM 171760), resulting in a reduced or absent TNAP activity and, subsequently, in increased levels of its substrates, such as PPi, PLP, and PEA [3]. The development of clinical manifestations is due to inactivating homozygous, heterozygous, or compound heterozygous mutations of the ALPL gene, which can either be dominant or recessive. The degree of loss of TNAP activity is variable among patients affected, and it has been associated with different mutations. Mutation analyses of HPP subjects have identified over 200 mutations in the coding region of the ALPL gene, 80% of which are missense mutations [8]. In addition, rare variants of the ALPL gene have been associated with serum TNAP levels and BMD [9, 10], and animal model studies have confirmed that substantial variations in TNAP expression and activity and in bone density and strength are the result of polymorphisms in the ALPL gene [10]. Polymorphisms of ALPL have been also associated with variations in circulating levels of PLP [11].
Disease severity is highly variable, ranging from stillbirth without mineralized bone to pathologic fractures developing only in adulthood [8, 12]. The presentation of HPP in adults has demonstrated a wide range of clinical manifestations, many of which are nonspecific [7]. Due to the rarity of adult HPP, and considering the lack of information regarding clinical course, systemic manifestations, progression of disease, and difficult specific diagnosis patterns, the correct and differential diagnosis of adult HPP is currently difficult, and the disease is often misdiagnosed or not-diagnosed, and, subsequently, wrongly or not treated. The most common presenting features include musculoskeletal pain and fractures, mild and often overlapping symptoms of other rare or common metabolic bone disorders [7]. In most cases, diagnosis of adult HPP is made after a low serum ALP level is casually detected during routine blood screening, or when tested after a direct family member was diagnosed with the condition. In the appropriate clinical setting, HPP can be diagnosed by the presence of a persistently low serum ALP and elevated ALP substrates in blood (PLP or PEA) or in urine (PEA or PPi), although urine PPi testing remains investigational [4, 13].
Bilateral femoral pseudo-fractures (Looser’s zones), occurring laterally in the sub-trochanteric diaphysis, are a distinctive sign of adult HPP [14]; their occurrence has been associated with the long-term amino-bisphosphonate (NBP) treatment in HPP patients wrongly diagnosed with osteoporosis [15]. Because NBPs are analogs of PPi, it has been speculated that HPP patients may be sensitized to the effects of NBPs, causing a higher prevalence of bisphosphonate-associated atypical sub-trochanteric femur fractures (ASFFs). Perhaps patients misdiagnosed with osteoporosis, but with a genetic profile of ALPL altering the normal expression and activity of TNAP, are prone to develop these fractures from NBP treatment [15].
The genetic screening of the ALPL gene favors the differential diagnosis. The assessment of the effective role of polymorphic variants in regulation of TNAP activity, and thus in the manifestation of adult and mild forms of HPP, is mandatory to improve the strength of the genetic differential diagnosis. A correct differential diagnosis between adult HPP and osteoporosis is certainly necessary to choose the right therapy and avoid damage due to incorrect pharmacological treatment. An effective enzyme replacement therapy (Asfotase Alpha) is now available and currently approved for treatment of perinatal, infantile and juvenile-onset HPP in the USA, the European Union, Canada, and Japan.
Here, we screened the polymorphic variants of ALPL (as single variants and/or haplotypes) in 56 subjects with low serum levels of TNAP and/or clinical signs of adult HPP, selected from a population of 12,000 subjects of the outpatient of the Metabolic Bone Diseases Unit of Florence, and then associated different genotypes with clinical manifestations of adult HPP.
Materials and methods
Patients
The study was conducted according to the approval of the Ethical Committee of the “Azienda Ospedaliero-Universitaria Careggi.” Recruited patients were requested to sign an informed consent form to participate into the genetic study, and data collection is performed accordingly the Declaration of Helsinki. All clinical and genetic data were analyzed anonymously and published as aggregate data.
Patients were retrospectively selected for this genetic study from 12,000 subjects who presented to the Outpatient Clinic of the Metabolic Bone Disease Unit of “Azienda Ospedaliero-Universitaria Careggi” from 2014 to 2019 for the evaluation of bone and mineral metabolism in suspected osteoporosis.
Bone turnover markers, assessed by standard procedures, were extracted from the medical records: serum calcium (Ca), serum phosphate (Pi), 24-h urinary Ca and Pi, TNAP, bone-specific alkaline phosphatase (BAP), parathyroid hormone (PTH), vitamin D (25-hydroxy-vitamin D), and urinary pyridinoline.
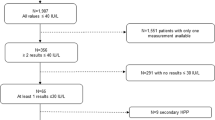
After reviewing all the medical records, we selected 56 unrelated individuals who presented low serum levels of TNAP (the normal reference value of our hospital laboratory was 50–126 IU/L; patients presenting two or more measurements of TNAP less than 50 IU/L were selected for the study) and/or history of clinical signs of adult HPP.
