
Abstract
Summary
Drugs used for the prevention and the treatment of osteoporosis exert various favourable and unfavourable extra-skeletal effects whose importance is increasingly recognized notably for treatment selection.
Introduction
The therapeutic armamentarium for the prevention and the treatment of osteoporosis is increasingly large, and possible extra-skeletal effects of available drugs could influence the choice of a particular compound.
Methods
The present document is the result of a national consensus, based on a systematic and critical review of the literature.
Results
Observational research has suggested an inverse relationship between calcium intake and cardiovascular diseases, notably through an effect on blood pressure, but recent data suggest a possible deleterious effect of calcium supplements on cardiovascular risk. Many diverse studies have implicated vitamin D in the pathogenesis of clinically important non-skeletal functions or diseases, especially muscle function, cardiovascular disease, autoimmune diseases and common cancers. The possible effects of oral or intravenous bisphosphonates are well-known. They have been associated with an increased risk of oesophageal cancer or atrial fibrillation, but large-scale studies have not found any association with bisphosphonate use. Selective oestrogen receptor modulators have demonstrated favourable or unfavourable extra-skeletal effects that vary between compounds. Strontium ranelate has a limited number of non-skeletal effects. A reported increase in the risk of venous thromboembolism is not found in observational studies, and very rare cases of cutaneous hypersensitivity reactions have been reported. Denosumab has been introduced recently, and its extra-skeletal effects still have to be assessed.
Conclusion
Several non-skeletal effects of bone drugs are well demonstrated and influence treatment choices.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The pharmacological armamentarium for the management of osteoporosis has considerably expanded. Indeed, ability to substantially reduce fracture risk with a generally favourable risk–benefit ratio is now documented in well-conducted large clinical trials for a series of different molecules encompassing different pharmacological classes and different modes of action [1]. Osteoporosis is a highly prevalent problem in the ageing population, and the absolute number of affected subjects increases as a consequence of demographic evolutions. Albeit at present only a fraction of these patients at risk are treated, progress is being made and awareness increases of the consequences of osteoporotic fractures in terms of personal suffering and burden for the public health. Therefore, a large and steadily increasing number of patients are likely to be exposed for prolonged periods of treatment to osteoporosis medication. Availability of several treatment alternatives confronts the clinician with the difficulty to make the best choice for the individual patient, whereas the large-scale and prolonged prescription of osteoporosis medication puts much emphasis on safety issues.
To compare treatments, there is little evidence available from direct comparative trials, and no direct comparisons are available with fracture incidence as primary evaluation criterion. To select the ‘best choice treatment’ for their individual patient, clinicians thus depend on indirect comparisons, with little possibility of reliable differentiation in terms of efficacy, taking into account a variety of drug characteristics in relation to the patient’s clinical profile and preferences. In this context, consideration of the non-skeletal actions of the osteoporosis medications will not seldom intervene in the final choice, be it positively in terms of perceived potential ‘added value’ or negatively because of perceived potential risk for the patient. Aside from controversies related to potential long-term osseous adverse effects of osteoporosis treatments, a number of alleged extra-skeletal safety issues have been raised in the recent literature concerning as widely prescribed treatments as calcium and bisphosphonates (BPs).
The present document is the result of a national consensus based on a systematic review and a critical appraisal of the literature. It aims at providing the clinicians with an overview of what is the state of our knowledge on potentially deleterious or beneficial non-skeletal actions of the main pharmacological treatments of osteoporosis.
Methods
We included randomised controlled trials(RCTs), meta-analyses as well as epidemiologic retrospective or prospective studies and well documented case reports considering non-skeletal actions of osteoporosis treatments. Relevant articles related to treatment with calcium, vitamin D, bisphosphonates, selective oestrogen receptor modulators (SERMs), strontium ranelate, teriparatide, parathyroid hormone (PTH) and denosumab were identified through a systematic search, from 1966 to 2011, in MEDLINE and databases such as Cochrane Controlled Register. Following this extensive search of the literature, a critical appraisal was obtained through a consensus expert meeting.
Calcium
In the elderly, low calcium intake and vitamin D deficiency result in a negative calcium balance. This stimulates the secretion of PTH and induces age-associated secondary hyperparathyroidism, which enhances bone turnover and accelerates bone loss [2]. Adequate intake of calcium and vitamin D, through diet and/or supplements, reverses this secondary hyperparathyroidism and is recommended in the prevention of osteoporotic fractures [1, 3]. More specifically, the National Institutes of Health (NIH) in the USA proposes a recommended dietary allowance for calcium of 1,000 mg in men aged 50–70 years and 1,200 mg in men older than 70 years and women older than 50 years.
In combination with vitamin D substitution, calcium supplements have proven anti-fracture efficacy when targeted to persons at risk of calcium and/or vitamin D insufficiency, including elderly or institutionalized individuals, osteoporosis patients on antiresorptive or anabolic medication and persons receiving glucocorticoids [4–8]. Benefits are most apparent when a daily dose of 1,000–1,200 mg calcium is complemented with 800 IU vitamin D [6, 8]. This section reviews the evidence for the positive and negative non-skeletal effects of calcium [9].
Calcium as potentially protective against cardiovascular events
Observational research has suggested an inverse relationship between calcium intake and vascular diseases. In the Iowa Women’s Health Study in 34,486 postmenopausal women aged 55 to 69 years, Bostick and colleagues found that the highest quartile of total calcium intake (>1,425 mg/day), when compared to the lowest quartile (<696 calcium/day), was associated with a 33% reduction in ischaemic heart disease mortality (risk ratio (RR) 0.67, 95% confidence interval (CI) 0.47 to 0.94). According to the analysis, this risk reduction was dependent of the high total intake of calcium and could be attained by diet, supplements or both [10]. Similarly, Knox found a strong negative correlation between dietary calcium intake and mortality ratios for ischemic heart disease [11]. In the Nurses’ Health Study cohort of 85,764 women aged 39 to 59 years followed for 14 years, women in the highest quintile of total calcium intake (median calcium 1,145 mg/day) had a lower risk of stroke (RR 0.69, 95% CI 0.50–0.95) than those in the lowest quintile (median calcium 395 mg/day) [12].
To explain this observed protection against vascular diseases, potential beneficial effects of calcium on a number of vascular risk factors have been postulated. In particular, reductions in blood pressure, serum lipid concentration and body weight might be involved, although the data, to some extent, remain inconsistent [9].
An inverse relationship between calcium and blood pressure has been observed in several studies. In a meta-analysis of randomised controlled trials, both dietary calcium intake and calcium supplements were associated with reduced blood pressure, with a trend towards larger effects with dietary intake. However, the effect size was relatively small, with a mean reduction in systolic and diastolic blood pressure of −1.44 mmHg (95% CI −2.20 to −0.68) and −0.84 mmHg (95% CI −1.44 to −0.24), respectively [13]. In line with these findings, a recent trial showed significantly lower rates of hypertension amongst women aged over 45 years with a dietary calcium intake of at least 679 mg/day. In women in the highest quintile of dietary calcium intake (1,000 to 2,560 mg calcium/day), the relative risk reduction was 13% (RR 0.87, 95% CI 0.81 to 0.93). However, in women taking calcium supplements, even in the highest dosed quintile (1,000–2,100 mg), the risk of hypertension was unchanged (RR 1.07, 95% CI 0.97 to 1.18) [14]. A recent Cochrane review concluded that any association between calcium supplements and reduction in blood pressure is uncertain and that poor quality of individual trials and heterogeneity between trials do not allow any firm conclusions [15]. Any antihypertensive effect, if real, is at best small and transient [16].
Another potential cardioprotective mechanism might be a reduction in serum lipid concentration, due to the binding of calcium to fatty acids and bile acids in the gut, resulting in malabsorption of fat, and a direct effect on adipocytes with increased lipolysis [17–19]. In a randomised controlled trial in men, a diet fortified with calcium significantly reduced total cholesterol, LDL cholesterol and apolipoprotein B [18]. Similarly, in a randomised placebo-controlled trial in postmenopausal women, a supplement of 1,000 mg calcium during 12 months increased high-density lipoprotein (HDL) cholesterol levels and HDL to low-density lipoprotein (LDL) cholesterol ratio [20]. In another randomised study in men and women, however, no significant effect of calcium supplements (1,000–2,000 mg) was seen on total cholesterol or HDL cholesterol [21]. It is unclear, therefore, if and to what extent calcium determines lipid profile.
Reduced body weight has been implicated as well. Several large epidemiological studies have suggested that dietary calcium intake and calcium supplements may be associated with weight loss [22, 23], an effect that might be mediated by the same mechanisms affecting lipid profile [23]. However, several systematic reviews of randomised controlled trials argued against an inverse relationship between calcium (both dietary intake and supplements) and body weight [24–26], suggesting that any conclusions are preliminary and that the implications of calcium intake for body weight remain to be clarified.
Calcium supplements potentially associated with an increase in cardiovascular risk
Whereas spontaneous calcium intake, up to 800 mg/day, was not related to any cardiovascular deleterious effects, the cardiovascular safety of calcium supplements has been questioned. Rather than having a neutral or even beneficial effect, increased exposure to calcium might actually increase cardiovascular risk. In a meta-analysis published in 2010 by Bolland and colleagues in the British Medical Journal, more than 12,000 individuals from 15 double-blind placebo-controlled randomised trials were enrolled, and an increase in the incidence of myocardial infarction of about 30% was seen in individuals on calcium supplements (≥500 mg daily) compared to those on placebo [27]. More specifically, the analysis of patient level data showed that the relative risk of incident myocardial infarction in individuals allocated to calcium increased by 31% (HR 1.31, 95% CI 1.02 to 1.67) and trial level analysis showed a similar increase in risk by 27% (HR 1.27, 95% CI 1.01 to 1.59). However, no significant increase was observed in the incidence of a number of related vascular endpoints, including the incidence of stroke (HR 1.20, 95% CI 0.96 to 1.50), death (HR 1.09, 95% CI 0.96 to 1.23) and the composite end point of myocardial infarction, stroke and sudden death (HR 1.18, 95% CI 1.00 to 1.39).
The findings of this meta-analysis were partly driven by a previous randomised placebo-controlled trial from the same group that contributed 17% to the overall weight [28]. In this trial, calcium supplements were associated with a significant increase in HDL cholesterol levels but, nevertheless, also an increase in the risk of myocardial infarction [20, 28]. The authors postulated that calcium supplements may acutely elevate serum calcium levels [29] and, as a result, may enhance vascular calcification [28]. In fact, in a number of observational studies, high serum calcium levels have been associated with vascular calcification and an increased risk of vascular events, including myocardial infarction, stroke and death [30, 31]. Further support for a potentially deleterious effect of an acute increase in serum calcium comes from the observation that, in the meta-analysis, dietary intake was not associated with myocardial infarction, in line with observations that calcium from dairy products hardly affects serum calcium levels [27].
Whilst the meta-analysis of Bolland and colleagues should be interpreted as a strong signal that calcium supplements (without vitamin D) may potentially increase the risk of myocardial infarction, several limitations and even inconsistencies should be taken into account as well. First, the statistical outcome was only borderline significant (HR 1.31, 95% CI 1.02 to 1.67; p = 0.035), with a broad 95% confidence interval that approached 1 in the lower limit, suggesting that the findings have to be interpreted with caution. Also, the studies included in the analysis had been designed to assess the effects of calcium on bone density and fracture risk. None of the included trials had cardiovascular outcomes as primary or even secondary endpoint. As a result, cardiovascular events had not been adjudicated in a standardized manner, which may have resulted in over- or underreporting. Third, whilst the meta-analysis provided evidence for an increased risk of myocardial infarction, no increase was observed in the incidence of stroke, death or the composite end point of myocardial infarction, stroke and sudden death. In addition, trials that combined calcium and vitamin D supplements, the recommend strategy to prevent fractures in most elderly individuals, were excluded. In this context, it should be noted that a number of large-scale studies of calcium combined with vitamin D did not document an increase in cardiovascular risk [32, 33]. It is possible but not known if correction of vitamin D deficiency might counteract any potential detrimental vascular effect of calcium supplements [34, 35]. Finally, with the exception of the relatively small-sized trial from the same group [28], individual trials with calcium supplements did not show a significant increase in cardiovascular risk. In fact, a recent randomised placebo-controlled trial by Lewis et al., not included in the meta-analysis, did not find a higher risk of death or first-time hospitalization from atherosclerotic vascular disease in patients on calcium supplements [36]. A subset analysis even suggested a cardioprotective effect of calcium supplements in patients with pre-existing cardiovascular diseases. Nevertheless, the meta-analysis by Bolland et al. should be taken seriously, not as conclusive evidence but as a significant safety signal. Future studies with calcium should be designed to include careful assessment of cardiovascular endpoints, preferably by independent and blinded adjudication.
Calcium and cancer risk
There is also much controversy about the effect of calcium on the risk of cancer, with observational studies showing no effect, a protective effect or even an increased cancer risk [37]. Because the topic is diverse and the findings inconsistent, this section will only briefly discuss the association between calcium exposure and colorectal cancer, breast cancer and prostate cancer, since these have received most attention in recent years [9].
Whilst several observational studies concluded that calcium intake does not affect the risk of colorectal cancer [38], a number of cohort studies did find evidence for a protective effect of high total calcium intake (dietary intake plus supplements) [37, 39, 40]. In one of the main studies, a NIH-funded 7-year prospective trial in 293,907 men and 198,903 women aged 50 to 71 years, the risk reduction for colorectal cancer in the highest compared to the lowest quintile of total calcium intake was 0.79 (95% CI 0.70 to 0.89) in men and 0.72 (95% CI 0.61 to 0.86) in women [37]. Moreover, in a meta-analysis of randomised controlled trials in patients with previously removed colorectal adenomas and randomly assigned to calcium (1,200, 1,600 or 2,000 mg) or placebo, calcium supplements were significantly associated with a reduction in the risk of recurrent adenomas, considered as the precursors of colorectal cancer [41]. In line with these findings, the American College of Gastroenterology recommends daily dietary supplementation with 3 g calcium carbonate (1,200 mg calcium) in the prevention of recurrent colorectal adenomas [42].
Despite these data from observational studies and adenoma prevention trials, it is still uncertain if calcium supplements prevent colorectal cancer because large-scale long-term randomised controlled trials are not available. The only major randomised placebo-controlled study, the Women’s Health Initiative (WHI) trial in 36,282 postmenopausal women, found no effect of daily supplementation with 1,000 mg calcium and 400 IU of vitamin D for 7 years on colorectal cancer risk [43]. A Cochrane review concluded that there is not sufficient evidence to currently recommend the general use of calcium supplements in the prevention of colorectal cancer and that more research is needed [44].
The relationship between calcium exposure and breast cancer is not clear either. Some observational studies in premenopausal women found an inverse relationship between calcium intake and breast cancer [45–47], but some did not [37, 48]. Similarly, in trials in postmenopausal women, a protective effect has been reported [47], but most studies were negative [37, 45, 46, 48]. If and to what extent the source of calcium intake (dietary intake versus supplements) plays any role is not known [48]. Overall, an independent effect of calcium on the incidence of breast cancer remains uncertain.
In men, epidemiological studies have suggested that a higher total intake of calcium might be associated with an increased risk of developing prostate cancer. In these studies, total intake of calcium varied from more than 1,500 mg to more than 2,000 mg/day [49–51]. Calcium could potentially suppress the active form of vitamin D (1,25-OH2-D3), known to have an antiproliferative effect on prostate cancer cells [50, 52]. However, other studies could not confirm this association and found no or only a weak relationship between calcium intake and prostate risk [37, 53–55], even at very high intakes of calcium [37, 54]. As with colon cancer and breast cancer, conclusive evidence is lacking and more studies are required.
