
Abstract
Purpose
To test whether the polymyxin B hemoperfusion (PMX HP) fiber column reduces mortality and organ failure in peritonitis-induced septic shock (SS) from abdominal infections.
Method
Prospective, multicenter, randomized controlled trial in 18 French intensive care units from October 2010 to March 2013, enrolling 243 patients with SS within 12 h after emergency surgery for peritonitis related to organ perforation. The PMX HP group received conventional therapy plus two sessions of PMX HP. Primary outcome was mortality on day 28; secondary outcomes were mortality on day 90 and a reduction in the severity of organ failures based on Sequential Organ Failure Assessment (SOFA) scores.
Results
Primary outcome: day 28 mortality in the PMX HP group (n = 119) was 27.7 versus 19.5 % in the conventional group (n = 113), p = 0.14 (OR 1.5872, 95 % CI 0.8583–2.935). Secondary endpoints: mortality rate at day 90 was 33.6 % in PMX-HP versus 24 % in conventional groups, p = 0.10 (OR 1.6128, 95 % CI 0.9067–2.8685); reduction in SOFA score from day 0 to day 7 was −5 (−11 to 6) in PMX-HP versus −5 (−11 to 9), p = 0.78. Comparable results were observed in the predefined subgroups (presence of comorbidity; adequacy of surgery, <2 sessions of hemoperfusion) and for SOFA reduction from day 0 to day 3.
Conclusion
This multicenter randomized controlled study demonstrated a non-significant increase in mortality and no improvement in organ failure with PMX HP treatment compared to conventional treatment of peritonitis-induced SS.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Among the etiologies of severe sepsis or septic shock (SS), pulmonary and the abdomen infection are the most frequent [1]. The mortality of peritonitis remains high when associated with SS [2, 3]. Experimental models of peritonitis using rodent cecal ligation and puncture (i.e., peritonitis) have been largely investigated to dissect the septic-induced mechanisms with potentials for “adjuvant therapies”. Among these mechanisms, presence of endotoxin (ET) is well established to induced systemic inflammatory response in Gram-negative infection [4] but also after injection in a non-septic animal model [5] or in human [6]. Randomized clinical trials (RCT) testing treatment interfering with different steps of this pathway have been completed with negative results [4, 7]. ET plasma removal, neutralization or blocking ET-receptors have also been tested. In the same line, extra-corporeal device to remove ET or inflammatory mediators have been tested [8–10]. The benefit to remove ET by hemoperfusion with polymyxin B membrane membranes (PMX-HP; Toraymyxin; Toray Industries, Tokyo, Japan) using an extra-corporeal device has been developed and is currently largely used in patients with severe sepsis or SS in Japan [11–15]. The reviewed 28 clinical studies have reported some benefits in membrane-treated patients, such as a hemodynamic failure improvement and a reduction in mortality [16]. However, most of these studies were not randomized, selecting heterogeneous patients for the source of infection, with limited sample size, poor or no characterization of inflammation, and most often with no measurement of ET levels. A recent RCT enrolling 64 severe sepsis or SS relayed to intra-abdominal infection showed a hemodynamic improvement at day 3 with a significant reduction in mortality in the PMX-HP arm in a post hoc analysis [17]. The small size of the cohort not initially powered to test the impact on outcome justified the design of a larger RCT on SS related to peritonitis. This present randomized controlled study tested the benefit to use the PMX-HP membrane on mortality at day 28 as a primary outcome.
Methods
The registered protocol (Clinical trials.gov NCT01222663) was approved by the French Ethical committee “Comité de Protection des Personnes” (CPP Ile de France IV; 2010-A0004039) fixing the primary end-point on day 28 mortality. A written informed consent was obtained from patients or their surrogates according to the French Ethical Law for clinical investigation. This law allows the use of “emergency enrollment” when the delay for inclusion is too short to obtain informed consent from surrogates. In this case, a doctor independent from a unit not related to the protocol can authorize inclusion after checking the medical history and ethical conditions. After enrollment, the investigator has to subsequently validate the agreement to use the data by a surrogate or the patient himself.