In detail, the following data concerning clinical signs of adult HPP were retrieved from the medical records: (1) history of low-trauma fractures [classified as metatarsal fractures, spine fractures, classical femur fractures, “atypical” femur fractures (hip or subtrochanteric localization), wrist fractures and other fractures (fractures in other bone sites)]; (2) history of non-healing bone fractures; (3) premature tooth loss and/or periodontal diseases; (4) presence of chondrocalcinosis; (5) presence of nephrocalcinosis; and (6) presence of fatigue with chronic musculoskeletal pain.
Following the report of persistent bone pain, foot, rib, and spine fractures were ascertained by X-ray evaluation, followed by a visit to the patient’s general practitioner or to our academic hospital. Conversely, femur, wrist, tibia, and humerus fractures were prevalently assessed by X-ray evaluation at emergency rooms.
A detailed medical history obtained from the patients included the evaluation of dietary phosphate and calcium intake assessed by a validated food questionnaire [16].
The evaluation of serum vitamin B6 was performed in all the 56 patients as marker of reduced TNAP activity, dosed by HPLC method and expressed as μg/L (normal value 3.6–18 μg/L).
A population of 36 individuals with normal TNAP serum levels and no clinical signs of suspected adult HPP was used as control for the distribution of frequencies of the less expressed alleles of ALPL polymorphic variants.
Genetic analysis
Genomic DNA was extracted from peripheral blood collected from patients using the microvolume extraction method QIAamp DNA Mini Kit (Qiagen GmbH, Hilden, Germany), according to the manufacturer’s instructions. The primer sequences utilized were described by Mornet et al. [17] and are designed to cover all the coding regions of the ALPL gene and include the flanking intronic regions containing all the described ALPL polymorphisms. Each PCR reaction was performed in a final volume of 30 μL with approximately 30 pmol of each primer, 250 μM each of dATP, dCTP, dGTP and dTTP, 1 unit of Gold Taq DNA polymerase (Perkin-Elmer, Foster City, CA, USA), and 1–3 mM MgCl2, depending on the exon amplified. Reactions were heated at 94 °C for 1 min and subjected to 30 cycles of 1 min at 94 °C, 30 s at the annealing temperature (60–66 °C) and 40 s at 72 °C, and another final cycle at 72 °C per 7 min [17].
PCR products were checked by electrophoresis on a 2% agarose gel and purified, using DNA Gel Extraction Kit (QIAGEN). Approximately 100 ng of each PCR amplicon were directly sequenced using the Sanger-based BigDye Terminator Purification Kit 1.1 (Applied Biosystems, Foster City, CA, USA) and analyzed on the ABI Prism 3130 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). The amplified DNA fragments, containing the ALPL polymorphisms, were sequenced for both the forward and reverse primers, to increase the strength of sequencing analysis in identifying the polymorphic variants. The obtained sequences were compared to wild type reference sequence of the ALPL gene published on GenBank Database (NC_000001) and NCBI reference sequence NG_008940.1.
Statistical analysis
Differences in allele frequencies between study and control populations were analyzed by chi-square test with Yates’ correction.
Anova and Bonferroni post hoc tests were used to compare categorical with continuous variable. Kruskal-Wallis test was used to compare categorical variable with more than three continuous variables.
Spearman’s rank-order correlation coefficient (Sperman’s rho) was used to correlate the concentration of serum TNAP and vitamin B6.
Pearson’s χ2 and Fisher’s exact tests were used as a measure of association between the characteristics of the samples.
For all the performed statistical tests, a nominal value of significance equal to 0.05 was assumed. A correction for multiple comparisons was applied to set up the level statistical significance with a p-value equal or less to 0.002.
Assuming a nominal alpha value of .05, with a sample of 56 patients, the following analyses are able to detect a large effect size (w = .5 and f = .45) with a power of .80.
Results
Our study population consisted of 56 unrelated subjects (40 women with a median age of 45.04 years and 16 men with a median age of 56.6 years; median age of the total population was 45.04 ± 16.80 years) presenting a low serum level of TNAP and a personal history of clinical signs of suspected adult HPP.
Out of the 56 patients, 21 showed a history of non-healing fractures (37.5%), 20 manifested musculoskeletal pain (35.7%), 6 reported premature tooth loss (10.7%), and 10 periodontal diseases (17.9%). Nephrocalcinosis was reported in 3 patients (5.4%). No cases of chondrocalcinosis were reported in our patients.
Of the total population, 40 patients had a personal history of fragility fractures (12 males and 28 females), as detailed in Fig. 1.