Calcium and the risk of kidney stones
Since most kidney stones are composed of calcium oxalate, an association with calcium intake is a theoretical concern. In the prospective Nurses’ Health Study, women who took supplemental calcium (1 to ≥500 mg/day) had a small but significant increase in the risk of incident symptomatic kidney stones (RR 1.20, 95% CI 1.02–1.41) compared to those who did not take supplements [56]. Women in the highest quintile of dietary calcium intake (median calcium 1,303 mg/day had, however, a lower risk (RR 0.65, 95% CI 0.50–0.83) compared to those in the lowest quintile (median calcium 391 mg/day). Other trials also showed a slightly increased risk of kidney stones in individuals on supplemental calcium (1,000 mg/day) [32] and a lower risk in individuals on a diet rich in calcium [57, 58].
The lower incidence of kidney stones in individuals on high dietary calcium intake is likely due to binding of dietary calcium with dietary oxalate in the gut, with reduced intestinal absorption and urinary excretion of oxalate. Calcium supplements, on the other hand, do not bind dietary oxalate when taken without meals. A combination of maintained oxalate excretion and increased calcium absorption and excretion from supplements increases the risk of stone formation [59].
In addition to beneficial musculoskeletal effects, especially when combined with vitamin D, calcium supplements have been suggested to protect against colorectal and breast cancer and to reduce some vascular risk factors. At the same time, safety questions have been raised about the role of calcium supplements in potentially increasing cardiovascular events, prostate cancer and kidney stones. Whilst these safety concerns have to be taken seriously, currently available evidence is not conclusive. In future research, priority should be given to well-designed long-term studies to assess cardiovascular and other safety endpoints.
Vitamin D
Rickets and osteomalacia are the diseases traditionally associated with severe vitamin D deficiency, defined as 25(OH) vitamin D levels below 10 ng/ml (25 nmol/l). A growing body of evidence has emerged indicating that less severe degrees of vitamin D deficiency between 10 and 20 ng/ml (25 and 50 nmol/l) and even vitamin D insufficiency, defined as 25(OH) vitamin D levels between 20 and 30 ng/ml (50 and 75 nmol/l), impair gastrointestinal absorption of calcium and bone mineralization, contributing to the pathogenesis of osteoporosis in older people [60]. Vitamin D has an impact on bone density and bone quality. In addition, by increasing muscle strength, adequate vitamin D status reduces the risk of falling in older individuals (see below). Therefore, vitamin D has a dual benefit for prevention of fractures in the elderly, a benefit on bone density and on muscle strength [61]. The importance of vitamin D for the prevention and treatment of osteoporosis has notably been reviewed in a previous Consensus of the Belgian Bone Club [1].
Furthermore, many studies have implicated vitamin D and its metabolites in the pathogenesis of a wide variety of clinically important non-skeletal functions or diseases, especially muscle function, cardiovascular disease, autoimmune diseases and several common cancers. The principal non-classical targets will be reviewed in this section. Whilst the evidence on bone and muscle health is based on randomised clinical trials, the evidence on other disease areas is nevertheless of a lower level. Most trials are small to moderate sized, and the outcomes of interest are only secondary outcomes. Interestingly, a meta-analysis of 18 randomised clinical trials including 57,311 individuals nevertheless concluded that vitamin D supplementation was associated with a decrease in total mortality (RR 0.93; 95% CI 0.77–0.96 compared to the control group) that could be due to effects of vitamin D on the musculoskeletal system or, as summarized below, on various non-skeletal diseases [35].
Vitamin D and muscular function
Vitamin D receptors have been shown to be present in muscle tissue [62], and a direct effect of vitamin D on muscle physiology is probable [63]. In muscle, vitamin D activates protein kinase C, which promotes calcium release, increasing the calcium pool that is essential for muscle contraction [64]. The potential cell signalling pathways affected by vitamin D in muscle have been recently reviewed [65]. Vitamin D deficiency has long been clinically associated with impaired muscle strength [66] and is also associated with loss of muscle mass [67]. With ageing, the number of vitamin D receptors in muscle decreases and the number of type II fibres, the first to be recruited to avoid falls, also decreases [68]. Treatment of elderly stroke survivors with 1,000 IU of vitamin D2 daily increases mean type II muscle fibre diameter by 2.5-fold over a 2-year period [69]. Because muscle weakness is a major risk factor for falls, it is not surprising that low vitamin D status is associated with an increased falls risk, as notably shown in a longitudinal study [70]. A meta-analysis including seven randomised, double-blind trials evaluating a daily dose of 700–1,000 IU/day of vitamin D demonstrated that falling was significantly reduced by 19% (RR 0.81; 95% CI 0.71–0.92) in vitamin D supplemented individuals compared with those receiving calcium or placebo [71]. This benefit may not depend on additional calcium supplementation, was significant within 2–5 months of treatment and extended beyond 12 months of treatment.
Vitamin D insufficiency and deficiency are associated with an increase in muscle fat as demonstrated by a significant negative relationship between circulating 25(OH) vitamin D levels and computed tomography measures of percent muscle fat (p < 0.001) [72]. Most studies have not found a significant relationship between baseline 25(OH) vitamin D levels and muscle strength [73]. However, correction of vitamin D deficiency has most often been associated with an improvement in muscle strength. Vitamin D supplementation in vitamin D-deficient Asian Indians during 6 months has thus shown an enhancement in skeletal muscle strength and physical performance [74]. A recent randomised, placebo-controlled, double-blind trial of 1,000 IU/day of vitamin D for 1 year showed a significant increase in muscle strength and mobility in subjects in the lowest tertile of baseline 25(OH) vitamin D values [75]. A longer duration trial showed that vitamin D and calcium supplementation during 20 months were superior to calcium alone in reducing fall frequency and improving muscle function in community-dwelling elderly subjects with 25(OH) vitamin D levels below 31 ng/ml [76]. These studies are in agreement with a recent systematic review and meta-analysis where the authors confirmed a beneficial effect of vitamin D supplementation on proximal muscle strength in adults with vitamin D deficiency but no significant effect on muscle strength in vitamin D replete adults [77].
Vitamin D and cardiovascular risk
A low level of 25(OH) vitamin D could be an independent risk factor for cardiovascular events, although a causal relationship has yet to be supported by large interventional trials. The evidence supporting a link between vitamin D deficiency and myocardial diseases has recently been reviewed [78]. In addition to possible direct effects due to the presence of the vitamin D receptor and of the 1-alpha hydroxylase enzyme in cardiac myocytes and other cells of the cardiovascular system [79], vitamin D has significant effects on several cardiovascular risk factors. Studies, ranging from animal studies to clinical trials, have shown that pharmacological doses of vitamin D notably reduce inflammation [80], improve endothelial function [81], control the secretion of insulin and improve insulin sensitivity [82]. Furthermore, as recently reviewed, vitamin D status has been linked to arterial hypertension [83].
Several observational studies suggest that 25(OH) vitamin D levels less than 15 ng/ml are associated with an excess risk of cardiovascular events when compared to levels >30–40 ng/ml. A nested case–control study in 18,225 men in the Health Professionals Follow-up Study (men aged 40–75 years, free of cardiovascular disease at baseline) showed that men with a 25(OH) vitamin D level ≤15 ng/ml had an increased risk for myocardial infarction relative to men with a level ≥30 ng/ml (RR 2.42; 95% CI 1.35–3.84) [84]. Even men with a 25(OH) vitamin D level 22.6–29.9 ng/ml had an increased risk (RR 1.60; 95% CI 1.10–2.32) compared with those with a level ≥30 ng/ml. In the Framingham offspring cohort study, 25(OH) vitamin D was measured in 1,739 participants without prior heart disease. At a mean follow-up of 5.4 years, amongst those with hypertension, there was a 2-fold increase in the risk of cardiovascular events for the participants with a 25(OH) vitamin D level <15 ng/ml compared to those with a level ≥15 ng/ml [34]. The Ludwigshafen Risk and Cardiovascular Health Study, a prospective cohort comprising 3,300 patients referred to coronary angiography and followed for 7.7 years, demonstrated a strong association between vitamin D status and several cardiovascular outcomes, such as cardiovascular mortality [85], stroke [86], heart failure and sudden cardiac death with the lowest risk amongst those with the highest 25(OH) vitamin D levels [87]. However, such associations have not been found in other studies. In the Osteoporotic Fractures in Men Study, vitamin D intake was evaluated in 3,094 men and 25(OH) vitamin D was measured in 813 men. The authors found no association between vitamin D intake or 25(OH) vitamin D levels and incidence of cardiovascular disease during a median follow-up of 4.4 years [88]. Similarly, serum levels of 25(OH) vitamin D levels were not independently associated with cardiovascular mortality in the prospective Rancho Bernardo study including 1,073 community-dwelling older adults followed up to 10.4 years [89]. On the other hand, in a cross-sectional study of 2,722 subjects, the prevalence of hypertension was found to be increased in subjects with 25(OH) vitamin D levels <40 ng/ml; odds ratios were 2.7 (1.4–5.2), 2.0 (1.4–5.2) and 1.3 (1.2–1.6) for 25(OH) vitamin D levels <15, 15–29 and 30–39 ng/ml, respectively, compared with the >40-ng/ml group [90]. This inverse relationship between 25(OH) vitamin D levels and hypertension has been recently confirmed in a meta-analysis of 18 studies [91]. These various sets of data raise the question of whether vitamin D supplementation can prevent hypertension and cardiovascular events.
The evidence of benefit of vitamin D supplementation from randomised trials is, however, scarce. In a small trial, 8 weeks of supplementation with vitamin D3 (800 UI/day) and calcium was reportedly more effective in reducing systolic blood pressure than calcium alone [92]. In the Women’s Health Initiative trial, including 36,282 postmenopausal women, vitamin D3 plus calcium supplementation did not reduce blood pressure, nor the risk of developing hypertension over 7 years of follow-up; however, in this trial, supplementation consisted only of 400 IU/day and adherence to supplementation was only around 60% [93]. A recent meta-analysis of eight randomised clinical trials in patients with a mean baseline blood pressure above 140/90 mmHg concluded that vitamin D reduces blood pressure modestly but significantly [94]. In summary, results from different studies are conflicting and trials specifically assessing effects of vitamin D on cardiovascular diseases as a primary endpoint are lacking. It is therefore premature to recommend supplemental vitamin D intake for the prevention of cardiovascular diseases or hypertension [95].
Vitamin D and the immune system
Vitamin D receptors are present in almost all immune cells, including activated T and B lymphocytes and antigen-presenting cells. Immune cells also express vitamin D-activating enzymes, allowing local conversion of inactive vitamin D into calcitriol within the immune system [96]. Several autoimmune diseases such as type 1 diabetes mellitus or multiple sclerosis are more frequent in countries with less sunshine, and vitamin D deficiency in early life increases the risk of autoimmune diseases and infections later on [96, 97]. There are several epidemiological studies that have reported an association between vitamin D deficiency and susceptibility to respiratory infections, especially tuberculosis and Gram-negative infections [98]. Studies using animal models of autoimmune diseases have identified vitamin D as a potential modulator of differentiation, proliferation and secretion processes in autoimmune reaction [96]. Supplementation in humans might thus be preventive in a number of autoimmune disorders.
A Finnish birth-cohort study, including >10,000 children born in 1966, showed that vitamin D supplementation during the first year of life (2,000 IU/day) was associated with a risk reduction of 78% for developing type 1 diabetes (followed up until end 1997) compared to no supplementation or use of lower doses [99]. A meta-analysis of data from four case–control studies and one cohort study support the beneficial effects of vitamin D in prevention of type 1 diabetes [100]. A more recent supplementation study, however, was negative [101]. Data indicate that treatment with vitamin D could be beneficial in reducing the risk of developing multiple sclerosis and diminishing its exacerbations [102]. Although contradictory data exist concerning supplementation benefits in rheumatoid arthritis (RA) and systemic lupus erythematosus, an association between low levels of 25(OH) vitamin D levels and activity of both diseases has been reported [103, 104]. Furthermore, an inverse association between higher intake of vitamin D and risk of rheumatoid arthritis was demonstrated in the Iowa Women’s Health Study [105]. However, we still lack non-biased large cohort studies that can sustain the proposed benefits of vitamin D supplementation for optimal immune function. Large-scale intervention trials in humans that support the findings in preclinical or observational studies are lacking [96].
Vitamin D and cancer treatment and prevention
Many experimental data show that calcitriol stimulates apoptosis and differentiation and inhibits angiogenesis and proliferation in tumour cells [106]. Numerous association studies suggest that serum 25(OH) vitamin D levels are inversely associated with the risk of many types of cancer. Further, in some studies of patients with cancer, an association between low 25(OH) vitamin D levels and poor prognosis has been observed [107, 108]. A meta-analysis of available studies indicated that there is a trend for lower incidence of colorectal carcinoma and adenoma with 25(OH) vitamin D levels >20 ng/ml in a dose–response association [109]. For breast cancer, a pooled analysis of two studies with 880 cases and 880 controls demonstrated that individuals with sufficient serum 25(OH) vitamin D levels had 50% lower risk of breast cancer than those with levels <13 ng/ml [110]. In addition, a large case–control study on 1,394 post-menopausal breast cancer patients and 1,365 controls also showed that the 25(OH) vitamin D level was significantly associated with lower breast cancer risk, particularly at levels above 20 ng/ml [111]. Most evidence concerning the link between vitamin D and cancer is derived from laboratory studies and observational investigations of 25(OH) vitamin D levels in association with cancer incidence and outcome. There are, however, several possible confounding factors and association cannot prove causation. Moreover, results from prospective studies only are more heterogeneous and do not support a significant association between vitamin D status and breast cancer [112].
There have been no clinical trials with cancer incidence or mortality as a primary outcome to support causality between vitamin D status and cancer. One population-based randomised clinical trial found that calcium plus vitamin D supplementation decreased cancer incidence as a secondary outcome. In that study including 1,179 healthy postmenopausal women aged >55 years, the mean level of 25(OH) vitamin D at baseline was 29 ng/ml. Supplementation with 1,100 IU vitamin D/day increased serum 25(OH) vitamin D to 38 ng/ml. After 4 years of treatment, the supplemented group had a 60% lower risk of developing cancer than the placebo group [113]. However, a recent re-analysis has indicated that this inverse association between vitamin D levels and cancer incidence disappeared after adjustment for BMI and physical activity [9, 112]. In another randomised trial, the Women’s Health Initiative, no effect of calcium and 400 IU vitamin D/day was found on the incidence of colorectal or breast cancer, which were secondary outcomes [114]. However, the dose of 400 IU used in that trial may have been inadequate to raise 25(OH) vitamin D blood levels significantly, particularly after factoring in adherence levels. A recent review of randomised vitamin D supplementation trials with cancer incidence as a secondary endpoint concluded that the results were null [112]. Moreover, the recent large-scale “Cohort Consortium Vitamin D Pooling Project of Rarer Cancers” showed no evidence linking higher serum 25(OH) vitamin D levels to reduced risks of less common cancers, including endometrial, gastric, kidney, pancreatic and ovarian cancers [115]. In summary, the available evidence that vitamin D reduces cancer incidence is inconsistent and inconclusive. Randomised controlled trials assessing vitamin D supplementation for cancer prevention are in progress. Their results are to be awaited before promoting vitamin D supplementation to reduce cancer risk.