Study patients
Adult patients were eligible for inclusion if they had SS and underwent emergency surgery to treat visually confirmed peritonitis. In order to distinguish between hypotension resulting from the effect of sedation, shock had to occur or persist within 10 h after surgical procedure with a duration of at least 2 h. Shock was classically defined as a hypotension resistant to fluid administration requiring norepinephrine or other vasopressor [18]. The minimal amount of fluid administration was fixed on the protocol at 20 mL/kg of body weight during the 4 h interval including the onset of vasopressor infusion (see inclusion and exclusion criteria in ESM). The confirmation of peritonitis was made de visu observing the peritoneal cavity infection (purulent fluid with a detected perforation of the gut or biliary tract). Routine laboratory samples done on a day-to-day basis allowed to calculate the Sequential Organ Failure Assessment (SOFA) score. The remainder volume of blood samples at day 1 was used to measure plasma IL-6 levels as a marker of systemic inflammation [19].
Since the quality of surgery was shown to be essential for prognosis [20], the surgical procedure quality was evaluated by an independent and blinded general surgeon (see ESM). Reports were classified into adequate, non-adequate, or non-evaluable procedures. The adequacies of antimicrobial therapy for administration were also collected.
Study treatments
After fulfilling the entry criteria and satisfying the ethical agreement, the patient was enrolled and randomized from a centralized system (allocation 1:1 stratified by center with block size of 4) to receive PMX-HP treatment or a conventional treatment. By nature, PMX-HP-treated pateients could not realistically be blinded from investigators and clinicians in charge. Hemoperfusion was performed with a circuit allowing the blood to circulate through a PMX membrane connected to a double-lumen venous catheter. The membrane used was an adsorbent column containing 5 mg of polymyxin B per gram of polystyrene fiber (Toraymyxin; Toray Industries). Hemoperfusion consisted of two sessions of perfusion for 2 h each as previously reported [10, 17].
The protocol steps are summarized in Fig. 1. The first PMX-HP session had to start within 12 h after abdominal surgery followed by a second PMX-HP session 22–24 h after completion of the first session. A PMX-HP session was defined as complete when at least 1 h 30 min of perfusion were achieved. When the first session had to be interrupted for any reason, the delay for the second session was unchanged.

Protocol design of the trial from the surgery until the second session of hemoperfusion
PMX-HP sessions were performed on different renal replacement therapy (RRT) machines depending on availability in the participating centers. The circuit anticoagulation was made with unfractionated heparin at a recommended (manufacturer and previous investigators) 2,000–4,000 U dose range. If a premature clotting of the circuit occurred during the first session, increasing the heparin dosage for the second session can be considered. The blood flow rate in the circuit had to range from 80 to 120 mL/min. All other treatments were at the discretion of the clinician in charge essentially applying the Surviving Sepsis Campaign recommendations, especially for RRT.
The primary endpoint was the rate of death at day 28.
The secondary endpoints were the mortality at days 7, 14, 21 and 90; the SOFA score variation within the first 3 days, excluding the neurology component of SOFA difficult to analyze during sedation and/or anesthesia; the SOFA score variation and adverse events; time to withdraw catecholamine; and adverse events related to hemoperfusion.
The post hoc analysis investigated: the SOFA score variation at day 7; the incidence of RRT; the impact on day 28 and day 90 mortality of the presence or not of comorbidity (≤1 or >1 value of McCabe score), the quality of surgery, and the presence of positive blood culture in pre-specified sub-groups. Adverse events, especially hemorrhage and/or necessity to perform an additional surgery, were also collected.
Study oversight
The principal investigators (D.P.) and the coordinator (R.R.) wrote the protocol, managed the trial, obtained the CPP agreement and wrote the manuscript. The Steering Committee amended the protocol independently from the sponsors Toray Medical and Meditor SA. The statistical analysis was performed by an independent statistician (co-author of the paper). The article was submitted for publication after the agreement of the Steering Committee and all investigators. All authors had full and independent access to all data and assure the integrity, accuracy and completeness of the analysis and the fidelity to the study protocol. A Data Safety Monitoring Board (DSMB) of three independent experts reviewed the data to assure the quality, the safety and the faithfulness to the protocol. A predefined blinded interim analysis after the 80th patient aimed to verify the feasibility and safety of the trial. A second blinded interim analysis after enrolling 160 patients evaluated the safety, the efficiency and/or futility, which may lead to decide to stop or continue the trial.