Distribution of fragility fractures in our population
We reported, in our patients, the presence of fourteen different polymorphic variants of the ALPL gene, all previously described in the literature [18,19,20,21,22]. No ALPL gene mutation was found in our cases. Forty-six patients of 56 (82.1%) were carriers of at least one of the minor frequency alleles (MFA) of ALPL polymorphic variants. No MFAs were found in 10 patients (17.9%) for any of the ALPL polymorphic variants. Identified polymorphisms and the distribution of frequencies of their MFAs in our population, in the control population, and in previously published studies, are shown in Table 1.
Statistical comparison, by chi-squared test with Yates’ correction, of MFA frequencies between the study population and the control group showed no significant differences for all the ALPL polymorphic variants.
The MFAs of the variants IVS5 c.472+12delG, IVS8 c.862+20G>T, IVS8 c.862+51G>A, IVS8 c.862+58C>T, were the most represented in our population (20/56; frequency 0.35), followed by IVS7 c.793-31C>T (17/56; frequency 0.30), Ex.7 c.787T>C (13/56; frequency 0.23), Ex.9 c.876A>G and Ex.12 c.1565T>C (7/56; frequency 0.1), Ex.4 c.455G>A, IVS8 c.863-7T>C and IVS8 c.863-12C>G (4/56; frequency 0.07), Ex.5 c.330C>T (3/56; frequency 0.05), Ex.12 c.1542T>G (2/56; frequency 0.03), and finally IVS10 c.1189+19G>T (1/56; frequency 0.02).
On the basis of the number of MFAs of the ALPL polymorphic variants found in each patient, we divided the study population into four different groups, in order to reduce the dispersion of the data: (1) group 1: patients without MFA of the ALPL gene polymorphic variants; (2) group 2: patients with one MFA of the ALPL polymorphic variants; (3) group 3: patients with two MFAs of the ALPL polymorphic variants, and (4) group 4: patients with three or more MFAs of the ALPL polymorphic variants.
Table 2 shows the distribution of individuals in the four groups.
Looking at the distribution of the total fractures in the 4 groups, we cannot observe any statistically significant difference (Pearson’s χ2 test 2.07; df 3; p = 0.556). However, we can observe a trend characterized by higher frequency of fragility fractures in patients of group 4 [12 fractured patients (85.7%) and 2 non-fractured patients (14.3%)]. Table 3 shows the distribution of total and metatarsal fractures among the 4 groups and percentages of fractures “intra-group” (comparison between patients with or without fractures within a single group) and “intra-fractures” (percentages of patients with or without fractures within total population). Patients with ≥ 3 MFAs (group 4) had a statistically higher frequency of metatarsal fractures, with respect to patients carrying only one or two MFAs and patients without any MFA. Indeed, 9 (64.3% intra-fractures) of the total 13 patients with history of metatarsal fractures carried ≥3 MFAs (Pearson χ2 15.48; df 3; p = 0.001).
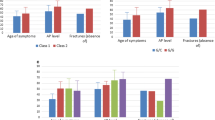
Gender distribution of HPP-related clinical signs investigated in our patients with persistent low TNAP is shown in Fig. 2. No statistically significant differences were observed between the 4 groups. Regarding the “history of tooth loss” variable, we observed, by applying the Pearson’s χ2 test, that this manifestation was not referred in the majority of the study population, and that, when the clinical sign was present, it was reported only by females (Pearson’s χ2 test 3.83; df = 1; p = 0.05) (Fig. 2A). Regarding the musculoskeletal pain variable, by applying the Pearson’s χ2 test, we observed that this manifestation was significantly more represented in males (64.5% intra-sex) than in females; 80% of women did not refer musculoskeletal pain (Pearson’s χ2 test 11.5; df 1; p = 0.03) (Fig. 2B).

HPP-related clinical signs in our population. Main table on the left summarizes all the HPP-related clinical signs investigated in our patients with persistent low TNAP. (A and B) On the right, report significant gender differences in two of the main clinical signs of adult HPP. (A) “History of tooth loss” was present only in females (Pearson’s χ2 test 3.83; df = 1; p = 0.05). (B) Musculoskeletal pain was significantly more present in men (Pearson’s χ2 test 8.90; df 1; p = 0.03)
Analyzing the distribution of MFAs of ALPL polymorphic variants, we observed a statistically significant difference in the distribution of some of them between the four groups of patients.
Indeed, the C allele of the Ex.7 c.787 T>C polymorphism was present in 13 of a total 46 patients included in groups 2/3 and 4. We observed that 11 patients, carrying this variant, were in group 4, and only 2 in group 2/3. (Pearson χ2 25.12; df 1; p = 0.001).
For the variants IVS8 c.862+20G>T, IVS8 c.862+51G>A, and IVS8 c.862+58C>T, we observed that, in the group 4, 12 patients carried the MFAs of these 3 polymorphisms and the 3 variants were always present together, suggesting a possible linkage disequilibrium (Pearson χ2 29.07; df 3; p = 0.001).