As a general conclusion, the importance of vitamin D for bone health and the prevention of osteomalacia and osteoporosis are well recognized. More recently, vitamin D deficiency has been associated with other chronic conditions, including cardiovascular disease, autoimmune diseases and cancer. However, most evidence for the importance of vitamin D in these conditions comes from laboratory studies and observational investigations. Randomised controlled trials are needed to determine whether long-term supplementation with vitamin D has a favourable impact on the development or clinical course of non-skeletal diseases [116].
Bisphosphonates
BPs are the mainstay in the treatment of osteoporosis and other metabolic bone diseases such as Paget’s disease, as well as in tumoural conditions such as multiple myeloma, bone metastases and cancer-induced hypercalcaemia. Their efficacy and safety have been thoroughly established on the basis of multiple large pivotal trials dealing with their main indications. Their daily use in clinical medicine since 1969 has confirmed the general conclusions of the trials. Their strong affinity for the skeleton partially explains their excellent safety profile for other systems of the body. Even at high pharmacologic doses, their bone affinity grossly precludes tissue uptake outside the skeleton. First of all, intestinal absorption after oral administration is weak, on the order of less than 1%, even under ideal conditions (after a prolonged fast, with a full glass of water, and remaining fasting for at least 30 min in an upright position before any other food or beverage intake), leading to very low peak values in the plasma. After intravenous administration, however, if the plasma peak levels are higher, these levels are transient and short-lived. Similarly to what is observed after oral administration, serum levels rapidly decrease due to their rapid adsorption on the surface of bone (±50%). The rest is cleared by both glomerular filtration and proximal tubular secretion (± the remaining 50%) [117]. The retention time in the skeleton is extremely long and depends on the individual bone affinity of the various BPs. Part of the released BPs from the skeleton can be re-uptaken, and part is eliminated in the urine. Even if their terminal half-life is long, plasma levels remain very low. However, small amounts have been detected in body fluids up to 8 years after stopping the drug [118, 119]. This justified some warning regarding the use of BPs in premenopausal women of child bearing age. Even if there has been no demonstrated adverse foetal events in humans, large controlled studies are lacking to confirm their widespread safe use [120]. Some caution to restrict the use BPs to severe condition is still justified.
Bisphosphonate and acute phase reaction
After the first intravenous administration of a nitrogen-containing bisphosphonate (n-BP) (e.g. disodium pamidronate, zoledronic acid, ibandronate), about 25% of patients experienced flu-like symptoms, consisting of transient and self-limited fever, myalgias and/or arthralgias for 2 to 3 days. Acute phase reaction (APR) has been associated with the release of serum inflammatory cytokines such as tumour necrosis factor (TNFα) and IL-6, but not IL-1 [121]. The origin of these pro-inflammatory agents was homed on monocytes and/or macrophages [122] but also in human peripheral blood γδ T cells, which could constitute the trigger for activation of the former cells [123]. The APRs were absent or at least strongly attenuated with subsequent infusions with n-BPs. The APR has also been observed after high-dose oral monthly ibandronate [124]. The post-infusion syndrome can be reduced by acetaminophen [125]. It has been suggested that the co-administration of statins could prevent this reaction [123, 126], but this preventative effect does not seem to be systematic [127]. On the contrary, concomitant glucocorticoid (GC) therapy did not alleviate it [128]. Depletion in 25(OH)D could constitute a factor favouring the occurrence of APR after n-BPs infusion in n-BP-naive patients, but this remains to be confirmed [129].
Bisphosphonate and musculoskeletal pain
Some cases of prolonged musculoskeletal pain have been reported [130] in up to 20% to 25% of patients on alendronate and risedronate, as well as zoledronic acid [128, 131]. The majority of patients experienced gradual relief of pain after discontinuation of the drug. A few patients redeveloped pain following re-challenge of the drug. No plausible explanation has been proposed for their occurrence, and the association between BPs and musculoskeletal pain has therefore been questioned [132].
Bisphosphonate and the risk of renal failure
In line with the renal elimination of BPs, it is not recommended to prescribe BPs to patients with a creatinine clearance less than 30 ml/min, and this is specified in the Summary of Products Characteristics of BP who were granted an European Marketing Authorisation. In all pivotal studies of BPs, chronic kidney diseases (CKD) constituted an exclusion criterion, based on the calculated estimated glomerular filtration rate using the formula of Miller et al. [133]. In these large studies, however, several patients with CKD, but without other calcium metabolism abnormalities, notably in serum calcium, phosphate, alkaline phosphatase, vitamin D and PTH were included. Some exceptions to this 30-ml/min rule could therefore be theoretically possible [133–135]. Even if clinical trials and clear recommendations in the population with CKD are lacking, many clinicians suggested to halve the dose or reduce the frequency of administration of BPs in CKD [135]. Potential indications of BPs in CKD are the prevention of bone loss in kidney after transplantation. However, in these cases, no antifracture efficacy has so far been demonstrated with BP use [136–138]. Moreover, some patients treated with IV pamidronate developed low-bone turnover adynamic bone [137]. Calciphylaxis is a rare complication of CKD. Case reports have suggested the potential usefulness of BPs in its treatment [139, 140]. Proteinuria and proximal tubular necrosis has been described in mice and rats after parenteral doses of pamidronate sodium and clodronate five to 20 times higher than clinical doses used in humans [141]. However, acute renal toxicity was also reported in humans after rapid infusion of high doses of non-n-BPs [142]. Renal function deterioration, defined by elevations in the serum creatinine level, was observed in up to 15% of the patients receiving 4 mg of zoledronic acid over 15 min in trials of treatment for bone metastases (compared with 6.7% to 11.5% in patients on placebo) [143]. In the doses registered for the treatment of postmenopausal osteoporosis, oral BPs did not adversely affect the renal function. With intravenous zoledronic acid infusions, with infusion times of 15 min, short-term increases in serum creatinine have been observed for 9 to 11 days in a small subset of patients [144]. It seems therefore justified that patients be well hydrated and avoid simultaneous therapeutic agents at risk of impairing renal function. Patients with a glomerular filtration rate less than 30 ml/min should ideally be excluded, the precise diagnosis of bone loss in such patients being uncertain. Other kinds of bone disease than osteoporosis could be present [144]. As there exists no head-to-head comparative trial, it is not possible to determine whether intravenous n-BPs such as pamidronate disodium or ibandronate would have a different renal safety profile than zoledronic acid [144].
Bisphosphonate and ocular risk
Cases of iritis, episcleritis and scleritis, but also conjunctivitis, have been reported after therapy with n-BPs (mainly alendronate, pamidronate disodium and zoledronic acid) in up to 1% [145–147]. This does not seem to constitute an exclusive complication for n-BPs, but they were rarely reported with first-generation BPs [148]. Eye inflammation can resolve after local GC administration, but some patients can recur after BP rechallenge. In severe cases of uveitis and scleritis, it could be better to discontinue IV BP [149].
Bisphosphonate and the gastrointestinal tract
Digestive problems are at the origin of most drug withdrawals with oral n-BPs, mainly due to oesophageal irritation and upper gastrointestinal side effects [150]. They are poorly absorbed by the gastrointestinal tract, of the order of about 1%. Moreover, their absorption is further reduced if they are taken with food and beverage such as coffee, milk, orange juice etc. Hence, the recommendation is to take them in a fasting condition with a glass of water and to remain fasting in an upright position for at least 30 min after swallowing the drug until the first meal of the day. These precautions help to prevent most upper gastrointestinal side effects [151]. Moreover, the availability of weekly and monthly BPs has further decreased the frequency of the upper gastrointestinal tract symptoms [152–157]. It has been suggested that a lot of adverse events in upper gastrointestinal tract might be already present prior to start BPs therapy [158] and that clinicians and patients may sometimes inappropriately attribute gastrointestinal complaints to therapy [159]. Irrespective of whether gastrointestinal symptoms in individual patients are linked with oral BPs or not, it should be remembered that such a link has not been reported with intravenous therapy.
A study based on the General Practice Research Database containing anonymised patient records of about six million people in UK suggested a doubling of the incidence of oesophageal cancer with 5 years’ use of oral BPs [160], but this was not confirmed in another analysis of the same database [161]. No excess of gastric and colorectal cancer was found. Moreover, in patients with Barrett’s oesophagus on oral BPs, no increased risk of oesophageal adenocarcinoma was observed [162]. Even if no definitive conclusion can be drawn from these studies, upper gastrointestinal investigation is recommended if a patient on BPs develops dysphagia and pain.
Bisphosphonates and cardiovascular risk
In the pivotal study of zoledronic acid versus placebo in postmenopausal osteoporotic women, atrial fibrillation reported as serious adverse events (SAEs) was more frequent in the actively treated patients (1.3% versus 0.5%; p < 0.001). This was not observed in the HORIZON recurrent fracture trial, in which a similar frequency of ‘serious’ atrial fibrillation was observed both in actively treated and placebo-treated patients (1.1% versus 1.3%) [163]. Post hoc analyses of previous main trials on alendronate, risedronate and ibandronate having involved about 30,000 patients did not show any clear-cut association with atrial fibrillation [164–166]. It is possible that a lot of BP-treated patients have increased risks of cardiovascular events already before the start of therapy [167, 168]. Also, any potential cardiovascular risk should be weighted against the benefits of BP therapy. These include the well-documented antifracture efficacy, of course, but may also include additional benefits like the mortality benefit after hip fracture with zoledronic acid therapy, a 30% mortality reduction not simply attributable to anti-fracture efficacy [163, 169].
Bisphosphonate and hypocalcaemia
BPs and in particular n-BPs are potent inhibitors of osteoclastic bone resorption. They can therefore provoke hypocalcaemia, hypocalciuria and PTH reaction in some cases. Etidronate, however, did not induce any fall in serum and urine calcium because it acutely impaired the accretion of calcium into bone, offsetting a hypocalcaemic response [170]. Even with intravenous potent n-BPs, symptomatic hypocalcaemia rarely occurs in the treatment of osteoporosis under usual conditions, i.e. with supplemental calcium and vitamin D, lack of pre-existing hypoparathyroidism and/or renal failure.
Miscellaneous
-
Skin reactions like rash, pruritus and urticaria have been rarely reported with BP use. Re-challenge was positive in some cases [171]. Change of BP was not always accompanied by resurgence of symptoms, suggesting that BP-induced cutaneous reactions are probably not attributable to a class effect [171].
-
Extremely rare case reports of damage to the oral mucosa, apparently not related to osteonecrosis of the jaw, have been reported with the incorrect administration of n-BPs. Discontinuation of the inappropriate use allowed healing of the mucosa ulcers, even with maintained oral intake, but taken according to the prescription instructions [172].
-
A few reports of transient hepatitis after months to years of alendronate and/or risedronate, with liver biopsies compatible with a drug-induced toxicity, have been described [173, 174]. Healing occurred soon or later after stopping the drug.
Bisphosphonates and cancer
BPs constitute an efficacious therapy in order to prevent skeletal complications in patients with bone metastases. They might help to maintain functional independence and quality of life [175]. Several BPs have shown some efficacy in this regard, but owing to its easy mode of administration and its potency, zoledronic acid became the most used drug. Improved quality of life and prolonged disease-free survival have been observed with adjuvant therapy with zoledronic acid. In addition, zoledronic acid has shown a direct inhibition of tumorigenesis and cellular growth in preclinical models. So far, clinical results remain controversial [160, 176–183].
SAPHO syndrome
Synovitis, acne, pustulosis, hyperostosis and osteitis syndrome is a rare condition consisting of sterile inflammatory osteoarticular disorders, frequently associated with skin lesions resistant to conventional anti-inflammatory therapy [184]. Several case reports have shown successful therapy with infusions of pamidronate disodium and zoledronic acid [185, 186].
Multicentric reticulohistiocytosis
Multicentric reticulohistiocytosis is a rare systemic condition characterized by erosive polyarthritis frequently progressing to arthritis mutilans and papulonodular lesions on the skin. Alleviation of the arthritis and concurrent reduction of the size and number of cutaneous nodules have been observed in single case reports with therapy with alendronate, pamidronate and zoledronic acid [187].
Hypertrophic osteoarthropathy
Hypertrophic osteoarthropathy can be disabling and resistant to analgesic and anti-inflammatory drugs. Clubbing, arthralgias, cutaneous and osseous (periosteal) proliferation in the upper and lower extremities are frequently associated with bronchogenic carcinoma and right-to-left cardiac shunts. A few case reports have shown an effective alleviation of symptoms after pamidronate disodium and zoledronic acid in both benign and malignant conditions [188].
There are potentially other indications for BPs such as periodontitis leading to local bone loss. However, there is not yet enough evidence to recommend a wide use of BPs in the treatment of this condition. Moreover, the theoretical albeit questioned risk of osteonecrosis of the jaw could deter clinicians to use them thoughtlessly [189].
Selective oestrogen receptor modulators (SERMs)
SERMs and the risk of stroke
Several meta-analyses have reported an increased risk of stroke with tamoxifen use. Braithwaite et al. [190] observed a 49% increased stroke risk (RR 1.49; 95% CI 1.16 to 1.90). Similarly, Bushnell and Goldstein [191] found an OR of 1.82 (95% CI 1.41 to 2.36) for ischemic stroke and 1.40 (1.14 to 1.72) for any stroke. During a mean follow-up period of 4.9 years, the frequency of ischemic stroke was 0.71% with tamoxifen versus 0.39% for controls (absolute increased risk, 0.32%; number needed to harm, 313).
In the Ruth study, the incidence of all strokes did not differ between raloxifene (incidence rate per 100 woman-years = 0.95) and placebo (incidence rate = 0.86) treatment groups (p = 0.30). There was, however, in the group of women assigned to raloxifene a higher incidence of fatal strokes than amongst placebo users (incidence rates = 0.22 and 0.15, respectively, p = 0.0499). No significant subgroup interactions were found except that there was a higher incidence of stroke associated with raloxifene use amongst current smokers [192]. Lasofoxifene, contrary to other SERMs, at a dose of 0.5 mg/day, as compared with placebo, was associated with reduced stroke risk (2.5 versus 3.9 cases per 1,000 person-years; hazard ratio 0.64; 95% CI 0.41 to 0.99) in a randomised osteoporosis trial (8,556 women) [193].
SERMs and cardiovascular risk
In the meta-analysis conducted by Braithwaite et al. [190], tamoxifen was associated with significantly decreased myocardial infarction deaths (RR 0.62; 95% CI 0.41 to 0.93) but not myocardial infarction incidence (RR 0.90; 95% CI 0.66 to 1.23). Five years of treatment with tamoxifen was associated with reduced mortality from coronary heart disease compared with that in the 2-year group (hazard ratio = 0.67, 95% confidence interval = 0.47 to 0.94. Ten years after surgery, 2.1% of the patients in the 5-year group and 3.5% of those in the 2-year group had died from coronary heart disease.
Initial results from the breast prevention studies reported that tamoxifen was associated with a doubling of the risk of deep-vein thrombosis and pulmonary embolism. This was reported for instance during the active treatment of the IBIS-I trial (52 versus 23 cases, RR = 2.26, 95% CI = 1.36 to 3.87), but not after tamoxifen was stopped (16 versus 14 cases, RR = 1.14, 95% CI = 0.52 to 2.53) [194]. Similarly, Braithwaite et al., observed a 88% increased pulmonary emboli risk (RR 1.88; 95% CI 1.77 to 3.01).
The Raloxifene Use for The Heart (RUTH) trial showed that raloxifene had no overall effect on the incidence of coronary events in women with established coronary heart disease or coronary heart disease risk factors. In addition, raloxifene had no effect on the incidence of coronary events in any subgroup except in the case of a post hoc age subgroup analysis using age categories defined in the Women’s Health Initiative randomised trials. The effect of raloxifene on the incidence of coronary events differed significantly by age (interaction p = 0.0118). The incidence of coronary events in women <60 years of age was significantly lower in those assigned raloxifene (50 events) compared with placebo (84 events; hazard ratio 0.59; 95% confidence interval, 0.41 to 0.83; p = 0.003; absolute risk reduction, 36 per 1,000 women treated for 1 year). No difference was found between treatment groups in the incidence of coronary events in women > or =60 and <70 or > or =70 years of age [195].
