
Abstract
Purpose
Cellular immune dysfunctions, which are common in intensive care patients, predict a number of significant complications. In order to effectively target treatments, clinically applicable measures need to be developed to detect dysfunction. The objective was to confirm the ability of cellular markers associated with immune dysfunction to stratify risk of secondary infection in critically ill patients.
Methods
Multi-centre, prospective observational cohort study of critically ill patients in four UK intensive care units. Serial blood samples were taken, and three cell surface markers associated with immune cell dysfunction [neutrophil CD88, monocyte human leucocyte antigen-DR (HLA-DR) and percentage of regulatory T cells (Tregs)] were assayed on-site using standardized flow cytometric measures. Patients were followed up for the development of secondary infections.
Results
A total of 148 patients were recruited, with data available from 138. Reduced neutrophil CD88, reduced monocyte HLA-DR and elevated proportions of Tregs were all associated with subsequent development of infection with odds ratios (95% CI) of 2.18 (1.00–4.74), 3.44 (1.58–7.47) and 2.41 (1.14–5.11), respectively. Burden of immune dysfunction predicted a progressive increase in risk of infection, from 14% for patients with no dysfunction to 59% for patients with dysfunction of all three markers. The tests failed to risk stratify patients shortly after ICU admission but were effective between days 3 and 9.
Conclusions
This study confirms our previous findings that three cell surface markers can predict risk of subsequent secondary infection, demonstrates the feasibility of standardized multisite flow cytometry and presents a tool which can be used to target future immunomodulatory therapies.
Trial registration
The study was registered with clinicaltrials.gov (NCT02186522).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
ΓÇÿSecondary infections are a major concern in intensive care and have been convincingly associated with immune dysfunction occurring as a result of critical illnesses. Effective targeting of immunomodulatory therapies requires the ability to identify patients who are at risk, and this study presents three immune cell markers which additively predict the risk of subsequent secondary infectionΓÇÖ |
Introduction
Critical illness occurs when a sterile [1, 2] or infective [3] insult leads to organ dysfunction [4]. A hallmark of critical illness is systemic inflammation and immune-mediated organ damage [5]. However it is increasingly clear that this immune activation is accompanied by an equally pronounced immunodepression [6], and that a persistence of this immune duality is associated with a complicated intensive care unit (ICU) course [7]. If effective treatments for this complex state of immune dysfunction are to be developed, we need to identify clinical and laboratory markers for it.
A significant complication in intensive care is the development of nosocomial infection, which occurs in 20–40% of patients [8]. The development of such infections is relevant to patients and the healthcare system, as they are associated with increased morbidity, mortality, prolonged need for organ support, and greater overall illness costs [8]. ICU-acquired infections are also a major driver of antibiotic use, which contributes to increased antimicrobial resistance and the risk of drug toxicity [9]. We, and others, have previously demonstrated a strong association between immune dysfunction and the subsequent development of nosocomial infection [1, 3].
We previously published the findings of a single-centre study examining three markers associated with immune dysfunction [3], namely neutrophil CD88 (as a marker of C5a-mediated neutrophil dysfunction [10, 11]), monocyte human leucocyte antigen-DR (HLA-DR) (as a marker of monocyte deactivation [12, 13]) and the proportion of regulatory T cells (Tregs, associated with an increased risk of secondary infection [14]). These three markers independently, and additively, predicted the subsequent development of nosocomial infection [3].
The results of single-centre studies can be hard to replicate, but development of clinically useable measures of immune function to guide trials of immunomodulatory therapy in critical illness [15] requires accurate, reproducible immunophenotyping tools. The aim of the ImmuNe FailurE in Critical Therapy study was to confirm our previously identified markers associated with immune dysfunction, and their ability to identify risk of subsequent nosocomial infection in a multicentre study, using standardized flow cytometry. Some of the results of these studies have been previously reported in the form of an abstract [16].
Methods
The study protocol has been published [17]. The study was approved by National Health Service Research Ethics Committees and was performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
Setting and participants
Patients were recruited from the four participating ICUs, based in the UK. Details of the recruiting units are shown in the supplemental section (Table S1). Inclusion criteria were adult patients admitted to ICU and receiving either invasive mechanical ventilation or two or more other organ systems support, who were predicted to remain in ICU for at least 48 h [17]. Written, informed assent was obtained from the nearest relative or personal consultee, and retrospective consent was sought from all patients who regained capacity. A full list of exclusion criteria is in the supplemental section.