Finally, we observed that of a total 10 patients carried the G allele of the Ex.9 c.876A>G variant, 9 were in group 4 (Pearson χ2: 21.4; df 1; p = 0.001).
Distribution of the MFAs of these polymorphisms in groups 2+3 and group 4 is reported in Table 4 (intra-group percentage indicates the percentage of differences inside of a single group; intra-polymorphic percentage indicates the percentage of patients with or without the MFA of a polymorphic variant within the total population).
Applying Pearson’s χ2 test, we did not find statistically significant differences in the frequency of fractures and other clinical signs in patients carrying or not any of these polymorphic variants (data not shown).
The ANOVA analysis was applied to evaluate the correlation between bone biomarkers and polymorphic variants. We did not observe any statistically significant differences among the 4 groups regarding the levels of serum Ca, Pi, 24-h urine Ca and Pi, BAP, PTH, 25-OH vitamin D and urinary pyridinoline. However, we did observe a trend characterized by a lower level of BAP in group 4 in comparison with the other three groups (ANOVA and Bonferroni post hoc test: p = 0.08). Table 5 summarizes the distribution of biochemical parameters among the four genetic groups.
Regarding TNAP, we observed a statistically significant higher serum level in patients without any MFA of ALPL polymorphisms (50.6 ± 19 IU/L), patients with one MFA (45.7 ± 2.1 IU/L), and patients with 2 MFAs (41 ± 2.3 IU/L), all with respect to patients with ≥ 3 MFAs (25.4 ± 2.1 IU/L) (ANOVA and Bonferroni post hoc test: p = 0.001) (Fig. 3a). Conversely, we observed that vitamin B6 was significantly lower in patients without any MFA or with one MFA with respect to patients with ≥ 3 MFAs (ANOVA and Bonferroni post hoc test: p = 0.01 for group 1 vs. 4 and p = 0.005 for group 2 vs. 4) (Fig. 3b). To confirm these data, we applied the Pearson’s correlation test, which showed a significant reverse correlation between serum TNAP and B6 (p = 0.001; r = − 0.452) (Fig. 3c).

Correlation between ALPL genotypes and serum level of TNAP, vitamin B6 in the study population. a Serum TNAP was statistically significantly higher in patients without polymorphisms (50.6 ± 19 IU/L; normal value 50–126 IU/L), patients with 1 polymorphism (45.7 ± 2.1 IU/L), and patients with 2 polymorphisms (41 ± 2.3 IU/L), all with respect to patients with ≥ 3 polymorphic variants (25.4 ± 2.1 IU/L) (ANOVA and Bonferroni post hoc test: p = 0.001). b Serum vitamin B6 was statistically significantly lower in patients without any polymorphism or with only one polymorphism with respect to patients with ≥ 3 polymorphic variants (ANOVA and Bonferroni post hoc test: p = 0.01 for groups 1 vs. 4 and p = 0.005 for groups 2 vs. 4). c Reverse correlation between serum level of TNAP and vitamin B6 in the study population. d Correlation between the most frequent polymorphic variants (Ex7 c.787T>C; IVS7 793-31 C>T; Ex9 c.876A>G and the haplotype IVS8 c.862+20G>T, IVS8 c.862+51G>A, and IVS8 c.862+58C>T) in the group 4 and HPP-related biochemical parameters
To evaluate the correlation between the most represented polymorphic variants in group 4 and main HPP-related biochemical parameters, we applied the Kruskal–Wallis analysis, which showed a statistically significant difference between patients carrying one MFA of these polymorphic variants and patient non-carrying any MFA of these variants for all serum levels of TNAP, vitamin B6 and 25(OH) vitamin D. In particular, TNAP and 25(OH) vitamin D were lower and vitamin B6 was higher in patients carrying at least one MFA of these variants in comparison with individuals without them (Kruskal–Wallis analysis p = 0.002, 0.05, and 0.024, respectively) (Fig. 3d).
Discussion
Mineralization of hard tissues is a fundamental process, and alteration of its normal development leads to severe disorders. HPP is a heritable disorder characterized by defective bone and teeth mineralization and a deficiency of TNAP. Despite a continuum of severity, six clinical forms are currently recognized, depending on the age at diagnosis and the severity of the symptoms [6], but the presentation of HPP in adults has demonstrated a wide range of clinical manifestations, many of which are nonspecific [7]. In the present study, we evaluated the genetic pattern of the ALPL gene in a population of 56 subjects with low levels of serum TNAP, measured out before any treatment for osteoporosis, and retrospectively selected from a large cohort of 12,000 individuals referred to our hospital clinic for suspected osteoporosis. A similar retrospective genetic study has recently been performed by Alonso et al. [24] who screened for ALPL mutation patients with low ALP serum level selected from a large cohort of patients referred to a clinic referral center for bone diseases over a period of ten years. Their study identified 0.49% of individuals with low levels of ALP and found an ALPL mutation in 10/16 (62.5%) of them. Moreover, they identified the presence of the c.455G>A polymorphism in exon 5, affecting a residue involved in odonto-HPP, in 4/16 (25%) patients. Unlike this study, we failed to find any ALPL mutation in our cohort of patients with low serum level of TNAP. In our study, only 4/56 (7.1%) of patients bear the A allele of the c.455G>A polymorphic variant, only one of them presenting a history of periodontal diseases.