Adomaityte et al. [196] assessed the risk of raloxifene on venous thromboembolism using a meta-analysis (nine trials, 24,523 postmenopausal women) and found a 62% increase in odds of either DVT or PE (odds ratio 1.62; 95% CI 1.25 to 2.09). Similarly, raloxifene therapy was associated with 54% increase in odds of DVT (odds ratio 1.54; 95% CI 1.13 to 2.11) and 91% increase in odds of PE alone (odds ratio 1.91; 95% CI 1.05 to 3.47). The excess event rate, in the More trial, was 1.8 per 1,000 woman-years (95% CI −0.5–4.1), and the number needed to treat to cause one event was 170 (95% CI 100–582) over 3.3 years [197]. Similarly to what is observed with tamoxifen and with menopause hormone therapy, the excess of risk is more pronounced during the first 2 years of use. Similar results were seen in the RUTH trial. Overall, raloxifene use was associated with an increased VTE risk (HR 1.44, 95% CI 1.06–1.95) versus placebo. Concomitant use of aspirin or non-aspirin antiplatelet agents along with raloxifene did not change VTE risk [198]. Still the risk with raloxifene seems lower than with tamoxifen, since in the updated report of the STAR trial (TAM versus RALOX), Toxicity RRs (raloxifene/tamoxifen) were 0.75 (95% CI 0.60–0.93) for thromboembolic events.
Lasofoxifene was associated with reduced risks of coronary heart disease events (5.1 versus 7.5 cases per 1,000 person-years; hazard ratio 0.68; 95% CI 0.50 to 0.93) [193]. There was a reduced risk of coronary revascularization (hazard ratio 0.56; 95% CI 0.32 to 0.98), hospitalization for unstable angina (hazard ratio 0.55; 95% CI 0.29 to 1.04) but no reduction of coronary death or nonfatal myocardial infarction [199].
SERMs and global mortality and morbidity
In a post hoc analysis of the MORE osteoporosis treatment trial (7,705 postmenopausal women), the global index outcome (defined as described for the WHI trial; i.e. occurrence of coronary heart disease, stroke, pulmonary embolism, invasive breast cancer, endometrial cancer, colorectal cancer, hip fracture or death because of other causes) resulted in annual rates of 1.39% and 1.83% in the raloxifene and placebo groups, respectively (HR 0.75; 95% CI 0.62–0.92), which were compatible with a favourable risk–benefit profile for raloxifene [200]. A pooled analysis of mortality data was performed from large clinical trials of raloxifene (60 mg/day) versus placebo, including the MORE/CORE trials (7,705 postmenopausal osteoporotic women followed for 4 years and a subset of 4,011 participants followed for an additional 4 years; 110 deaths) and the RUTH trial (10,101 postmenopausal women with coronary disease or multiple risk factors for coronary disease followed for 5.6 years; 1,149 deaths). All-cause mortality was 10% lower amongst women assigned to raloxifene 60 mg/day versus placebo (relative hazard 0.90; 95% CI 0.80–1.00; p = 0.05). Lower overall mortality was primarily due to lower rates of non-cardiovascular deaths, especially a lower rate of non-cardiovascular, non-cancer deaths [201]. The mechanism whereby raloxifene might reduce the risk of non-cardiovascular death remains unclear.
SERMs and cancer risk
It is well-known that tamoxifen is associated with significantly increased risks of endometrial cancer (RR 2.70; 95% CI 1.94 to 3.75) [190]. SERMS like tamoxifen and raloxifene are approved in the USA, but not in Europe, for reducing breast cancer risk in patients at risk of breast cancer. It has been repeatedly shown that tamoxifen reduces the risk of invasive ER-positive tumours [194].
On the hand, raloxifene did not increase risk for endometrial hyperplasia (RR 1.3; 95% CI 0.4–5.1), or endometrial cancer (RR 0.9; 95% CI 0.3–2.7) [197]. In the updated report of the STAR trial (TAM versus RALOX), Toxicity RRs (raloxifene/ tamoxifen) were 0.55 (95% CI 0.36–0.83; p = 0.003) for endometrial cancer (this difference was not significant in the initial results) [202].
The MORE trial found that 4 years of raloxifene therapy also decreased the incidence of invasive breast cancer amongst postmenopausal women with osteoporosis by 72% compared with placebo. The CORE (an extension trial) examined the effect of four additional years of raloxifene therapy. Incidences of invasive breast cancer and ER-positive invasive breast cancer were reduced by 59% (HR = 0.41; 95% CI = 0.24 to 0.71) and 66% (HR = 0.34; 95% CI = 0.18 to 0.66), respectively, in the raloxifene group compared with the placebo group. There was no difference between the two groups in incidence of ER-negative invasive breast cancer. Over the 8 years of both trials, the incidences of invasive breast cancer and ER-positive invasive breast cancer were reduced by 66% (HR = 0.34; 95% CI = 0.22 to 0.50) and 76% (HR = 0.24; 95% CI = 0.15 to 0.40), respectively, in the raloxifene group compared with the placebo group [203]. It has further been suggested that breast cancer risk reduction persists for some time in patients who discontinue raloxifene although this conclusion is limited by the post hoc analyses in unrandomised patients and the small sample sizes [204]. Raloxifene reduced also the incidence of invasive breast cancer by 44% (HR = 0.56; 95% CI = 0.38 to 0.83; absolute risk reduction = 1.2 invasive breast cancers per 1,000 women treated for 1 year) in the RUTH trial [205]. The lower incidence of invasive breast cancer reflected a 55% lower incidence of invasive ER-positive tumours (HR = 0.45; 95% CI = 0.28 to 0.72). However, raloxifene treatment did not reduce the incidence of non-invasive breast cancer or of invasive ER-negative breast cancer. The reduced incidence of invasive breast cancer was similar across subgroups, including those defined by age, body mass index, family history of breast cancer, prior use of postmenopausal hormones and 5-year estimated risk of invasive breast cancer. An updated analysis with an 81-month median follow-up of the STAR trial (tamoxifen (20 mg/day) or raloxifene (60 mg/day) for 5 years in women at high-risk breast cancer) was published in 2010 [202]. The RR (raloxifene/ tamoxifen) for invasive breast cancer was 1.24 (95% CI 1.05–1.47) and for non-invasive disease, 1.22 (95% CI 0.95–1.59). Compared with initial results, the RRs widened for invasive and narrowed for non-invasive breast cancer [202]. There were no significant mortality differences. Long-term raloxifene retained 76% of the effectiveness of tamoxifen in preventing invasive disease and grew closer over time to tamoxifen in preventing non-invasive disease.
In the PEARL trial (n = 8,556), lasofoxifene 0.5 mg reduced the risk of total breast cancer by 79% (hazard ratio 0.21; 95% CI 0.08 to 0.55) and ER+ invasive breast cancer by 83% (hazard ratio 0.17; 95% CI 0.05 to 0.57) compared with placebo. This effect was similar regardless of Gail score, whereas the effects were markedly stronger for women with higher baseline estradiol levels [206].
SERMs and menopausal symptoms
In breast cancer patients, it has been well documented that tamoxifen increases both severity and frequency of hot flushes.
The situation is likely less severe when using raloxifene. Some RCTs did not report an increased frequency or severity of vasomotor symptoms in women discontinuing oestrogen–progestin as compared with placebo [207, 208]. Nevertheless, other studies reported an increase in hot flushes when using raloxifene [209], which led to the suggestion of a gradual conversion to raloxifene from low-dose oestrogen, with a progression from 60 mg every alternate day to 60 mg/day.
It has been showed in short duration studies that it is possible to avoid SERMs associated hot flushes and menopausal symptoms, using a combination of a SEM (bazedoxifene) and estrogens (conjugated estrogens) [210].
Some non-skeletal side effects are favourable (breast cancer protection); others on the other hand are unfavourable (stroke risk, thromboembolism and endometrial cancer). The presence and the magnitude of these side effects vary between SERMs concluding that women with breast cancer treated with tamoxifen have an 82% increased risk of ischemic stroke and a 29% increased risk of any stroke, although the absolute risk remains small.
Strontium ranelate
Strontium ranelate is a first-line treatment for the management of postmenopausal osteoporosis. Its dual mode of action simultaneously reduces bone resorption and increases bone formation [211]. Strontium ranelate has a limited number of non-skeletal effects, for which most of the evidence comes from post hoc analyses of these two trials.
Strontium and cartilage
Osteoarthritis involves the degeneration of joint cartilage and the adjacent bone, which leads to joint pain and stiffness.
There is some preclinical evidence for an effect of strontium ranelate on cartilage degradation. Strontium ranelate has been demonstrated to stimulate the production of proteoglycans in isolated human chondrocytes, leading to cartilage formation without affecting cartilage resorption [212]. There is also evidence for an impact on biomarkers of cartilage degradation. Treatment with strontium ranelate was associated with significantly lower levels of urinary excretion of a marker of cartilage degradation (CTX-II) (p < 0.0001) [213, 214].
The potential for a clinical effect of strontium ranelate in osteoarthritis indicated that 3 years’ treatment with strontium ranelate was associated with a 42% lower overall osteoarthritis score (p = 0.0005 versus placebo) and a 33% reduction in disc space narrowing score (p = 0.03 versus placebo). These changes were concomitant to a 34% increase in the number of patients free of back pain (p = 0.03 versus placebo) [215].
Strontium ranelate and cardiovascular risk
The possibility of a vascular effect was raised following a pooled analysis of results in the SOTI and TROPOS populations, which found a higher annual incidence of venous thromboembolism over 5 years with strontium ranelate than with placebo (0.9% versus 0.6%; relative risk 1.4; 95% CI 1.0–2.0) [216]. Although these rates of venous thromboembolism were similar to those in the age-matched general population [217–219], they merited further investigation. The possibility of an impact was therefore explored in a retrospective study in the General Practice Research Database (GPRD) [220]. The GPRD was used to identify 11,546 women with osteoporosis but no treatment, 20,084 women with osteoporosis treated with alendronate and 2,408 women with osteoporosis treated with strontium ranelate; 115,009 women without osteoporosis were used as a comparator group [220]. Women with osteoporosis but no treatment were at greater risk for venous thromboembolism than women without osteoporosis (hazard ratio 1.43; 95% CI 1.10–1.86; p = 0.007; age-adjusted model), possibly due to the reduced mobility associated with bone disease. On the other hand, there was no difference in the rates of venous thromboembolism in the samples of women with osteoporosis (no treatment, strontium ranelate or alendronate). Similar findings have been reported from other observational studies [221, 222], which allays to a great extent the concerns.
Strontium ranelate and cutaneous adverse reactions
The other non-skeletal effect of concern with strontium ranelate is the occurrence of rare cases of cutaneous hypersensitivity reactions, which are manifested as drug reaction with eosinophilia and systemic symptoms (DRESS) or toxic epidermal necrolysis [223–226] (19-22). The pathogenesis of these hypersensitivity reactions remains unclear. Early recognition and appropriate management, including drug withdrawal, can improve the prognosis. The incidence of these adverse reactions is extremely low, estimated at 1/54,000 patient-years of treatment. This is most likely why no cases were detected in the phase 3 clinical trials. Similarly, no cases were reported in the observational study following over 13,000 patients receiving strontium ranelate over 2 years [222].
In conclusion, strontium ranelate has few non-skeletal effects. A possible beneficial effect on cartilage degradation and formation may translate into a new therapy for osteoarthritis. Observational studies suggest no cause for concern over possible vascular effects, whilst the rate of hypersensitivity reactions with cutaneous effects remains very low.
Denosumab
Denosumab is a fully human monoclonal antibody that inhibits the activity of the ligand for receptor activating NFκB (RANKL), the main stimulator of osteoclastogenesis and of osteoclast activity [227].
The potential extra skeletal effects of denosumab concern its interaction with RANK function in non-skeletal tissues, as RANK is largely expressed in several cell types, mainly of the immunological and vascular systems [228].
Denosumab and the immune and inflammatory response
Besides its major role to regulate bone resorption, the RANK/RANKL/OPG system is also an important regulator of the immune system where it is produced by T cells and enhances dendritic cells survival and antigen presentation [229]. A theoretical concern is the possible effect of denosumab on the susceptibility to infectious diseases and on the risk of cancer. A deregulation of the immune system could also lead to the appearance of atopic disease or autoimmune diseases. Conversely, there could be a benefit in inflammatory diseases. However, though RANK and RANK-L are essential in mice for ontogeny of the lymphoid tissues [227], patients with a mutation of the RANKL gene did not present immunological defects [230]. Suppression of RANKL does not interfere with inflammatory or immune response in mature individuals, and RANKL inhibition did not prevent inflammatory disease in several rat and mice models, except in the IL-2-deficient mice whose lymphocytes over express RANKL [229, 231].
The only human model of inflammatory disease in which denosumab has been used is RA. The authors followed at MRI for 12 months 143 patients receiving 60 or 180 mg injections of denosumab every 6 months. All patients were treated with methotrexate. At 12 months, the MRI erosion score was less increased from baseline in both denosumab groups than in the patients receiving a placebo (p < 0.012 and 0.007, respectively), but there was no evidence of an effect of denosumab on joint space narrowing or on measures of RA disease activity [232]. Thus, denosumab cannot substitute for DMARDs or anti-TNF in RA but could be an interesting adjuvant in patients with progression of bone erosions; beside, it could prevent osteoporosis associated with RA, particularly in patients requiring glucocorticoid treatment [233].
Concerning the problem of atopic disease and susceptibility to infections, Stolina et al. have shown that mice treated with OPG, the natural inhibitor of RANKL signalling, did not differ from controls with regard to contact hypersensitivity or infectious load induced by mycobacterial infection [234]. There was no decrease of humoral or cellular immunity. Another study in mice showed that inhibition of RANK signalling by a single dose of RANK-Fc 100 or 500 μg, which inhibits hypercalcaemia induced by 1, 25-dihydroxyvitamin D, did not decrease the immune response to influenza infection [235].
In the first clinical study in postmenopausal women with low bone density [236], the 1.9% of neoplasms in the denosumab group versus none in the placebo or alendronate groups was intriguing though not significant. However, in the FREEDOM study, including nearly 4,000 patients treated for 3 years with denosumab, the incidence of neoplasia did not differ significantly from the placebo group (3.7% versus 3.2%) [237]. In this study, the authors found a significant increase of eczema (3.0% versus 1.7%) and of cellulitis (0.3% versus <0.1%) reported as SAEs in the denosumab group but no difference in the overall proportion of patients with skin infection. Other clinical trials did not provide evidence for an increased risk of infectious complications either [238–240]. Because denosumab is a relatively recent treatment option, continued follow-up of any potential safety signals will be required, as with other agents in osteoporosis.