Sampling schedule
Tripotassium ethylenediaminetetraacetate (K3-EDTA) anticoagulated blood samples were taken on the day of study enrolment, and then at 2-day intervals until day 12 with patients being followed for determination of infection to day 16. Patients discharged to hospital wards remained in the study. Patients discharged home during the study period were followed up using hospital records to ascertain re-admission with infection.
Definition of infection
We used methods developed in our previous study to identify infection [3, 17]. Briefly, researchers blinded to the immune phenotyping data assessed patients for the development of infections which were defined using the Hospitals in Europe Link for Infection Control through Surveillance criteria [18] (see supplemental methods). Suspected infections that did not meet the criteria for confirmed infection underwent independent, blinded review by two expert intensive care clinicians (see supplemental methods). The onset of infection was defined as the time the relevant microbiology sample was taken. Only the first infection was defined as the outcome of interest.
Flow cytometric standardization and sample staining
All sites used the same flow cytometry analyser, the FACS Canto II (BD Biosciences, San Jose, CA, USA), and antibodies from the same batch, using study standard operating procedures. Machines were standardized by monthly matching of target values using a common batch of Cytometry Setup and Tracking (CS&T) beads (BDB), and daily internal quality control using the same common batch of CS&T beads. Details of the fluorophores and antibodies are reported in Table S2.
Immunophenotyping measures
The primary measures were those used in the published study [3], namely surface neutrophil CD88 (nCD88, as a marker associated with C5a-mediated neutrophil dysfunction [10, 11]), monocyte HLA-DR (mHLA-DR, as a marker associated with monocyte deactivation [19]) and the proportion of CD4 T cells expressing a regulatory phenotype (CD4+/CD25++/CD127− Tregs) [3, 20]. These were compared against simple enumeration of leucocyte populations and lymphocyte to neutrophil ratios which have previously been reported to be predictive of subsequent infection [21, 22].
A study of the reliability and reproducibility of the measures of cell surface markers was conducted as set out in the published protocol [17] (see supplemental section).
Defining cut-off for ‘dysfunction’
Changes in antibody clones, fluorophores and flow cytometers necessitated by the multicentre standardization process meant we were unable to directly compare the marker values from the current study and the previous study [3]. Therefore, we re-derived optimum cut-off values for cell-surface markers using receiver operating characteristic (ROC) curve analysis, defining ‘dysfunction’ and ‘no dysfunction’ using the same method [3]. Briefly, patients were categorized using values from the sample taken most proximally to the relevant clinical end-point (death, infection or discharge without infection). For patients who acquired an infection, the sample used was the one taken closest to but preceding the 48-h time point before the infection. This was chosen to identify markers that changed prior to infection developing.
Statistical analysis
Statistical analysis followed the statistical analysis plan that was finalized prior to database lock (see supplemental section). The sample size calculation is set out in the protocol paper [17]. Assuming a 35% nosocomial infection rate [3], to estimate a 50% positive predictive value with 95% confidence interval width of 39–61% would require 200 patients. Optimum cut-offs were defined by constructing ROC curves comparing values from samples 48 h before an infection with samples from patients who did not develop infection, using Youden’s index [23]. Post hoc analyses to adjust for clinical and demographic predictors of infection were undertaken; these were not prespecified in the analysis plan but were included in response to independent review.
Modelling potential clinical use
In a pre-planned evaluation of the clinical potential for these markers to alter decision-making [17], we examined the ability of the immune dysfunction measurements to predict subsequent infection, ICU length of stay and duration of organ support. We used the optimum cut-offs from ROC analysis and calculated their predictive ability when measured at baseline, and days 2–4, 6–8 and 10–12 post study enrolment. We chose these time points as clinically relevant times at which the immune phenotyping might be undertaken and operationalised in routine care. We modelled predictive performance with a two-step process: first, using the best-performing test (monocyte HLA-DR) to select patients with low HLA-DR; and second, then classifying patients with either low neutrophil CD88 or elevated Tregs (or both) as ‘highest risk’.
Results
Recruitment and development of infection
We recruited 148 patients. Time and budgetary restraints limited recruitment of the predefined numbers. Supplemental Fig. S1 shows the recruitment diagram. A total of 138 patients were available for analysis. Patients were enrolled a median 1.5 days (IQR 0.7–2 days) after admission to ICU. Demographic and clinical data are shown in Table 1.