We observed a high variability in the expression of ALPL polymorphic variants, with patients ranging from bearing no MFA of ALPL polymorphisms to carrying one or more MFAs of different polymorphic variants. No significant difference of MFA distribution was found for any ALPL polymorphism with respect to a control population with normal levels of serum ALP. Our study population has been divided into four genetic groups on the basis of number of MFAs of ALPL polymorphic variants. No statistical differences were observed in the frequency of fractures and other clinical signs between the groups. However, in patients with ≥ 3 MFAs we observed a statistically higher incidence of metatarsal fractures, a typical clinical sign of HPP.
The most frequent MFAs present in our patients were those of, in order of frequency, Ex.7 c.787T>C (Y263H), IVS7 793-31 C>T, IVS8 c.862+20G>T, IVS8 c.862+51G>A, IVS8 c.862+58C>T, and Ex.9 c.876A>G polymorphisms (Fig. 1). Frequency of MFAs of ALPL polymorphisms in our study population did not significantly differ from those indicated in the Tissue Nonspecific Alkaline Phosphatase Gene Mutations Database (http://www.sesep.uvsq.fr/03_hypo_polymorphismes.php), for seven polymorphic variants. pt?>A higher MFA frequency was found in our population for the IVS5 c.472+12delG, IVS8 862+20G>T, and IVS8 c.862+58C>T variants, in comparison with the data published by Mornet et al. (0.35 vs. 0.07, 0.35 vs 0.07, and 0.35 vs 0.12, respectively) [19]. In contrast, a lower frequency was found for the G allele of the Ex.9 c.876A>G variant (0.1 vs. 0.47). Finally, the C allele of the Ex.12 c.1565T>C variant was lower in our population in comparison with the frequency found by Greenberg et al. 1993 (0.1 vs. 0.26) [20] and more near to the frequency observed by Mornet et al. in the French population (0.06) [19].
In the literature, the C allele of the Ex.7 c.787T>C variant has been described to be frequent in the North American population [21]. Henthorn et al. [21] showed that that the frequencies of this Ex.7 c.787T>C allele do not differ significantly between normal and HPP-affected populations, being 0.33 and 0.43, respectively. In our study population, we found a lower frequency in comparison with the data in the literature with the main expression of this variant in patients with ≥3 MFAs. Two studies of postmenopausal women in Japan have demonstrated that there was a significant difference in bone mineral density (BMD) among Ex.7 c.787T>C genotypes (lowest in c.787T homozygotes, highest in the c.787C homozygotes, and intermediate among heterozygotes) [25, 26]. Moreover, they studied the effect of different Ex.7 c.787T>C genotypes in the expression of the ALPL gene by transfection of opposite Ex.7 c.787T>C alleles, respectively in ST2 mouse marrow stromal cell line and COS-1 monkey fibroblast-like cell line [25, 26]. The polymorphic 787T>C nucleotide change causes an amino acid substitution of a tyrosine with a histidine at position 263 in TNAP enzyme (Y263H). Both these studies demonstrated that expression of TNAP protein was not modified by the presence of 787T or 787C allele, and 263H protein had ALP-specific activity similar to that of 263Y enzyme. However, the measure of TNAP affinity for its substrates (Km value) resulted to be significantly decreased in cells transfected with 787C allele (263H) compared to those bearing the 787T genotype (263Y), suggesting that the significant difference in Km may contribute to regulatory effects on bone metabolism.
Regarding the IVS8-c862+58C>T and IVS8-c862+20C>T variants, Mornet et al. [20] observed that their MFA frequencies were 0.12 and 0.07, respectively. In our population frequencies of MFAs of these two polymorphic variants, both were higher (0.25 and 0.21, respectively) and statistically more represented in group 4. The frequency of the T allele of the IVS8-c862+51C>T variant is not available in literature.
Interesting, MFAs of these variants were mainly present in group 4, all three presented together in our patients and in linkage disequilibrium also with the C allele of the Ex.7 c.787T>C variant.
Taillandier et al. [12] analyzed the ALPL gene in patients with severe HPP and described the IVS8 c.862+5G>A mutation, affecting the splice donor site of exon 8. This mutation was the third most common of 32 different described splicing mutations in the ALPL gene. Since IVS8-c862+20G>T, IVS8 c.862+51G>A, and IVS8 c.862+58C>T polymorphisms are all closed to splicing mutation described by Taillandier et al. [12], it is plausible to speculate that this region in the intron 8 of the gene may be a “hot spot”, and that polymorphic variants in this region could have a functional role in influencing the activity of the enzyme.