Denosumab and cardiovascular risks
RANKL and OPG could also play a role in the regulation of vascular calcification. Mice knocked out for OPG developed extensive vascular calcifications [241]. OPG produced locally by endothelial cells could promote endothelial survival and decrease atherotic plate mineralisation [228]. Several clinical studies have shown that circulating OPG was higher in patients with cardiovascular diseases, particularly in terminal renal failure [242, 243], an increase considered as a reaction to the inflammatory signal [244]. One human study has shown conversely an inverse relationship between OPG and echogenicity of carotid plaques, thus that individuals with more fibrous and calcified plates had a lower serum OPG concentration [245]. Inhibiting RANKL decreased vascular calcifications in human RANKL knocked-in mice with glucocorticoid induced osteoporosis [246]. Thus, one could expect that besides protecting bone, denosumab could decrease the risk of atherosclerosis. The clinical trials on bone efficacy in osteoporosis and osteopenia did not show differences in cardiovascular accidents in the denosumab-treated patients. However, these studies were not designed to study this end point, and the cardiovascular risk in the patients included was not high (6.8% of the patients in the placebo group of the FREEDOM study had a cardiovascular event, stroke, coronary heart disease or peripheral vascular disease). It would be interesting to look at high-risk subgroups and to include cardiovascular events as an end point in osteopenia or osteoporosis studies conducted in patients at increased risk of atheromatosis, like those with glucocorticoid induced osteoporosis.
Teriparatide and parathyroid hormone(1–84)
The biological activity of the intact human PTH, i.e. PTH(1–84), resides in its N-terminal sequence. Within the PTH peptide family, teriparatide, the recombinant human PTH(1–34) fragment has been most extensively developed for clinical use in osteoporosis.
Miscellaneous effects
In clinical trials, commonly reported mild side effects have been headaches (8%), nausea (8%), dizziness (9%) and leg cramps (3%), with only for the latter two a significantly higher incidence compared to placebo. These side effects tend to occur within the first few hours following subcutaneous injection [247, 248].
Subcutaneous injection of 20 μg of teriparatide results in a limited increase (around 0.8 mg/dl) of serum calcium, peaking after 4 to 6 h, followed by a progressive return to baseline before the next injection. These changes occur usually within the physiologic range, with occasional, mild hypercalcaemia having been observed in 11% of patients in the pivotal clinical trial. Repeated or persistent hypercalcaemia necessitating reduction or cessation of concomitant calcium supplementation and/or teriparatide dose reduction occurred in about 3% of patients. In this trial, the 24-h urinary calcium excretion showed a modest increase with a median of 30 mg/24 h. There were no clinical consequences, but patients with history of hypercalciuria or of urinary calculi in the past 5 years were excluded from the trial. Significant increases of serum uric acid have been observed in about 3% of patients. Although these biochemical changes are generally mild, it has been suggested that treatment with teriparatide should be avoided in subjects with a history of nephrolithiasis or gout, unless close monitoring is undertaken of serum and urinary calcium excretion or serum uric acid [247, 248].
The more limited data available on treatment with PTH(1–84) suggests that at a proposed dose of 100 μg/day, transient hypercalcaemia might be more frequent and mild hypercalciuria observed in up to 10% of patients [249, 250]. Mild local irritation with erythema at the injection site can occur with teriparatide and PTH(1–84) [226, 247].
Recently, teriparatide and PTH(1–84) have been proposed as a possible therapeutic option for hypoparathyroidism [251, 252].
Conclusions
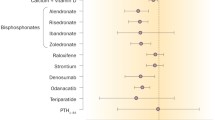
There is no doubt about the skeletal efficacy of bone drugs as used in their registered indications: treatment of osteoporosis in males and females, Paget’s disease of bone, multiple myeloma, bone metastases, cancer-induced hypercalcaemia, prevention and treatment of glucocorticoid induced osteoporosis or bone loss after hormonal deprivation in hormone sensitive cancers as, e.g. prostate or breast. Fractures can be prevented and bone pain and progressive bone disease limited. In this manuscript, an extensive review of non-skeletal effects of these drugs is presented. These can be either beneficial or deleterious.
Beneficial non-skeletal effects are proven for vitamin D and SERMs. Fall reduction, improved muscular function and physical performance are observed for substitution with adequate doses of vitamin D (800 IU/day) in deficient populations. As the health impact of falls is broader than for fractures only, fall reduction is a separate, valuable clinical outcome. For SERMs, long-term (up to 8 years) primary chemoprevention of oestrogen receptor positive breast cancers in postmenopausal women is documented. Viewing the lower level of evidence of non-vertebral fracture reduction by SERMs compared to other anti-resorptive bone drugs, breast cancer prevention contributes to the preferred use of SERMs in a specific therapeutic niche determined by younger age, axial osteoporosis and increased breast cancer risk.
More recently, some studies illustrated a reduction in mortality (with vitamin D, SERMs, IV bisphosphonate), which was probably not related to the fracture reduction. This interesting observation requires confirmation by additional large scaled and long-term studies including specific endpoints on cardiovascular risk factors and events and cancer.
Other promising beneficial effects are described for strontium on cartilage and spinal osteoarthritis and for denosumab on the prevention of bone erosions in rheumatoid arthritis. More clinical trials are needed to validate a potential use in these therapeutic areas. Furthermore animal or observational data support some speculation on potential benefits of calcium on ischemic cardiac mortality and stroke; of vitamin D on cardiovascular outcomes, autoimmune diseases and cancer prevention and of SERMs on coronary events and of denosumab on the prevention of vascular calcification.
The most frequent non-skeletal side effects of bone drugs are the gastrointestinal intolerance of calcium supplements and oral bisphosphonates, contributing in part to the reported low adherence of these drugs, and the acute phase reactions following intravenous amino-bisphosphonates applications. More important side effects in terms of severity, but fortunately infrequent, are stroke and venous thromboembolic events for SERMs and endometrium cancer for tamoxifen. A severe cutaneous hypersensitivity reaction, described as DRESS syndrome, has been reported in extremely rare case (only 16 reported) in clinical practice with strontium ranelate, although etiologic linkage remains doubtful. Hypocalcaemia has rarely been observed in bisphosphonate and denosumab trials (including calcium and vitamin D repleted patients); moreover, it was mild, transient and asymptomatic. Some studies, but not all, report kidney stones and myocardial infarction as side effects of calcium supplements and renal toxicity for iv pamidronate and zoledronate. Speculative side effects are discussed: musculoskeletal pain, uveitis, scleritis and oesophageal cancer for oral bisphosphonates and atrial fibrillation for iv zoledronate, coronary disease for SERMs, venous thromboembolism of strontium ranelate and skin infections for denosumab.
In conclusion, some of the non-skeletal effects of bone drugs, either beneficial or deleterious, may influence treatment choices, whereas others still require more studies to reveal additional insights into remaining questions concerning the clinical management of patients with bone diseases.
References
Body JJ, Bergmann P, Boonen S, Boutsen Y, Devogelaer JP, Goemaere S, Kaufman JM, Rozenberg S, Reginster JY (2010) Evidence-based guidelines for the pharmacological treatment of postmenopausal osteoporosis: a consensus document by the Belgian Bone Club. Osteoporos Int 21:1657–1680
Boonen S, Vanderschueren D, Geusens P, Bouillon R (1997) Age-associated endocrine deficiencies as potential determinants of femoral neck (type II) osteoporotic fracture occurrence in elderly men. Int J Androl 20:134–143
Boonen S, Bischoff-Ferrari HA, Cooper C, Lips P, Ljunggren O, Meunier PJ, Reginster JY (2006) Addressing the musculoskeletal components of fracture risk with calcium and vitamin D: a review of the evidence. Calcif Tissue Int 78:257–270
Boonen S, Vanderschueren D, Haentjens P, Lips P (2006) Calcium and vitamin D in the prevention and treatment of osteoporosis—a clinical update. J Intern Med 259:539–552
Group D (2010) Patient level pooled analysis of 68 500 patients from seven major vitamin D fracture trials in US and Europe. BMJ 340:b5463
Tang BM, Eslick GD, Nowson C, Smith C, Bensoussan A (2007) Use of calcium or calcium in combination with vitamin D supplementation to prevent fractures and bone loss in people aged 50 years and older: a meta-analysis. Lancet 370:657–666
Avenell A, Gillespie WJ, Gillespie LD, O’Connell D (2009) Vitamin D and vitamin D analogues for preventing fractures associated with involutional and post-menopausal osteoporosis. Cochrane Database Syst Rev CD000227
Boonen S, Lips P, Bouillon R, Bischoff-Ferrari HA, Vanderschueren D, Haentjens P (2007) Need for additional calcium to reduce the risk of hip fracture with vitamin d supplementation: evidence from a comparative metaanalysis of randomized controlled trials. J Clin Endocrinol Metab 92:1415–1423
Chung M, Balk EM, Brendel M et al (2009) Vitamin D and calcium: a systematic review of health outcomes. Evid Rep Technol Assess (Full Rep) 1–420
Bostick RM, Kushi LH, Wu Y, Meyer KA, Sellers TA, Folsom AR (1999) Relation of calcium, vitamin D, and dairy food intake to ischemic heart disease mortality among postmenopausal women. Am J Epidemiol 149:151–161
Knox EG (1973) Ischaemic-heart-disease mortality and dietary intake of calcium. Lancet 1:1465–1467
Iso H, Stampfer MJ, Manson JE, Rexrode K, Hennekens CH, Colditz GA, Speizer FE, Willett WC (1999) Prospective study of calcium, potassium, and magnesium intake and risk of stroke in women. Stroke 30:1772–1779
Griffith LE, Guyatt GH, Cook RJ, Bucher HC, Cook DJ (1999) The influence of dietary and nondietary calcium supplementation on blood pressure: an updated metaanalysis of randomized controlled trials. Am J Hypertens 12:84–92
Wang L, Manson JE, Buring JE, Lee IM, Sesso HD (2008) Dietary intake of dairy products, calcium, and vitamin D and the risk of hypertension in middle-aged and older women. Hypertension 51:1073–1079
Dickinson HO, Nicolson DJ, Cook JV, Campbell F, Beyer FR, Ford GA, Mason J (2006) Calcium supplementation for the management of primary hypertension in adults. Cochrane Database Syst Rev CD004639
Reid IR, Horne A, Mason B, Ames R, Bava U, Gamble GD (2005) Effects of calcium supplementation on body weight and blood pressure in normal older women: a randomized controlled trial. J Clin Endocrinol Metab 90:3824–3829
Govers MJ, Van der Meet R (1993) Effects of dietary calcium and phosphate on the intestinal interactions between calcium, phosphate, fatty acids, and bile acids. Gut 34:365–370
Denke MA, Fox MM, Schulte MC (1993) Short-term dietary calcium fortification increases fecal saturated fat content and reduces serum lipids in men. J Nutr 123:1047–1053
Zemel MB, Shi H, Greer B, Dirienzo D, Zemel PC (2000) Regulation of adiposity by dietary calcium. FASEB J 14:1132–1138
Reid IR, Mason B, Horne A, Ames R, Clearwater J, Bava U, Orr-Walker B, Wu F, Evans MC, Gamble GD (2002) Effects of calcium supplementation on serum lipid concentrations in normal older women: a randomized controlled trial. Am J Med 112:343–347
Bostick RM, Fosdick L, Grandits GA, Grambsch P, Gross M, Louis TA (2000) Effect of calcium supplementation on serum cholesterol and blood pressure. A randomized, double-blind, placebo-controlled, clinical trial. Arch Fam Med 9:31–38, discussion 39
Heaney RP (2003) Normalizing calcium intake: projected population effects for body weight. J Nutr 133:268S–270S
Parikh SJ, Yanovski JA (2003) Calcium intake and adiposity. Am J Clin Nutr 77:281–287
Barr SI (2003) Increased dairy product or calcium intake: is body weight or composition affected in humans? The Journal of nutrition 133:245S–248S
Trowman R, Dumville JC, Hahn S, Torgerson DJ (2006) A systematic review of the effects of calcium supplementation on body weight. Br J Nutr 95:1033–1038
Lanou AJ, Barnard ND (2008) Dairy and weight loss hypothesis: an evaluation of the clinical trials. Nutr Rev 66:272–279
Bolland MJ, Avenell A, Baron JA, Grey A, MacLennan GS, Gamble GD, Reid IR (2010) Effect of calcium supplements on risk of myocardial infarction and cardiovascular events: meta-analysis. BMJ 341:c3691
Bolland MJ, Barber PA, Doughty RN, Mason B, Horne A, Ames R, Gamble GD, Grey A, Reid IR (2008) Vascular events in healthy older women receiving calcium supplementation: randomised controlled trial. BMJ 336:262–266
Reid IR, Schooler BA, Hannan SF, Ibbertson HK (1986) The acute biochemical effects of four proprietary calcium preparations. Aust N Z J Med 16:193–197
Foley RN, Collins AJ, Ishani A, Kalra PA (2008) Calcium-phosphate levels and cardiovascular disease in community-dwelling adults: the Atherosclerosis Risk in Communities (ARIC) Study. Am Heart J 156:556–563
Vestergaard P, Mollerup CL, Frokjaer VG, Christiansen P, Blichert-Toft M, Mosekilde L (2003) Cardiovascular events before and after surgery for primary hyperparathyroidism. World J Surg 27:216–222
Jackson RD, LaCroix AZ, Gass M et al (2006) Calcium plus vitamin D supplementation and the risk of fractures. N Engl J Med 354:669–683
Hsia J, Heiss G, Ren H et al (2007) Calcium/vitamin D supplementation and cardiovascular events. Circulation 115:846–854
Wang TJ, Pencina MJ, Booth SL, Jacques PF, Ingelsson E, Lanier K, Benjamin EJ, D’Agostino RB, Wolf M, Vasan RS (2008) Vitamin D deficiency and risk of cardiovascular disease. Circulation 117:503–511