In total 51 (37%) patients developed secondary infections after study entry; 34 (67%) met the criteria for confirmed infection whilst 17 (33%) were deemed “highly likely” on expert review. Infection sites, organisms and relationship to primary infection are summarised in supplemental Table S3. Infection occurred a median 7 days (IQR 3–11) after ICU admission. In nine patients, infection developed within 48 h of study entry; these patients were excluded from the ROC analysis, but included in a sensitivity analysis.
Association of markers of immune dysfunction with subsequent infection
The three tests all demonstrated high intra- and inter-rater reliability (see supplemental results section).
Reduced nCD88, reduced mHLA-DR and elevated proportions of Tregs were all associated with subsequent development of infection when applying optimal ROC cut-offs (Table 2); odds ratios (95% CI) were 2.18 (1.00–4.74), 3.44 (1.58–7.47) and 2.41 (1.14–5.11), respectively. Area under the ROC curve for each marker is reported in the supplemental section (Table S4). None of the clinical or demographic variables were significant independent predictors of subsequent infection. Adjustment for potential confounding by these variables did not result in significant changes to the unadjusted odds ratios (Table S5). Values plotted for each marker dichotomised by group (did versus did not develop infection) on the different days before infection events occurred showed that changes in CD88 and HLA-DR tended to occur 2–3 days prior to the events (supplemental results Fig. S2a, b).
Marker performance in survival analysis
In survival analysis, all three measures showed a significant association with the hazard of infection (Fig. 1a–c). As for the odds ratios, adjustment for clinical/demographic variables did not result in significant changes to the hazard ratios (see Table S5).

Survival curves for patients dichotomised by markers at the cut-offs shown. a Neutrophil CD88 expression, b total monocyte HLA-DR expression, c Tregs as a percentage of all CD4+ cells. d Additive combination of markers p value by log-rank test (panels a–c) and log-rank test for trend (panel d). Hazard ratios for combined markers are shown in Table 3
Baseline marker values did not predict subsequent infection, with or without adjustment for clinical variables (supplemental Table S6).
Additional and sensitivity analyses
Sensitivity analyses that included patients who developed secondary infection within the 48-h window of first blood sampling are shown in the supplement (Table S7). The relationships between the markers and other clinical outcomes, namely length of stay, days alive and free of organ support, and SOFA scores are shown in the supplement (Tables S8, S9).
We found no predictive value for total leucocyte or differential counts (supplemental Table S10).
Marker combinations for predicting subsequent infection
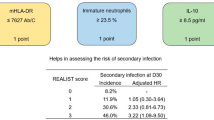
Using the best cut-offs derived from the ROC analysis for each of the three biomarkers, we assigned each ‘dysfunction’ a score of 1 (no dysfunction scored 0). The number of dysfunctions was summed to give a cumulative total (range 0–3). With this approach, an increase in the number of immune dysfunctions was associated with a progressive increase in the risk of subsequent infection (Table 3, Fig. 1d). When the combined markers were used, the area under ROC curve (AUC ROC) was 0.72 (95% CI 0.62–0.81); this was larger than each individual biomarker AUC (range 0.57–0.64; Table S4). Adjustment for potential clinical confounders did not significantly alter this relationship (Table S11).
Modelling of clinical use
The modelling of potential clinical use of these tests, which would most likely be to guide immunomodulatory therapy, aimed to dichotomise patients into particularly ‘low’ risk (unlikely to benefit from immunomodulation) and ‘high’ risk (immunomodulation potentially could modify risk) for subsequent infection.
When the clinical modelling strategy set out in the “Methods” was used, the sample taken at study baseline demonstrated no significant ability to predict subsequent infection (see Table 4, Figure S3a), similar to the analysis of single markers at baseline (Table S6). In sensitivity analyses that selected higher sensitivity thresholds for ‘low’ monocyte HLA-DR, there was no improvement in the predictive ability of the test at baseline (data not shown).
In contrast, the sample taken between study days 2 and 4 had potentially useful discriminant value for patients at low and high risk of subsequent secondary infection (see Table 4, Fig. S3b). ‘High-risk’ patients also had lower infection-free survival on survival analysis [HR 2.8 (95% CI 1.4–5.7) p = 0.005 by log-rank test], and significantly longer ICU length of stay and duration of organ support (Table 4). This predictive ability was also found at days 6–8. A similar, but non-statistically significant, pattern was also found at days 10–12 (Table 4). Adjustment for potential clinical confounders did not significantly alter the predictive ability of these markers at any of the time points examined (Table S12). Selecting higher sensitivity thresholds for ‘low’ monocyte HLA-DR increased the overall sensitivity of the test but this occurred at the expense of specificity (data not shown).