Fauvert et al. [22] described a specific multipolymorphic haplotype in the ALPL gene (named haplotype E), defined by the allelic combination Ex.7 c.787C, IVS7 c.793-31C, IVS8 c.862+20T, IVS8 c.862+51A, IVS8 c.862+58T, IVS8 c.863-7C, IVS8 c.863-12G, and Ex.9 c.876G, three times more frequently in patients bearing an ALPL mutation (p = 1.4 10−3). Authors suggested a linkage disequilibrium between haplotype E and an undetected mutation in a subset of the population carrying this haplotype, indicating the presence of this specific haplotype as potential pathogenic for HPP. In addition, authors analyzed, by using semi-quantitative RT-PCR, the expression of the ALPL gene in cultured cells from 18 unrelated persons from the general population, 9 of them carrying the haplotype E and 9 carrying other haplotypes; no difference regarding both splicing regulation and RNA quantity was found between different haplotypes. However, it remains possible that these polymorphisms have an impact on the ALP activity itself [22]. In our population, we investigated a possible role of these polymorphic variants in the activity of TNAP by associating them with HPP- and bone turnover-related clinical and biochemical characteristics; no significant difference was found in the history of fractures and/or other clinical signs in patients bearing a MFA of one of these polymorphic variants in comparison with other patients. However, the serum levels of TNAP, vitamin B6 and 25 (OH) vitamin D were all significantly different in patients bearing a MFA of one of these polymorphic variants with respect to all the other patients. In particular, serum TNAP and vitamin B6 were, respectively, lower and higher with respect to subjects non-carrying any MFA of these ALPL genetic variants.
We could speculate that the modification of Km due to the Ex.7 c.787C allele and/or the possible alteration of the splicing due to variations in IVS8 may have a functional role in reducing the activity of the TNAP enzyme, which could be low enough to cause substrate accumulation with the development of mild HPP forms. In this light, the biochemical finding of a low level of serum TNAP and, contemporarily, an increased level of vitamin B6, should alert clinicians to carefully evaluate the clinical history of patients regarding signs and symptoms typically associated with adult HPP. Regarding vitamin D, it has been speculated that vitamin D deficiency rickets may complicate musculoskeletal health and bone mineralization in children [27]. However, low levels of vitamin D have not been correlated with ALPL gene mutations in the literature. So far, it is not possible to explain the results found in our study, which may represent a random association and need to be verified on a larger sample. Indeed, this is just a pilot study, evaluating only a small number of patients, certainly not enough to allow us promote a personalized therapeutic strategy of patients based on the ALPL polymorphic genotype. Data found in our population need to be expanded to a larger population, including the genetic screening of family members, in order to confirm results, to better study the correlation between the presence of multiple ALPL MAFs and response outcomes to anti-reabsorption therapies, and to better understand how the genetic data could be transferred to clinical practice. As in the study of Alonso et al. [24], we found no association of ALPL MAFs with fractures, neither did we find evidence of a different response to treatment with bisphosphonates or other anti-reabsorption drugs between individuals bearing or not multiple ALPL MAFs.
In addition, although all ALPL polymorphic variants are singularly referred to as “benign” in the ClinVar database and none have been directly associated with the development of adult HPP, the presence of multiple MFAs of these polymorphic variants may be an instrument to suspect adult HPP, confirm the clinical and biochemical data, and caution clinicians regarding the administration of anti-reabsorption drugs.
Data Availability
The dataset used and analyzed during the current study is available from the corresponding author on reasonable request.