Autier P, Gandini S (2007) Vitamin D supplementation and total mortality: a meta-analysis of randomized controlled trials. Arch Intern Med 167:1730–1737
Lewis JR, Calver J, Zhu K, Flicker L, Prince RL (2011) Calcium supplementation and the risks of atherosclerotic vascular disease in older women: results of a 5-year RCT and a 4.5-year follow-up. J Bone Miner Res 26:35–41
Park Y, Leitzmann MF, Subar AF, Hollenbeck A, Schatzkin A (2009) Dairy food, calcium, and risk of cancer in the NIH-AARP Diet and Health Study. Arch Intern Med 169:391–401
Martinez ME, Willett WC (1998) Calcium, vitamin D, and colorectal cancer: a review of the epidemiologic evidence. Cancer Epidemiol Biomarkers Prev 7:163–168
Wu K, Willett WC, Fuchs CS, Colditz GA, Giovannucci EL (2002) Calcium intake and risk of colon cancer in women and men. J Natl Cancer Inst 94:437–446
Cho E, Smith-Warner SA, Spiegelman D et al (2004) Dairy foods, calcium, and colorectal cancer: a pooled analysis of 10 cohort studies. J Natl Cancer Inst 96:1015–1022
Shaukat A, Scouras N, Schunemann HJ (2005) Role of supplemental calcium in the recurrence of colorectal adenomas: a metaanalysis of randomized controlled trials. Am J Gastroenterol 100:390–394
Bond JH (2000) Polyp guideline: diagnosis, treatment, and surveillance for patients with colorectal polyps. Practice Parameters Committee of the American College of Gastroenterology. Am J Gastroenterol 95:3053–3063
Wactawski-Wende J, Kotchen JM, Anderson GL et al (2006) Calcium plus vitamin D supplementation and the risk of colorectal cancer. N Engl J Med 354:684–696
Weingarten MA, Zalmanovici A, Yaphe J (2008) Dietary calcium supplementation for preventing colorectal cancer and adenomatous polyps. Cochrane Database Syst Rev CD003548
Shin MH, Holmes MD, Hankinson SE, Wu K, Colditz GA, Willett WC (2002) Intake of dairy products, calcium, and vitamin d and risk of breast cancer. J Natl Cancer Inst 94:1301–1311
Lin J, Manson JE, Lee IM, Cook NR, Buring JE, Zhang SM (2007) Intakes of calcium and vitamin D and breast cancer risk in women. Arch Intern Med 167:1050–1059
McCullough ML, Rodriguez C, Diver WR, Feigelson HS, Stevens VL, Thun MJ, Calle EE (2005) Dairy, calcium, and vitamin D intake and postmenopausal breast cancer risk in the Cancer Prevention Study II Nutrition Cohort. Cancer Epidemiol Biomarkers Prev 14:2898–2904
Larsson SC, Bergkvist L, Wolk A (2009) Long-term dietary calcium intake and breast cancer risk in a prospective cohort of women. Am J Clin Nutr 89:277–282
Rodriguez C, McCullough ML, Mondul AM, Jacobs EJ, Fakhrabadi-Shokoohi D, Giovannucci EL, Thun MJ, Calle EE (2003) Calcium, dairy products, and risk of prostate cancer in a prospective cohort of United States men. Cancer Epidemiol Biomarkers Prev 12:597–603
Mitrou PN, Albanes D, Weinstein SJ, Pietinen P, Taylor PR, Virtamo J, Leitzmann MF (2007) A prospective study of dietary calcium, dairy products and prostate cancer risk (Finland). Int J Cancer 120:2466–2473
Giovannucci E, Liu Y, Platz EA, Stampfer MJ, Willett WC (2007) Risk factors for prostate cancer incidence and progression in the health professionals follow-up study. Int J Cancer 121:1571–1578
Hedlund TE, Moffatt KA, Miller GJ (1996) Stable expression of the nuclear vitamin D receptor in the human prostatic carcinoma cell line JCA-1: evidence that the antiproliferative effects of 1 alpha, 25-dihydroxyvitamin D3 are mediated exclusively through the genomic signaling pathway. Endocrinology 137:1554–1561
Koh KA, Sesso HD, Paffenbarger RS Jr, Lee IM (2006) Dairy products, calcium and prostate cancer risk. Br J Cancer 95:1582–1585
Chan JM, Pietinen P, Virtanen M, Malila N, Tangrea J, Albanes D, Virtamo J (2000) Diet and prostate cancer risk in a cohort of smokers, with a specific focus on calcium and phosphorus (Finland). Cancer Causes Control 11:859–867
Rohrmann S, Platz EA, Kavanaugh CJ, Thuita L, Hoffman SC, Helzlsouer KJ (2007) Meat and dairy consumption and subsequent risk of prostate cancer in a US cohort study. Cancer Causes Control 18:41–50
Curhan GC, Willett WC, Speizer FE, Spiegelman D, Stampfer MJ (1997) Comparison of dietary calcium with supplemental calcium and other nutrients as factors affecting the risk for kidney stones in women. Ann Intern Med 126:497–504
Curhan GC, Willett WC, Rimm EB, Stampfer MJ (1993) A prospective study of dietary calcium and other nutrients and the risk of symptomatic kidney stones. N Engl J Med 328:833–838
Curhan GC, Willett WC, Knight EL, Stampfer MJ (2004) Dietary factors and the risk of incident kidney stones in younger women: Nurses’ Health Study II. Arch Intern Med 164:885–891
Bihl G, Meyers A (2001) Recurrent renal stone disease-advances in pathogenesis and clinical management. Lancet 358:651–656
Holick MF (2007) Vitamin D deficiency. N Engl J Med 357:266–281
Bischoff-Ferrari HA, Willett WC, Wong JB, Stuck AE, Staehelin HB, Orav EJ, Thoma A, Kiel DP, Henschkowski J (2009) Prevention of nonvertebral fractures with oral vitamin D and dose dependency: a meta-analysis of randomized controlled trials. Arch Intern Med 169:551–561
Bischoff HA, Borchers M, Gudat F, Duermueller U, Theiler R, Stahelin HB, Dick W (2001) In situ detection of 1,25-dihydroxyvitamin D3 receptor in human skeletal muscle tissue. Histochem J 33:19–24
Demay M (2003) Muscle: a nontraditional 1,25-dihydroxyvitamin D target tissue exhibiting classic hormone-dependent vitamin D receptor actions. Endocrinology 144:5135–5137
Capiati DA, Vazquez G, Boland RL (2001) Protein kinase C alpha modulates the Ca2+ influx phase of the Ca2+ response to 1alpha,25-dihydroxy-vitamin-D3 in skeletal muscle cells. Horm Metab Res 33:201–206
Dirks-Naylor AJ, Lennon-Edwards S (2011) The effects of vitamin D on skeletal muscle function and cellular signaling. J Steroid Biochem Mol Biol
Venning G (2005) Recent developments in vitamin D deficiency and muscle weakness among elderly people. BMJ 330:524–526
Visser M, Deeg DJ, Lips P (2003) Low vitamin D and high parathyroid hormone levels as determinants of loss of muscle strength and muscle mass (sarcopenia): the Longitudinal Aging Study Amsterdam. J Clin Endocrinol Metab 88:5766–5772
Bischoff-Ferrari HA, Borchers M, Gudat F, Durmuller U, Stahelin HB, Dick W (2004) Vitamin D receptor expression in human muscle tissue decreases with age. J Bone Miner Res 19:265–269
Sato Y, Iwamoto J, Kanoko T, Satoh K (2005) Low-dose vitamin D prevents muscular atrophy and reduces falls and hip fractures in women after stroke: a randomized controlled trial. Cerebrovasc Dis 20:187–192
Snijder MB, van Schoor NM, Pluijm SM, van Dam RM, Visser M, Lips P (2006) Vitamin D status in relation to one-year risk of recurrent falling in older men and women. J Clin Endocrinol Metab 91:2980–2985
Bischoff-Ferrari HA, Dawson-Hughes B, Staehelin HB, Orav JE, Stuck AE, Theiler R, Wong JB, Egli A, Kiel DP, Henschkowski J (2009) Fall prevention with supplemental and active forms of vitamin D: a meta-analysis of randomised controlled trials. BMJ 339:b3692
Gilsanz V, Kremer A, Mo AO, Wren TA, Kremer R (2010) Vitamin D status and its relation to muscle mass and muscle fat in young women. J Clin Endocrinol Metab 95:1595–1601
Annweiler C, Beauchet O, Berrut G, Fantino B, Bonnefoy M, Herrmann FR, Schott AM (2009) Is there an association between serum 25-hydroxyvitamin D concentration and muscle strength among older women? Results from baseline assessment of the EPIDOS study. J Nutr Health Aging 13:90–95
Gupta R, Sharma U, Gupta N, Kalaivani M, Singh U, Guleria R, Jagannathan NR, Goswami R (2010) Effect of cholecalciferol and calcium supplementation on muscle strength and energy metabolism in vitamin D-deficient Asian Indians: a randomized, controlled trial. Clin Endocrinol (Oxf) 73:445–451
Zhu K, Austin N, Devine A, Bruce D, Prince RL (2010) A randomized controlled trial of the effects of vitamin D on muscle strength and mobility in older women with vitamin D insufficiency. J Am Geriatr Soc 58:2063–2068
Pfeifer M, Begerow B, Minne HW, Suppan K, Fahrleitner-Pammer A, Dobnig H (2009) Effects of a long-term vitamin D and calcium supplementation on falls and parameters of muscle function in community-dwelling older individuals. Osteoporos Int 20:315–322
Stockton KA, Mengersen K, Paratz JD, Kandiah D, Bennell KL (2011) Effect of vitamin D supplementation on muscle strength: a systematic review and meta-analysis. Osteoporos Int 22:859–871
Pilz S, Tomaschitz A, Drechsler C, Dekker JM, Marz W (2010) Vitamin D deficiency and myocardial diseases. Mol Nutr Food Res 54:1103–1113
Tishkoff DX, Nibbelink KA, Holmberg KH, Dandu L, Simpson RU (2008) Functional vitamin D receptor (VDR) in the t-tubules of cardiac myocytes: VDR knockout cardiomyocyte contractility. Endocrinology 149:558–564
Schleithoff SS, Zittermann A, Tenderich G, Berthold HK, Stehle P, Koerfer R (2006) Vitamin D supplementation improves cytokine profiles in patients with congestive heart failure: a double-blind, randomized, placebo-controlled trial. Am J Clin Nutr 83:754–759
Sugden JA, Davies JI, Witham MD, Morris AD, Struthers AD (2008) Vitamin D improves endothelial function in patients with type 2 diabetes mellitus and low vitamin D levels. Diabet Med 25:320–325
Pittas AG, Lau J, Hu FB, Dawson-Hughes B (2007) The role of vitamin D and calcium in type 2 diabetes. A systematic review and meta-analysis. J Clin Endocrinol Metab 92:2017–2029
Pilz S, Tomaschitz A, Ritz E, Pieber TR (2009) Vitamin D status and arterial hypertension: a systematic review. Nat Rev Cardiol 6:621–630
Giovannucci E, Liu Y, Hollis BW, Rimm EB (2008) 25-Hydroxyvitamin D and risk of myocardial infarction in men: a prospective study. Arch Intern Med 168:1174–1180
Dobnig H, Pilz S, Scharnagl H, Renner W, Seelhorst U, Wellnitz B, Kinkeldei J, Boehm BO, Weihrauch G, Maerz W (2008) Independent association of low serum 25-hydroxyvitamin d and 1,25-dihydroxyvitamin d levels with all-cause and cardiovascular mortality. Arch Intern Med 168:1340–1349
Pilz S, Dobnig H, Fischer JE, Wellnitz B, Seelhorst U, Boehm BO, Marz W (2008) Low vitamin d levels predict stroke in patients referred to coronary angiography. Stroke 39:2611–2613
Pilz S, Marz W, Wellnitz B, Seelhorst U, Fahrleitner-Pammer A, Dimai HP, Boehm BO, Dobnig H (2008) Association of vitamin D deficiency with heart failure and sudden cardiac death in a large cross-sectional study of patients referred for coronary angiography. J Clin Endocrinol Metab 93:3927–3935
Messenger W, Nielson CM, Li H, Beer T, Barrett-Connor E, Stone K, Shannon J (2012) Serum and dietary vitamin D and cardiovascular disease risk in elderly men: a prospective cohort study. Nutr Metab Cardiovasc Dis. doi:10.1016/j.numecd.2011.10.019
Jassal SK, Chonchol M, von Muhlen D, Smits G, Barrett-Connor E (2010) Vitamin d, parathyroid hormone, and cardiovascular mortality in older adults: the Rancho Bernardo study. Am J Med 123:1114–1120
Bhandari SK, Pashayan S, Liu IL, Rasgon SA, Kujubu DA, Tom TY, Sim JJ (2011) 25-Hydroxyvitamin D levels and hypertension rates. J Clin Hypertens (Greenwich) 13:170–177
Burgaz A, Orsini N, Larsson SC, Wolk A (2011) Blood 25-hydroxyvitamin D concentration and hypertension: a meta-analysis. J Hypertens 29:636–645
Pfeifer M, Begerow B, Minne HW, Nachtigall D, Hansen C (2001) Effects of a short-term vitamin D(3) and calcium supplementation on blood pressure and parathyroid hormone levels in elderly women. J Clin Endocrinol Metab 86:1633–1637
Margolis KL, Ray RM, Van Horn L et al (2008) Effect of calcium and vitamin D supplementation on blood pressure: the Women’s Health Initiative Randomized Trial. Hypertension 52:847–855
Witham MD, Nadir MA, Struthers AD (2009) Effect of vitamin D on blood pressure: a systematic review and meta-analysis. J Hypertens 27:1948–1954
Geleijnse JM (2011) Vitamin D and the prevention of hypertension and cardiovascular diseases: a review of the current evidence. Am J Hypertens 24:253–262
Mathieu C (2011) Vitamin D and immune system: getting it right. IBMS BoneKEY 8:178–186
Shoenfeld N, Amital H, Shoenfeld Y (2009) The effect of melanism and vitamin D synthesis on the incidence of autoimmune disease. Nat Clin Pract Rheumatol 5:99–105
Hughes DA, Norton R (2009) Vitamin D and respiratory health. Clin Exp Immunol 158:20–25
Hypponen E, Laara E, Reunanen A, Jarvelin MR, Virtanen SM (2001) Intake of vitamin D and risk of type 1 diabetes: a birth-cohort study. Lancet 358:1500–1503
Zipitis CS, Akobeng AK (2008) Vitamin D supplementation in early childhood and risk of type 1 diabetes: a systematic review and meta-analysis. Arch Dis Child 93:512–517
Brekke HK, Ludvigsson J (2007) Vitamin D supplementation and diabetes-related autoimmunity in the ABIS study. Pediatr Diabetes 8:11–14
Pierrot-Deseilligny C (2009) Clinical implications of a possible role of vitamin D in multiple sclerosis. J Neurol 256:1468–1479
Amital H, Szekanecz Z, Szucs G et al (2010) Serum concentrations of 25-OH vitamin D in patients with systemic lupus erythematosus (SLE) are inversely related to disease activity: is it time to routinely supplement patients with SLE with vitamin D? Ann Rheum Dis 69:1155–1157
Cutolo M, Otsa K, Uprus M, Paolino S, Seriolo B (2007) Vitamin D in rheumatoid arthritis. Autoimmun Rev 7:59–64
Merlino LA, Curtis J, Mikuls TR, Cerhan JR, Criswell LA, Saag KG (2004) Vitamin D intake is inversely associated with rheumatoid arthritis: results from the Iowa Women’s Health Study. Arthritis Rheum 50:72–77
Fleet JC (2008) Molecular actions of vitamin D contributing to cancer prevention. Mol Aspects Med 29:388–396
Tretli S, Hernes E, Berg JP, Hestvik UE, Robsahm TE (2009) Association between serum 25(OH)D and death from prostate cancer. Br J Cancer 100:450–454
Goodwin PJ, Ennis M, Pritchard KI, Koo J, Hood N (2009) Prognostic effects of 25-hydroxyvitamin D levels in early breast cancer. J Clin Oncol 27:3757–3763
International Agency for Research on Cancer (IARC) (2008) Vitamin D and Cancer. In Cancer IAfro (ed) IARC Working Group reports. World Health Organisation, Lyon, pp 1–221
Garland CF, Gorham ED, Mohr SB, Grant WB, Giovannucci EL, Lipkin M, Newmark H, Holick MF, Garland FC (2007) Vitamin D and prevention of breast cancer: pooled analysis. J Steroid Biochem Mol Biol 103:708–711