In a hypothetical model of enriching a trial population with ‘high-risk’ patients for an immunomodulatory intervention, we found the numbers of patients needed to treat (and consequently the size of any trial) could likely be substantially reduced using this precision-medicine approach (see Tables S13, S14, supplemental section). For example, if this strategy was used to enrol high-risk patients based on a test carried out on ICU day 3–5 post admission, a 30% relative risk reduction in subsequent infection (80% power) could be detected with a sample size of 436, compared to 806 if ‘all comers’ were included.
Discussion
This is the first report of multicentre, multiparametric immunophenotyping for the determination of immune dysfunction amongst critically ill adults and one of only two studies [3] to examine multiple markers over time. Through this study we have confirmed the role of three markers associated with leucocyte dysfunction in predicting the development of nosocomial infection. Although each marker alone had relatively modest predictive ability, in combination they had stronger association with the risk of infection. These results are strikingly similar to our previous findings providing confirmatory evidence [3].
The demographic and clinical data (Table 1) demonstrate that the patients were broadly similar at the time of study enrolment regardless of whether they subsequently develop an infection, and had similar severity of illness scores and requirement for organ support on ICU admission. Adjustment for these factors did not alter the predictive ability of the cell-surface markers (see Tables S5, S11–12). These findings contrast with other studies which found that age and illness severity are strong predictors of subsequent nosocomial infection [24]. This discrepancy may be explained by differences between the cohorts of patients recruited. We deliberately recruited a group of patients who were already at high risk of secondary infection, confirmed by the 37% incidence observed. The markers we present have also been associated with severity of illness [25,26,27], and therefore their use will already encode similar information to that contained in severity scores. We contend that in the severely ill patient cohort we studied, clinical and demographic features are unlikely to reliably discriminate risk of subsequent infection.
We found that measuring immune dysfunction with the baseline sample (taken a median 1.5 days after ICU admission) shows poor predictive ability for subsequent infections. By contrast, samples taken between study days 2 and 8 (median 3–9 days after ICU admission) had better predictive ability for subsequent infections, and were also associated with longer ICU stay and greater need for organ support. The median time to infection from a positive test on the day 2–4 sample was 5 days, which appears a plausible interval in which an immunostimulatory therapy could work [28]. Using this approach would allow targeting of immunomodulatory therapies to patients who might have greatest risk–benefit balance from these therapies especially through reduction in secondary infections. This might also translate into reduced length of stay and infection-associated morbidity and mortality [28]. The impact of nosocomial infection on mortality is relatively limited [24], but the costs of prolonged hospital stay and organ support are significant and could be significantly substantially impacted on by targeted immunotherapy. A precision medicine approach has greatest chance of cost-effectiveness given the potential high cost of these therapies.
It is interesting to note that patients who developed secondary infections demonstrated an impaired recovery in both monocyte HLA-DR and neutrophil CD88 relative to those who did not develop infections (Fig. S2), suggesting that persistence of dysfunction is an important risk factor for subsequent infection, similar to an association reported in trauma patients [1]. Whether immunomodulatory therapies should be started at ICU admission and modified at days 3–5, or not started until days 3–5 will need to be determined by future randomized controlled trials.
Our study has a number of strengths. By using common protocols and reagents, the same model of flow cytometry analyser and cross-site standardization, we have been able to obtain comparable results from geographically and clinically diverse units. We have also been able to explore a number of putative markers of immune dysfunction, validating two, which we had established previously alongside the benchmark monocyte HLA-DR.
We used rigorous criteria for the determination of infection, and expert consensus for those suspected infections that did not meet the criteria. Sensitivity analyses demonstrated considerable overlap in the 95% confidence intervals for the predictive performance of the markers, either when early (< 48 h after enrolment, Table S7) or only confirmed infections were included (data not shown).
The fields of immunophenotyping and critical illness-induced immune dysfunction are relatively new, and as such there are no established benchmarks or levels for defining ‘normal’ and ‘abnormal’ measures. Although this study used development of infection to define cut-offs for immune dysfunction, further external validation of markers at these specific cut-offs is required. At present the assays need a high degree of machine standardization and further work is required to extend their usability to different flow cytometers or towards a near-patient test. The extension of the existing QuantiBRITE system (BD Biosciences, San Jose, CA) used for monocyte HLA-DR quantification to other fluorophores and other cell surface markers would be one potential solution to this issue.