References
Orimo H (2010) The mechanism of mineralization and the role of alkaline phosphatase in health and disease. J Nippon Med Sch 77:1–12. https://doi.org/10.1272/jnms.77.4
Harris H (1989) The human alkaline phosphatases: what we know and what we don’t know. Clin Chim Acta 186:133–150. https://doi.org/10.1016/0009-8981(90)90031-m
Hofmann C, Girschick HJ, Mentrup B, Graser S, Seefried L, Liese J, Jakob F (2013) Clinical aspect of hypophosphatasia: an update. Clin Rev Bone Miner Metab 1:60–70. https://doi.org/10.1007/s12018-013-9139-0
Millán JL (2006) Mammalian alkaline phosphatases: from biology to applications in medicine and biotechnology. Wiley-VCH, Weinheim
Fraser D (1957) Hypophosphatasia. Ann J Med 22:730–746. https://doi.org/10.1016/0002-9343(57)90124-9
Mornet E, Yvard A, Taillander A, Fauvert D, Simon-Bouy B (2011) A molecular-based estimation of the prevalence of hypophosphatasia in the European population. Ann Hum Genet 75:439–445. https://doi.org/10.1111/j.1469-1809.2011.00642.x
Berseth KE, Tebben PJ, Drake MT, Hefferan TE, Jewison DE, Wermers RA (2013) Clinical spectrum of hypophosphatasia diagnosed in adults. Bone 54:21–27. https://doi.org/10.1016/j.bone.2013.01.024
Whyte MP (1994) Hypophosphatasia and the role of alkaline phosphatase in skeletal mineralization. Endocr Rev 15:439–461. https://doi.org/10.1111/j.1749-6632.2010.05387.x
Nielson CM, Zmuda JM, Carlos AS, Wagoner WJ, Larson EA, Orwoll ES, Klein RF (2012) Rare coding variants in ALPL are associated with low serum alkaline phosphatase and low bone mineral density. J Bone Miner Res 27(1):93–103. https://doi.org/10.1002/jbmr.527
Sogabe N, Tanabe R, Haraikawa M, Maruoka Y, Orimo H, Hosoi T, Goseki-Sone M (2013) Associations between serum bone-specific alkaline phosphatase activity, biochemical parameters, and functional polymorphism of the tissue-nonspecific alkaline phosphatase gene in a Japanese population. Asia Pac J Clin Nutr 22(1):160–165. https://doi.org/10.6133/apjcn.2013.22.1.11
Carter TC, Pangilinan F, Molloy AM, Fan R, Wang Y, Shane B, Gibney ER, Midttun Ø, Ueland PM, Cropp CD, Kim Y, Wilson AF, Bailey-Wilson JE, Brody LC, Mills JL (2015) Common variants at putative regulatory sites of the tissue non-specific alkaline phosphatase gene influence circulating pyridoxal 5’-phosphate concentration in healthy adults. J Nutr 145:1386–1393. https://doi.org/10.3945/jn.114.208769
Taillander A, Zurutuza L, Muller F, Simon-Bouy B, Serre JL, Bird L, Brenner R, Boute O, Cousin J, Gaillard D, Heidemann PH, Steinmann B, Wallot M, Mornet E (1999) Characterization of eleven novel mutations (M45L, R119H, 544delG, G145V, H154Y, C184Y, D289V, 862+5A, 1172delC, R411X, E459K) in the Tissue- Nonspecific Alkaline Phosphatase (TNSALP) gene in patients with severe hypophosphatasia. Mutations in brief no. 217. Online Hum Mutat 13(2):171–172. https://doi.org/10.1002/(sici)1098-1004(1999)13:2<171::aid-humu16>3.0.co;2-t
Whyte MP (2013) Hypophosphatasia. In: Thakker RV, Whyte MP, Eisman J, Igarashi T (eds) Genetics of bone biology and skeletal disease. Elsevier (Academic Press), San Diego
Whyte M (2009) Atypical femoral fractures, bisphosphonates, and adult hypophosphatasia. J Bone Miner Res 24:1132–1134. https://doi.org/10.1359/jbmr.081253
Sutton RA, Mumm S, Coburn SP, Ericson KL, Whyte MP (2012) “Atypical femoral fractures” during bisphosphonate exposure in adult hypophosphatasia. J Bone Miner Res 27(5):987–994. https://doi.org/10.1002/jbmr.1565
Montomoli M, Gonnelli S, Giacchi M, Mattei R, Cuda C, Rossi S, Gennari C (2002) Validation of a food frequency questionnaire for nutritional calcium intake assessment in Italian women. Eur J Clin Nutr 56:21–30. https://doi.org/10.1038/sj.ejcn.1601278
Mornet E, Taillandier A, Peyramaure S, Kaper F, Muller F, Brenner R, Bussière P, Freisinger P, Godard J, Le Merrer M, Oury JF, Plauchu H, Puddu R, Rival JM, Superti-Furga A, Touraine RL, Serre JL, Simon-Bouy B (1998) Identification of fifteen novel mutations in the tissue-nonspecific alkaline phosphatase (TNSALP) gene in European patients with severe hypophosphatasia. Eur J Hum Genet 6:308–314. https://doi.org/10.1038/sj.ejhg.5200190
Mumm S, Jones J, Finnegan P, Henthorn PS, Podgornik MN, Whyte MP (2002) Denaturing gradient gel electrophoresis analysis of the tissue nonspecific alkaline phosphatase isoenzyme gene in hypophosphatasia. Mol Genet Metab 75(2):143–153. https://doi.org/10.1006/mgme.2001.3283
Mornet E (2000) Hypophosphatasia: the mutations in the tissue-nonspecific alkaline phosphatase gene. Hum Mutat 15(4):309–315. https://doi.org/10.1002/(SICI)1098-1004(200004)15:4<309::AID-HUMU2>3.0.CO;2-C
Greenberg CR, Taylor CL, Haworth JC, Seargeant LE, Philipps S, Triggs-Raine B, Chodirker BN (1993) A homoallelic Gly317-->Asp mutation in ALPL causes the perinatal (lethal) form of hypophosphatasia in Canadian mennonites. Genomics 17:215–217. https://doi.org/10.1006/geno.1993.1305