Abbas S, Linseisen J, Slanger T, Kropp S, Mutschelknauss EJ, Flesch-Janys D, Chang-Claude J (2008) Serum 25-hydroxyvitamin D and risk of post-menopausal breast cancer—results of a large case-control study. Carcinogenesis 29:93–99
Manson JE, Mayne ST, Clinton SK (2011) Vitamin D and prevention of cancer—ready for prime time? N Engl J Med 364:1385–1387
Lappe JM, Travers-Gustafson D, Davies KM, Recker RR, Heaney RP (2007) Vitamin D and calcium supplementation reduces cancer risk: results of a randomized trial. Am J Clin Nutr 85:1586–1591
Chlebowski RT, Johnson KC, Kooperberg C et al (2008) Calcium plus vitamin D supplementation and the risk of breast cancer. J Natl Cancer Inst 100:1581–1591
Helzlsouer KJ (2010) Overview of the Cohort Consortium Vitamin D Pooling Project of rarer cancers. Am J Epidemiol 172:4–9
Souberbielle JC, Body JJ, Lappe JM et al (2010) Vitamin D and musculoskeletal health, cardiovascular disease, autoimmunity and cancer: recommendations for clinical practice. Autoimmun Rev 9:709–715
Pazianas M, Cooper C, Ebetino FH, Russell RG (2010) Long-term treatment with bisphosphonates and their safety in postmenopausal osteoporosis. Ther Clin Risk Manag 6:325–343
Green JR, Rogers M (2002) Pharmacological profile of zoledronic acid: a highly potent inhibitor of bone resorption. Drug Dev Res 55:210–224
Papapoulos SE, Cremers SC (2007) Prolonged bisphosphonate release after treatment in children. N Engl J Med 356:1075–1076
McNicholl DM, Heaney LG (2010) The safety of bisphosphonate use in pre-menopausal women on corticosteroids. Curr Drug Saf 5:182–187
Thiebaud D, Sauty A, Burckhardt P, Leuenberger P, Sitzler L, Green JR, Kandra A, Zieschang J, Ibarra de Palacios P (1997) An in vitro and in vivo study of cytokines in the acute-phase response associated with bisphosphonates. Calcif Tissue Int 61:386–392
Sauty A, Pecherstorfer M, Zimmer-Roth I, Fioroni P, Juillerat L, Markert M, Ludwig H, Leuenberger P, Burckhardt P, Thiebaud D (1996) Interleukin-6 and tumor necrosis factor alpha levels after bisphosphonates treatment in vitro and in patients with malignancy. Bone 18:133–139
Hewitt RE, Lissina A, Green AE, Slay ES, Price DA, Sewell AK (2005) The bisphosphonate acute phase response: rapid and copious production of proinflammatory cytokines by peripheral blood gd T cells in response to aminobisphosphonates is inhibited by statins. Clin Exp Immunol 139:101–111
Miller PD, McClung MR, Macovei L et al (2005) Monthly oral ibandronate therapy in postmenopausal osteoporosis: 1-year results from the MOBILE study. J Bone Miner Res 20:1315–1322
Recker RR, Lewiecki EM, Miller PD, Reiffel J (2009) Safety of bisphosphonates in the treatment of osteoporosis. Am J Med 122:S22–32
Thompson K, Rogers MJ (2004) Statins prevent bisphosphonate-induced gamma, delta-T-cell proliferation and activation in vitro. J Bone Miner Res 19:278–288
Srivastava T, Haney CJ, Alon US (2009) Atorvastatin may have no effect on acute phase reaction in children after intravenous bisphosphonate infusion. J Bone Miner Res 24:334–337
Reid DM, Devogelaer JP, Saag K et al (2009) Zoledronic acid and risedronate in the prevention and treatment of glucocorticoid-induced osteoporosis (HORIZON): a multicentre, double-blind, double-dummy, randomised controlled trial. Lancet 373:1253–1263
Bertoldo F, Pancheri S, Zenari S, Boldini S, Giovanazzi B, Zanatta M, Valenti MT, Dalle Carbonare L, Lo Cascio V (2010) Serum 25-hydroxyvitamin D levels modulate the acute-phase response associated with the first nitrogen-containing bisphosphonate infusion. J Bone Miner Res 25:447–454
Bock O, Boerst H, Thomasius FE, Degner C, Stephan-Oelkers M, Valentine SM, Felsenberg D (2007) Common musculoskeletal adverse effects of oral treatment with once weekly alendronate and risedronate in patients with osteoporosis and ways for their prevention. J Musculoskelet Neuronal Interact 7:144–148
Black DM, Delmas PD, Eastell R et al (2007) Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. N Engl J Med 356:1809–1822
Caplan L, Pittman CB, Zeringue AL, Scherrer JF, Wehmeier KR, Cunningham FE, Eisen SA, McDonald JR (2010) An observational study of musculoskeletal pain among patients receiving bisphosphonate therapy. Mayo Clin Proc 85:341–348
Miller PD, Roux C, Boonen S, Barton IP, Dunlap LE, Burgio DE (2005) Safety and efficacy of risedronate in patients with age-related reduced renal function as estimated by the Cockcroft and Gault method: a pooled analysis of nine clinical trials. J Bone Miner Res 20:2105–2115
Jamal SA, Bauer DC, Ensrud KE, Cauley JA, Hochberg M, Ishani A, Cummings SR (2007) Alendronate treatment in women with normal to severely impaired renal function: an analysis of the fracture intervention trial. J Bone Miner Res 22:503–508
Toussaint ND, Elder GJ, Kerr PG (2009) Bisphosphonates in chronic kidney disease; balancing potential benefits and adverse effects on bone and soft tissue. Clin J Am Soc Nephrol 4:221–233
Fan SL, Almond MK, Ball E, Evans K, Cunningham J (2000) Pamidronate therapy as prevention of bone loss following renal transplantation. Kidney Int 57:684–690
Coco M, Glicklich D, Faugere MC et al (2003) Prevention of bone loss in renal transplant recipients: a prospective, randomized trial of intravenous pamidronate. J Am Soc Nephrol 14:2669–2676
Palmer SC, McGregor DO, Strippoli GF (2007) Interventions for preventing bone disease in kidney transplant recipients. Cochrane Database Syst Rev CD005015
Shiraishi N, Kitamura K, Miyoshi T et al (2006) Successful treatment of a patient with severe calcific uremic arteriolopathy (calciphylaxis) by etidronate disodium. Am J Kidney Dis 48:151–154
Monney P, Nguyen QV, Perroud H, Descombes E (2004) Rapid improvement of calciphylaxis after intravenous pamidronate therapy in a patient with chronic renal failure. Nephrol Dial Transplant 19:2130–2132
Body JJ (2006) The risk of cumulative renal effects of intravenous bisphosphonates. Support Cancer Ther 3:77–83
Bounameaux HM, Schifferli J, Montani JP, Jung A, Chatelanat F (1983) Renal failure associated with intravenous diphosphonates. Lancet 1:471
Ibrahim A, Scher N, Williams G et al (2003) Approval summary for zoledronic acid for treatment of multiple myeloma and cancer bone metastases. Clin Cancer Res 9:2394–2399
Miller PD (2011) The kidney and bisphosphonates. Bone 49:77–81
Rey J, Daumen-Legre V, Pham T, Bernard P, Dahan L, Acquaviva PC, Lafforgue P (2000) Uveitis, an under-recognized adverse effect of pamidronate. Case report and literature review. Joint Bone Spine 67:337–340
Malik AR, Campbell SH, Toma NM (2002) Bilateral acute anterior uveitis after alendronate. Br J Ophthalmol 86:1443
Durnian JM, Olujohungbe A, Kyle G (2005) Bilateral acute uveitis and conjunctivitis after zoledronic acid therapy. Eye (Lond) 19:221–222
Fietta P, Manganelli P, Lodigiani L (2003) Clodronate induced uveitis. Ann Rheum Dis 62:378
Fraunfelder FW, Fraunfelder FT, Jensvold B (2003) Scleritis and other ocular side effects associated with pamidronate disodium. Am J Ophthalmol 135:219–222
Lufkin EG, Argueta R, Whitaker MD, Cameron AL, Wong VH, Egan KS, O’Fallon WM, Riggs BL (1994) Pamidronate: an unrecognized problem in gastrointestinal tolerability. Osteoporos Int 4:320–322
de Groen PC, Lubbe DF, Hirsch LJ, Daifotis A, Stephenson W, Freedholm D, Pryor-Tillotson S, Seleznick MJ, Pinkas H, Wang KK (1996) Esophagitis associated with the use of alendronate. N Engl J Med 335:1016–1021
Cryer B, Miller P, Petruschke RA, Chen E, Geba GP, Papp AE (2005) Upper gastrointestinal tolerability of once weekly alendronate 70 mg with concomitant non-steroidal anti-inflammatory drug use. Aliment Pharmacol Ther 21:599–607
Greenspan S, Field-Munves E, Tonino R, Smith M, Petruschke R, Wang L, Yates J, de Papp AE, Palmisano J (2002) Tolerability of once-weekly alendronate in patients with osteoporosis: a randomized, double-blind, placebo-controlled study. Mayo Clin Proc 77:1044–1052
Eisman JA, Rizzoli R, Roman-Ivorra J, Lipschitz S, Verbruggen N, Gaines KA, Melton ME (2004) Upper gastrointestinal and overall tolerability of alendronate once weekly in patients with osteoporosis: results of a randomized, double-blind, placebo-controlled study. Curr Med Res Opin 20:699–705
Bobba RS, Beattie K, Parkinson B, Kumbhare D, Adachi JD (2006) Tolerability of different dosing regimens of bisphosphonates for the treatment of osteoporosis and malignant bone disease. Drug Saf 29:1133–1152
Cadarette SM, Katz JN, Brookhart MA, Sturmer T, Stedman MR, Levin R, Solomon DH (2009) Comparative gastrointestinal safety of weekly oral bisphosphonates. Osteoporos Int 20:1735–1747
Rosen CJ, Hochberg MC, Bonnick SL et al (2005) Treatment with once-weekly alendronate 70 mg compared with once-weekly risedronate 35 mg in women with postmenopausal osteoporosis: a randomized double-blind study. J Bone Miner Res 20:141–151
Vestergaard P, Schwartz K, Pinholt EM, Rejnmark L, Mosekilde L (2010) Gastric and esophagus events before and during treatment of osteoporosis. Calcif Tissue Int 86:110–115
Cryer B, Bauer DC (2002) Oral bisphosphonates and upper gastrointestinal tract problems: what is the evidence? Mayo Clin Proc 77:1031–1043
Green J, Czanner G, Reeves G, Watson J, Wise L, Beral V (2010) Oral bisphosphonates and risk of cancer of oesophagus, stomach, and colorectum: case–control analysis within a UK primary care cohort. BMJ 341:c4444
Cardwell CR, Abnet CC, Cantwell MM, Murray LJ (2010) Exposure to oral bisphosphonates and risk of esophageal cancer. JAMA 304:657–663
Nguyen DM, Schwartz J, Richardson P, El-Serag HB (2010) Oral bisphosphonate prescriptions and the risk of esophageal adenocarcinoma in patients with Barrett’s esophagus. Dig Dis Sci 55:3404–3407
Lyles KW, Colon-Emeric CS, Magaziner JS et al (2007) Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med 357:1799–1809
Cummings SR, Schwartz AV, Black DM (2007) Alendronate and atrial fibrillation. N Engl J Med 356:1895–1896
Karam R, Camm J, McClung M (2007) Yearly zoledronic acid in postmenopausal osteoporosis. N Engl J Med 357:712–713, author reply 714-715
Lewiecki EM, Cooper C, Thompson E, Hartl F, Mehta D, Papapoulos SE (2010) Ibandronate does not increase risk of atrial fibrillation in analysis of pivotal clinical trials. Int J Clin Pract 64:821–826
Varma R, Aronow WS, Basis Y, Singh T, Kalapatapu K, Weiss MB, Pucillo AL, Monsen CE (2008) Relation of bone mineral density to frequency of coronary heart disease. Am J Cardiol 101:1103–1104
Choi SH, An JH, Lim S et al (2009) Lower bone mineral density is associated with higher coronary calcification and coronary plaque burdens by multidetector row coronary computed tomography in pre- and postmenopausal women. Clin Endocrinol (Oxf) 71:644–651
Eriksen EF, Lyles KW, Colon-Emeric CS et al (2009) Antifracture efficacy and reduction of mortality in relation to timing of the first dose of zoledronic acid after hip fracture. J Bone Miner Res 24:1308–1313
McCloskey EV, Yates AJ, Beneton MN, Galloway J, Harris S, Kanis JA (1987) Comparative effects of intravenous diphosphonates on calcium and skeletal metabolism in man. Bone 8(Suppl 1):S35–41
Brinkmeier T, Kugler K, Lepoittevin JP, Frosch PJ (2007) Adverse cutaneous drug reaction to alendronate. Contact Dermatitis 57:123–125
Krasagakis K, Kruger-Krasagakis S, Ioannidou D, Tosca A (2004) Chronic erosive and ulcerative oral lesions caused by incorrect administration of alendronate. J Am Acad Dermatol 50:651–652
Yanik B, Turkay C, Atalar H (2007) Hepatotoxicity induced by alendronate therapy. Osteoporos Int 18:829–831
Phillips MB (2007) Risedronate-induced hepatitis. Am J Med 120:e1–2
Coleman R, Cook R, Hirsh V, Major P, Lipton A (2011) Zoledronic acid use in cancer patients: more than just supportive care? Cancer 117:11–23
Gnant M, Clezardin P (2012) Direct and indirect anticancer activity of bisphosphonates: a brief review of published literature. Cancer Treat Rev (in press)
Normanno N, De Luca A, Gallo M, Lamura L, Perrone F (2011) Zoledronic acid in early-stage breast cancer. Lancet Oncol 12:991
Coleman RE, Marshall H, Cameron D et al (2011) Breast-cancer adjuvant therapy with zoledronic acid. N Engl J Med 365:1396–1405
Gnant M (2011) Zoledronic acid in breast cancer: latest findings and interpretations. Ther Adv Med Oncol 3:293–301
Zhao X, Xu X, Zhang Q, Jia Z, Sun S, Zhang J, Wang B, Wang Z, Hu X (2011) Prognostic and predictive value of clinical and biochemical factors in breast cancer patients with bone metastases receiving “metronomic” zoledronic acid. BMC Cancer 11:403
Chlebowski RT, Col N (2011) Bisphosphonates and breast cancer prevention. Anticancer Agents Med Chem
Rennert G, Pinchev M, Rennert HS, Gruber SB (2011) Use of bisphosphonates and reduced risk of colorectal cancer. J Clin Oncol 29:1146–1150
Sassa S, Okabe H, Nemoto N, Kikuchi H, Kudo H, Sakamoto S (2009) Ibandronate may prevent colorectal carcinogenesis in mice with ulcerative colitis. Anticancer Res 29:4615–4619
Kahn MF, Chamot AM (1992) SAPHO syndrome. Rheum Dis Clin North Am 18:225–246
Kopterides P, Pikazis D, Koufos C (2004) Successful treatment of SAPHO syndrome with zoledronic acid. Arthritis Rheum 50:2970–2973
Siau K, Laversuch CJ (2010) SAPHO syndrome in an adult with ulcerative colitis responsive to intravenous pamidronate: a case report and review of the literature. Rheumatol Int 30:1085–1088
Codriansky KA, Runger TM, Bhawan J, Kantarci A, Kissin EY (2008) Multicentric reticulohistiocytosis: a systemic osteoclastic disease? Arthritis Rheum 59:444–448
King MM, Nelson DA (2008) Hypertrophic osteoarthropathy effectively treated with zoledronic acid. Clin Lung Cancer 9:179–182
Schonfeld SE (2010) Strategies for managing periodontal inflammation. J Calif Dent Assoc 38:272–283
Braithwaite RS, Chlebowski RT, Lau J, George S, Hess R, Col NF (2003) Meta-analysis of vascular and neoplastic events associated with tamoxifen. J Gen Intern Med 18:937–947
Bushnell CD, Goldstein LB (2004) Risk of ischemic stroke with tamoxifen treatment for breast cancer: a meta-analysis. Neurology 63:1230–1233
Mosca L, Grady D, Barrett-Connor E et al (2009) Effect of raloxifene on stroke and venous thromboembolism according to subgroups in postmenopausal women at increased risk of coronary heart disease. Stroke 40:147–155
Cummings SR, Ensrud K, Delmas PD et al (2010) Lasofoxifene in postmenopausal women with osteoporosis. N Engl J Med 362:686–696