Whilst the tests presented have relatively modest performance against typical ‘diagnostic’ tests, they are not designed to diagnose secondary infection, but rather to stratify the risk of developing secondary infection. As such the markers show comparable performance to risk markers used in other acute illnesses, such as predictors of mortality in acute exacerbations of chronic obstructive pulmonary disease [29] or major cardiac events in unstable angina [30]. Monocyte HLA-DR is in current use for risk stratification in clinical trials (NCT02361528), and we demonstrate in this study that its predictive ability is significantly enhanced by the addition of the two additional markers. As we demonstrate in modelling potential use (Tables S13, 14), the use of these markers could substantially alter the conduct of trials of immunomodulatory therapies, thus increasing the likelihood of successful trials and potentially increasing the cost-effectiveness of such novel therapies.
In conclusion, our study confirms our previous findings of the ability of neutrophil CD88, monocyte HLA-DR and the percentage of Tregs to predict secondary infection in patients requiring intensive care and organ support, and provides novel data on the time course of dysfunction and clinical relevance of the optimum timing of sampling in predicting risk of infection. The combination of markers allows for a more nuanced assessment of infection risk, with important implications for developing stratified medicine tools to support future immunomodulatory therapies amongst this group of patients.
References
Gouel-Chéron A, Allaouchiche B, Floccard B, Rimmelé T, Monneret G (2015) Early daily mHLA-DR monitoring predicts forthcoming sepsis in severe trauma patients. Intensive Care Med 41:2229–2230
Allen ML, Peters MJ, Goldman A et al (2002) Early postoperative monocyte deactivation predicts systemic inflammation and prolonged stay in pediatric cardiac intensive care. Crit Care Med 30:1140–1145
Conway Morris A, Anderson N, Brittan M et al (2013) Combined dysfunctions of immune cells predict nosocomial infection in critically ill patients. Brit J Anaes 11:778–787
Marshall JC (2010) Critical illness is an iatrogenic disorder. Crit Care Med 38:S582–S589
Lee WL, Downey GP (2001) Leukocyte elastase: physiological functions and role in acute lung injury. Am J Respir Crit Care Med 164:896–904
Pène F, Pickkers P, Hotchkiss RS (2016) Is this critically ill patient immunocompromised? Intensive Care Med 42:1051–1054
Vanzant EL, Lopez CM, Ozrazgat-Baslanti T et al (2014) Persistent inflammation, immunosuppression, and catabolism syndrome after severe blunt trauma. J Trauma Acute Care Surg 76:21–30
Vincent J-L, Rello J, Marshall J et al (2009) International study of the prevalence and outcomes of infection in intensive care units. JAMA 302:2323–2329
Kett DH, Cano E, Quartin AA et al (2011) Implementation of guidelines for management of possible multidrug-resistant pneumonia in intensive care: an observational, multicentre cohort study. Lancet Infect Dis 11:181–189
Morris AC, Brittan M, Wilkinson TS et al (2011) C5a-mediated neutrophil dysfunction is RhoA-dependent and predicts infection in critically ill patients. Blood 117:5178–5188
Conway Morris A, Kefala K, Wilkinson TS et al (2009) C5a mediates peripheral blood neutrophil dysfunction in critically ill patients. Am J Respir Crit Care Med 180:19–28
Döcke WD, Randow F, Syrbe U et al (1997) Monocyte deactivation in septic patients: restoration by IFN-gamma treatment. Nat Med 3:678–681
Lukaszewicz A-C, Grienay M, Resche-Rigon M et al (2009) Monocytic HLA-DR expression in intensive care patients: interest for prognosis and secondary infection prediction. Crit Care Med 37:2746–2752
Venet F, Chung C-S, Kherouf H et al (2009) Increased circulating regulatory T cells (CD4+CD25+CD127−) contribute to lymphocyte anergy in septic shock patients. Intensive Care Med 35:678–686
Leentjens J, Kox M, van der Hoeven JG, Netea MG, Pickkers P (2013) immunotherapy for the adjunctive treatment of sepsis: from immunosuppression to immunostimulation. Time for a paradigm change? Am J Respir Crit Care Med 187:1287–1293
Conway Morris A, Datta D, Shankar-Hari M, et al (2018) Predicting secondary infections using cell-surface markers of immune cell dysfunction: the INFECT study. J Intensive Care Soc (in press)