Henthorn PS, Raducha M, Fedde KN, Lafferty MA, Whyte MP (1992) Different missense mutations at the tissue-nonspecific alkaline phosphatase gene locus in autosomal recessively inherited forms of mild and severe hypophosphatasia. Proc Natl Acad Sci U S A 89:9924–9928. https://doi.org/10.1073/pnas.89.20.9924
Fauvert D, Brun-Heath I, Lia-Baldini AS, Bellazi L, Taillandier A, Serre JL, de Mazancourt P, Mornet E (2009) Mild forms of hypophosphatasia mostly result from dominant negative effect of severe alleles or from compound heterozygosity for severe and moderate alleles. BMC Med Genet 10:51. https://doi.org/10.1186/1471-2350-10-51
Orimo H, Goseki-Sone M, Sato S, Shimada T. (1997) Detection of deletion 1154-1156 hypophosphatasia mutation using TNSALP exon amplification. Genomics 42(2):364–366. https://doi.org/10.1006/geno.1997.4733
Alonso N, Larraz-Prieto B, Berg K, Lambert Z, Redmond P, Harris SE, Deary IJ, Pugh C, Prendergast J, Ralston SH (2020) Loss-of-function mutations in the ALPL gene presenting with adult onset osteoporosis and low serum concentrations of total alkaline phosphatase. J Bone Miner Res 35(4):657–661. https://doi.org/10.1002/jbmr.3928
Sogabe N, Oda K, Nakamura H, Orimo H, Watanabe H, Hosoi T, Goseki-Sone M (2008) Molecular effects of the tissue-nonspecific alkaline phosphatase gene polymorphism (787T > C) associated with bone mineral density. Biomed Res 29(4):213–219. https://doi.org/10.2220/biomedres.29.213
Goseki-Sone M, Sogabe N, Fukushi-Irie M, Mizoi L, Orimo H, Suzuki T, Nakamura H, Orimo H, Hosoi T (2005) Functional analysis of the single nucleotide polymorphism (787T>C) in the tissue-nonspecific alkaline phosphatase gene associated with BMD. J Bone Miner Res 20(5):773–782. https://doi.org/10.1359/JBMR.041229
Lin EL, Gottesman GS, McAlister WH, Bijanki VN, Mack KE, Griffin DM, Mumm S, Whyte MP (2020) Clinical report: Healing of vitamin D deficiency rickets complicating hypophosphatasia suggests a role beyond circulating mineral sufficiency for vitamin D in musculoskeletal health. Bone 136:115322. https://doi.org/10.1016/j.bone.2020.115322
Funding
Open access funding provided by Università degli Studi di Firenze within the CRUI-CARE Agreement. This work was fully supported by a grant from Alexion Investigator Sponsored Research Program to Maria Luisa Brandi.
Author information
Authors and Affiliations
Contributions
Laura Masi made substantial contributions to the design of the study, managed clinical data, performed the analysis of data, and drafted the manuscript. Francesca Marini made substantial contributions to the design of the study, managed genetic data, performed the analysis of data, and collaborated to draft the manuscript. Francesco Franceschelli and Giogliola Leoncini performed the genetic screening of patients and revised the manuscript. Luisella Cianferotti, Federica Cioppi, Francesca Giusti, and Gemma Marcucci managed patients, collected clinical data, and revised the manuscript. Giorgio Gronchi revised the statistical analyses. Maria Luisa Brandi designed and supervised the study, critically revised the manuscript for its content, ensuring that questions related to the accuracy or integrity of any part of the work were appropriately investigated and resolved, and gave her final approval of the version to be published. All the authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Ethics approval
Patients included in this study underwent genetic screening of the ALPL gene and the collection of bone and HPP-related clinical and biochemical data based on their diagnostic and clinical management, independently from the present study, performed after the signing of a specific informed consent form for genetic diagnosis, previously approved by the Internal Review Board of the “Azienda Ospedaliero-Universitaria Careggi, Firenze.”
Consent to participate
Each patient signed a specific informed consent form for the execution of the diagnostic ALPL genetic test, which was part of his/her normal diagnostic management, independent from the present study. All data in this manuscript were analyzed anonymously.
Consent for publication
This manuscript does not contain any individual person’s data in any form. All data were analyzed anonymously and published exclusively as aggregates.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Masi, L., Marini, F., Franceschelli, F. et al. Polymorphic variants of alkaline phosphatase gene correlate with clinical signs of adult hypophosphatasia?. Osteoporos Int 32, 2461–2472 (2021). https://doi.org/10.1007/s00198-021-05893-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-021-05893-8