Cuzick J, Forbes JF, Sestak I, Cawthorn S, Hamed H, Holli K, Howell A (2007) Long-term results of tamoxifen prophylaxis for breast cancer—96-month follow-up of the randomized IBIS-I trial. J Natl Cancer Inst 99:272–282
Collins P, Mosca L, Geiger MJ, Grady D, Kornitzer M, Amewou-Atisso MG, Effron MB, Dowsett SA, Barrett-Connor E, Wenger NK (2009) Effects of the selective estrogen receptor modulator raloxifene on coronary outcomes in the Raloxifene Use for The Heart trial: results of subgroup analyses by age and other factors. Circulation 119:922–930
Adomaityte J, Farooq M, Qayyum R (2008) Effect of raloxifene therapy on venous thromboembolism in postmenopausal women. A meta-analysis Thromb Haemost 99:338–342
Grady D, Ettinger B, Moscarelli E, Plouffe L Jr, Sarkar S, Ciaccia A, Cummings S (2004) Safety and adverse effects associated with raloxifene: multiple outcomes of raloxifene evaluation. Obstet Gynecol 104:837–844
Duvernoy CS, Yeo AA, Wong M, Cox DA, Kim HM (2010) Antiplatelet therapy use and the risk of venous thromboembolic events in the Raloxifene Use for the Heart (RUTH) trial. J Womens Health 19:1459–1465
Ensrud K, LaCroix A, Thompson JR et al (2010) Lasofoxifene and cardiovascular events in postmenopausal women with osteoporosis: five-year results from the Postmenopausal Evaluation and Risk Reduction with Lasofoxifene (PEARL) trial. Circulation 122:1716–1724
Barrett-Connor E, Cauley JA, Kulkarni PM, Sashegyi A, Cox DA, Geiger MJ (2004) Risk–benefit profile for raloxifene: 4-year data From the Multiple Outcomes of Raloxifene Evaluation (MORE) randomized trial. J Bone Miner Res 19:1270–1275
Grady D, Cauley JA, Stock JL, Cox DA, Mitlak BH, Song J, Cummings SR (2010) Effect of raloxifene on all-cause mortality. Am J Med 123(469):e461–467
Vogel VG, Costantino JP, Wickerham DL et al (2010) Update of the National Surgical Adjuvant Breast and Bowel Project Study of Tamoxifen and Raloxifene (STAR) P-2 Trial: preventing breast cancer. Cancer Prev Res (Phila) 3:696–706
Martino S, Cauley JA, Barrett-Connor E, Powles TJ, Mershon J, Disch D, Secrest RJ, Cummings SR (2004) Continuing outcomes relevant to Evista: breast cancer incidence in postmenopausal osteoporotic women in a randomized trial of raloxifene. J Natl Cancer Inst 96:1751–1761
Vogel VG, Qu Y, Wong M, Mitchell B, Mershon JL (2009) Incidence of invasive breast cancer in postmenopausal women after discontinuation of long-term raloxifene administration. Clin Breast Cancer 9:45–50
Grady D, Cauley JA, Geiger MJ, Kornitzer M, Mosca L, Collins P, Wenger NK, Song J, Mershon J, Barrett-Connor E (2008) Reduced incidence of invasive breast cancer with raloxifene among women at increased coronary risk. J Natl Cancer Inst 100:854–861
LaCroix AZ, Powles T, Osborne CK et al (2010) Breast cancer incidence in the randomized PEARL trial of lasofoxifene in postmenopausal osteoporotic women. J Natl Cancer Inst 102:1706–1715
Palacios S, Farias ML, Luebbert H et al (2004) Raloxifene is not associated with biologically relevant changes in hot flushes in postmenopausal women for whom therapy is appropriate. Am J Obstet Gynecol 191:121–131
Gordon S, Walsh BW, Ciaccia AV, Siddhanti S, Rosen AS, Plouffe L Jr (2004) Transition from estrogen–progestin to raloxifene in postmenopausal women: effect on vasomotor symptoms. Obstet Gynecol 103:267–273
Davis SR, O’Neill SM, Eden J, Baber R, Ekangaki A, Stocks JM, Thiebaud D (2004) Transition from estrogen therapy to raloxifene in postmenopausal women: effects on treatment satisfaction and the endometrium—a pilot study. Menopause 11:167–175
Utian W, Yu H, Bobula J, Mirkin S, Olivier S, Pickar JH (2009) Bazedoxifene/conjugated estrogens and quality of life in postmenopausal women. Maturitas 63:329–335
Marie PJ, Felsenberg D, Brandi ML (2010) How strontium ranelate, via opposite effects on bone resorption and formation, prevents osteoporosis. Osteoporos Int 22:1659–1667
Henrotin Y, Labasse A, Zheng SX, Galais P, Tsouderos Y, Crielaard JM, Reginster JY (2001) Strontium ranelate increases cartilage matrix formation. J Bone Miner Res 16:299–308
Alexandersen P, Karsdal MA, Qvist P, Reginster JY, Christiansen C (2007) Strontium ranelate reduces the urinary level of cartilage degradation biomarker CTX-II in postmenopausal women. Bone 40:218–222
Alexandersen P, Karsdal MA, Byrjalsen I, Christiansen C (2011) Strontium ranelate effect in postmenopausal women with different clinical levels of osteoarthritis. Climacteric 14:236–243
Bruyere O, Delferriere D, Roux C et al (2008) Effects of strontium ranelate on spinal osteoarthritis progression. Ann Rheum Dis 67:335–339
European Medicines Agency (2009) Strontium ranelate. Summary of product characteristics, 3 June 2010. European Medicines Agency, London
Naess IA, Christiansen SC, Romundstad P, Cannegieter SC, Rosendaal FR, Hammerstrom J (2007) Incidence and mortality of venous thrombosis: a population-based study. J Thromb Haemost 5:692–699
Oger E (2000) Incidence of venous thromboembolism: a community-based study in Western France. EPI-GETBP Study Group Groupe d’Etude de la Thrombose de Bretagne Occidentale Thromb Haemost 83:657–660
Silverstein MD, Heit JA, Mohr DN, Petterson TM, O’Fallon WM, Melton LJ 3rd (1998) Trends in the incidence of deep vein thrombosis and pulmonary embolism: a 25-year population-based study. Arch Intern Med 158:585–593
Breart G, Cooper C, Meyer O, Speirs C, Deltour N, Reginster JY (2010) Osteoporosis and venous thromboembolism: a retrospective cohort study in the UK General Practice Research Database. Osteoporos Int 21:1181–1187
Osborne V, Layton D, Perrio M, Wilton L, Shakir SA (2010) Incidence of venous thromboembolism in users of strontium ranelate: an analysis of data from a prescription-event monitoring study in England. Drug Saf 33:579–591
Breart G, Jakob FJ, Palacios S et al (2010) New interim analysis of a prospective observational cohort study of patients treated with strontium ranelate. Osteoporos Int S 1:S166
Jonville-Bera AP, Crickx B, Aaron L, Hartingh I, Autret-Leca E (2009) Strontium ranelate-induced DRESS syndrome: first two case reports. Allergy 64:658–659
Lee HY, Lie D, Lim KS, Thirumoorthy T, Pang SM (2009) Strontium ranelate-induced toxic epidermal necrolysis in a patient with post-menopausal osteoporosis. Osteoporos Int 20:161–162
Pernicova I, Middleton ET, Aye M (2008) Rash, strontium ranelate and DRESS syndrome put into perspective. European Medicine Agency on the alert Osteoporos Int 19:1811–1812
Musette P, Brandi ML, Cacoub P, Kaufman JM, Rizzoli R, Reginster JY (2010) Treatment of osteoporosis: recognizing and managing cutaneous adverse reactions and drug-induced hypersensitivity. Osteoporos Int 21:723–732
Kong YY, Yoshida H, Sarosi I et al (1999) OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 397:315–323
Baud’huin M, Lamoureux F, Duplomb L, Redini F, Heymann D (2007) RANKL, RANK, osteoprotegerin: key partners of osteoimmunology and vascular diseases. Cell Mol Life Sci 64:2334–2350
Ferrari-Lacraz S, Ferrari S (2011) Do RANKL inhibitors (denosumab) affect inflammation and immunity? Osteoporos Int 22:435–446
Sobacchi C, Frattini A, Guerrini MM et al (2007) Osteoclast-poor human osteopetrosis due to mutations in the gene encoding RANKL. Nat Genet 39:960–962
Ashcroft AJ, Cruickshank SM, Croucher PI et al (2003) Colonic dendritic cells, intestinal inflammation, and T cell-mediated bone destruction are modulated by recombinant osteoprotegerin. Immunity 19:849–861
Cohen SB, Dore RK, Lane NE, Ory PA, Peterfy CG, Sharp JT, van der Heijde D, Zhou L, Tsuji W, Newmark R (2008) Denosumab treatment effects on structural damage, bone mineral density, and bone turnover in rheumatoid arthritis: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, phase II clinical trial. Arthritis Rheum 58:1299–1309
Andrews NA (2008) Denosumab and the treatment of rheumatoid arthritis: in an occupied field, where will a RANKL inhibitor fit in? Bone Key 5:351–356
Stolina M, Guo J, Faggioni R, Brown H, Senaldi G (2003) Regulatory effects of osteoprotegerin on cellular and humoral immune responses. Clin Immunol 109:347–354
Miller RE, Branstetter D, Armstrong A, Kennedy B, Jones J, Cowan L, Bussiere J, Dougall WC (2007) Receptor activator of NF-kappa B ligand inhibition suppresses bone resorption and hypercalcemia but does not affect host immune responses to influenza infection. J Immunol 179:266–274
McClung MR, Lewiecki EM, Cohen SB et al (2006) Denosumab in postmenopausal women with low bone mineral density. N Engl J Med 354:821–831
Cummings SR, San Martin J, McClung MR et al (2009) Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med 361:756–765
Kendler DL, Roux C, Benhamou CL, Brown JP, Lillestol M, Siddhanti S, Man HS, San Martin J, Bone HG (2010) Effects of denosumab on bone mineral density and bone turnover in postmenopausal women transitioning from alendronate therapy. J Bone Miner Res 25:72–81
Brown JP, Prince RL, Deal C et al (2009) Comparison of the effect of denosumab and alendronate on BMD and biochemical markers of bone turnover in postmenopausal women with low bone mass: a randomized, blinded, phase 3 trial. J Bone Miner Res 24:153–161
Miller PD, Wagman RB, Peacock M, Lewiecki EM, Bolognese MA, Weinstein RL, Ding B, San Martin J, McClung MR (2011) Effect of denosumab on bone mineral density and biochemical markers of bone turnover: six-year results of a phase 2 clinical trial. J Clin Endocrinol Metab 96:394–402
Bucay N, Sarosi I, Dunstan CR et al (1998) Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev 12:1260–1268
Ziegler S, Kudlacek S, Luger A, Minar E (2005) Osteoprotegerin plasma concentrations correlate with severity of peripheral artery disease. Atherosclerosis 182:175–180
Mesquita M, Demulder A, Damry N, Melot C, Wittersheim E, Willems D, Dratwa M, Bergmann P (2009) Plasma osteoprotegerin is an independent risk factor for mortality and an early biomarker of coronary vascular calcification in chronic kidney disease. Clin Chem Lab Med 47:339–346
Kobayashi-Sakamoto M, Hirose K, Isogai E, Chiba I (2004) NF-kappaB-dependent induction of osteoprotegerin by Porphyromonas gingivalis in endothelial cells. Biochem Biophys Res Commun 315:107–112
Vik A, Mathiesen EB, Noto AT, Sveinbjornsson B, Brox J, Hansen JB (2007) Serum osteoprotegerin is inversely associated with carotid plaque echogenicity in humans. Atherosclerosis 191:128–134
Helas S, Goettsch C, Schoppet M, Zeitz U, Hempel U, Morawietz H, Kostenuik PJ, Erben RG, Hofbauer LC (2009) Inhibition of receptor activator of NF-kappaB ligand by denosumab attenuates vascular calcium deposition in mice. Am J Pathol 175:473–478
Hodsman AB, Bauer DC, Dempster DW et al (2005) Parathyroid hormone and teriparatide for the treatment of osteoporosis: a review of the evidence and suggested guidelines for its use. Endocr Rev 26:688–703
Neer RM, Arnaud CD, Zanchetta JR et al (2001) Effect of parathyroid hormone (1–34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med 344:1434–1441
Hodsman AB, Hanley DA, Ettinger MP, Bolognese MA, Fox J, Metcalfe AJ, Lindsay R (2003) Efficacy and safety of human parathyroid hormone-(1-84) in increasing bone mineral density in postmenopausal osteoporosis. J Clin Endocrinol Metab 88:5212–5220
Antoniucci DM, Sellmeyer DE, Bilezikian JP, Palermo L, Ensrud KE, Greenspan SL, Black DM (2007) Elevations in serum and urinary calcium with parathyroid hormone (1-84) with and without alendronate for osteoporosis. J Clin Endocrinol Metab 92:942–947
Winer KK, Sinaii N, Reynolds J, Peterson D, Dowdy K, Cutler GB Jr (2010) Long-term treatment of 12 children with chronic hypoparathyroidism: a randomized trial comparing synthetic human parathyroid hormone 1–34 versus calcitriol and calcium. J Clin Endocrinol Metab 95:2680–2688
Winer KK, Yanovski JA, Sarani B, Cutler GB Jr (1998) A randomized, cross-over trial of once-daily versus twice-daily parathyroid hormone 1–34 in treatment of hypoparathyroidism. J Clin Endocrinol Metab 83:3480–3486
Conflicts of interest
Jean-Jacques Body has received speaker and consultant fees from Amgen and Novartis and research support from Amgen, Daiichi Sankyo, GlaxoSmithKline, Merck Sharp & Dohme, Novartis, Nycomed, Servier and SMB.
Pierre Bergmann has received speaker fees from Servier and Roche.
Steven Boonen has received consulting fees and/or research support from Amgen, Merck, Novartis and Servier
Jean-Pierre Devogelaer has no conflict of interest.
Evelien Gielen has no conflict of interest.
Stephan Goemaere has received speakers fees and/or research support from Amgen, Daiichi Sankyo, Eli Lilly, Glaxo Smith Kline, Merck Sharp & Dohme, Novartis, Nycomed, Warner Chillcott, Sanofi-Aventis, Servier and Roche.
Jean-Marc Kaufman has received consulting fees, paid advisory boards, lecture fees and/or grant support from Amgen, Eli Lilly, Glaxo Smith Kline, Merck, Novartis, Procter & Gamble, Roche, Sanofi Aventis, Servier and Warner Chilcott.
Serge Rozenberg has received speakers or/and consultant fees from Amgen, Merck Sharp & Dohme and Pfizer.
Jean-Yves Reginster on behalf of the Department of Public Health, Epidemiology and Health Economics of the University of Liège, Liège, Belgium has received consulting fees or paid advisory boards from Servier, Novartis, Negma, Lilly, Wyeth, Amgen, GlaxoSmithKline, Roche, Merckle, Nycomed, NPS, Theramex and UCB; lecture fees when speaking at the invitation of a commercial sponsor from Merck Sharp and Dohme, Lilly, Rottapharm, IBSA, Genevrier, Novartis, Servier, Roche, GlaxoSmithKline, Teijin, Teva, Ebewee Pharma, Zodiac, Analis, Theramex, Nycomed and Novo-Nordisk and grant support from industries Bristol Myers Squibb, Merck Sharp & Dohme, Rottapharm, Teva, Lilly, Novartis, Roche, GlaxoSmithKline and Amgen, Servier.
Funding
This supplement was not sponsored by any outside commercial interests. It was funded entirely by the Belgian Bone Club, a non-profit scientific organisation.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Body, JJ., Bergmann, P., Boonen, S. et al. Extraskeletal benefits and risks of calcium, vitamin D and anti-osteoporosis medications. Osteoporos Int 23 (Suppl 1), 1–23 (2012). https://doi.org/10.1007/s00198-011-1891-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-011-1891-8