Conway Morris A, Datta D, Shankar-Hari M et al (2016) Predictive value of cell-surface markers in infections in critically ill patients: protocol for an observational study (ImmuNe FailurE in Critical Therapy (INFECT) study). BMJ Open 6:e011326
Suetens C, Morales I, Savey A et al (2007) European surveillance of ICU-acquired infections (HELICS-ICU): methods and main results. J Hosp Infect 65(Suppl 2):171–173
Drewry AM, Ablordeppey EA, Murray ET et al (2016) Comparison of monocyte human leukocyte antigen-DR expression and stimulated tumor necrosis factor alpha production as outcome predictors in severe sepsis: a prospective observational study. Crit Care 20:334
Seddiki N, Santner-Nanan B, Martinson J et al (2006) Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J Exp Med 203:1693–1700
Adrie C, Lugosi M, Sonneville R et al (2017) Persistent lymphopenia is a risk factor for ICU-acquired infections and for death in ICU patients with sustained hypotension at admission. Ann Intensive Care 7:30
Salciccioli JD, Marshall DC, Pimentel MAF et al (2015) The association between the neutrophil-to-lymphocyte ratio and mortality in critical illness: an observational cohort study. Crit Care 19:13
Youden WJ (1950) Index for rating diagnostic tests. Cancer 3:32–35
van Vught LA, Klein Klouwenberg PMC, Spitoni C et al (2016) Incidence, risk factors, and attributable mortality of secondary infections in the intensive care unit after admission for sepsis. JAMA 315:1469–1479
Unnewehr H, Rittirsch D, Sarma JV et al (2013) Changes and regulation of the C5a receptor on neutrophils during septic shock in humans. J Immunol 190:4215–4225
Huang L-F, Yao Y-M, Dong N, Yu Y, He L-X, Sheng Z-Y (2010) Association between regulatory T cell activity and sepsis and outcome of severely burned patients: a prospective, observational study. Crit Care 14:R3
Cheadle WG, Hershman MJ, Wellhausen SR, Polk HC (1991) HLA-DR antigen expression on peripheral blood monocytes correlates with surgical infection. Am J Surg 161:639–645
Meisel C, Schefold JC, Pschowski R et al (2009) Granulocyte-macrophage colony-stimulating factor to reverse sepsis-associated Immunosuppression: a double-blind, randomized, placebo-controlled multicenter trial. Am J Respir Crit Care Med 180:640–648
Singanayagam A, Schembri S, Chalmers JD (2013) Predictors of mortality in hospitalized adults with acute exacerbation of chronic obstructive pulmonary disease. A systematic review and meta-analysis. Ann ATS 10:81–89
Sabatine MS, Antman EM (2003) The thrombolysis in myocardial infarction risk score in unstable angina/non-ST-segment elevation myocardial infarction. J Am Coll Cardiol 41:S89–S95
Funding
The study was funded by Innovate UK (Sepsis 2: 101193), BD Biosciences and the National Institute for Academic Anaesthesia. Dr Conway Morris is supported by a Clinical Research Career Development Fellowship from the Wellcome Trust (WT 2055214/Z/16/Z). Dr Shankar-Hari is supported by the National Institute for Health Research Clinician Scientist Award (CS-2016-16-011). The views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the National Institute for Health Research or the Department of Health.
Author information
Authors and Affiliations
Contributions
Study design and protocol authorship: ACM, DD, MSH, CW, JR, JA, NW, KJ, JK, AW, KAB, SL, TM, ID, AGR, AJS, TSW. Obtaining funding: ACM, AB, NW, JK, KAB, AR, AJS, TSW. Recruiting patients: MSH, JA, AW, SL, TM, TSW. Conducting assays: DD, JR, TB, SL, TM, GH, AGR. Data analysis: ACM, DD, JS, CW, JR, NW, KJ, AW, KAB, SL, TM, GH, ID, AGR, TSW. Manuscript drafting: ACM, MSH, TB, AGR, TSW. Manuscript revision and approval: ACM, DD, MSH, JS, CW, JA, AB, NW, KJ, JK, AW, TB, SL, TM, AR, ID, AGR, AJS, TSW.
Corresponding author
Ethics declarations
Conflicts of interest
NW, KJ, JK and AW were all employees of BD biosciences whilst this work was being undertaken, and NW and KJ remain employees to date. All four authors hold stock in BD Biosciences. All other authors declare they have no conflict of interest.
Electronic supplementary material
This article has an online data supplement.
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Conway Morris, A., Datta, D., Shankar-Hari, M. et al. Cell-surface signatures of immune dysfunction risk-stratify critically ill patients: INFECT study. Intensive Care Med 44, 627–635 (2018). https://doi.org/10.1007/s00134-018-5247-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00134-018-5247-0