Sample size and statistical analysis
On the strict recommendations of the Ethical Committee, the primary endpoint was to target the mortality at day 28. The assumed mortality rate at day 28 in the control group was 37 %, a more stringent value than the one observed in the EUPHAS trial [17]. Such incidence corresponded to the death rate of a recent large study in patients with severe peritonitis [3]. The size of the cohort to test an absolute reduction in mortality at day 28 of 20 % (relative reduction of 54 %) shown by the EUPHAS study [17] was 240 patients (120 per group) to obtain a nominal two-sided p value of 0.045 and a power of 94 %.
For the first interim analysis, no-one was aware of the arm labels and the critical p value was fixed at a high level (p < 0.0005) to limit the risk of interruption. For the second interim analysis, the p value was fixed at a lower value (0.014).
Analyses were first performed on an intention to treat (ITT), i.e., all enrolled patients, and then on per protocol (PP). The PP population concerned patients treated at least with one session of PMX-HP and all the control group. Analyses were performed using SAS v.9 software (Cary, NC, USA). Mortality at different time points with adequate follow-up for all patients was compared by Chi square or Fisher’s exact test as appropriate. Main comparisons were performed without a first adjustment, and then after adjustment, on comorbidity at 24 h using the Cochran–Mantel–Haenszel method.
For the secondary endpoints and exploratory analyses, the p value was set at 0.05. The differences between the two arms were tested using Student’s t test, Mann–Whitney U test, Chi square or Fisher’s exact test as appropriate. The time variation of the SOFA score was analyzed using ANOVA or mixed models accounting for repeated measures. Time to events and overall mortality were estimated by the Kaplan–Meier method and compared within groups by the log-rank test.
Results
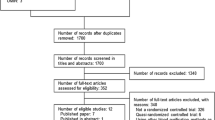
Among the 938 screened patients, 243 patients were randomly assigned to the study. Eleven patients were excluded due to refusal to participate by patient or family or legally protected adult (Fig. 2) (see detailed non-inclusion in e-Results). The 232 patients constituting the study group were followed up to 90 days.

Flow chart of the study
Baseline clinical characteristics were similar in both groups (Table 1). Peritonitis context and bacterial screening are shown in Table 2. Peritonitis mainly resulted from a lower gastrointestinal tract perforation with identified Gram-negative bacteria in 75 % of the cases. The incidence of negative cultures was similar in the two arms. The proportion of positive blood cultures was similar (26 %) in both groups. The blinded surgical treatment was considered adequate in 92 out 119 (77 %) of the (P) group and 89 out 113 (79 %) of the (C) group.
Primary endpoint
Thirty-three of 119 patients (27.7 %) died at day 28 in the PMX-HP group versus 22 of 113 patients (19.5 %) in the control group (p = 0.14, OR 1.5872, 95 % CI 0.8583–2.935).
Secondary endpoints
As shown in Table e3 in ESM, overall mortality at days 3, 7, 14 and 90 did not differ between the two arms. Cumulative incidence of death until day 90 was also not significantly different between the two groups (Fig. 3). After exclusion of the patients who did not complete at least one PMX-HP session (n = 6; 5 %), the per protocol analysis gave similar results for primary outcome at day 28: 24.8 % in PMX-HP group versus 19.5 % in control (p = 0.30). Again, mortalities at days 3, 7, 14 or 90 were not different in both groups (eTable 1 and eFig 2 in ESM). The paired analysis of SOFA score variation between day 0 and day 3 showed no significant difference in reduction [− 1 (−10–7) vs. −2 (−10–8)] in the control group (p = 0.08). Similarly, the incidence of SOFA score decrease by at least two points did not differ between the PMX-HP versus control (18.3 vs. 10.0 %, respectively; p = 0.09). The day 3 global SOFA score and its organ components were identical in both groups, except for hematology because of a significant decrease in platelet count in PMX-HP compared to the control group (Table 3). Similarly, per protocol analysis showed similar results at day 3 for the SOFA score values and variations (see eTables 3, 4, 5, 6 in ESM). The number of patients with at least one adverse event were 92 in PMX-HP and 82 in the control group (including 6 and 3 severe adverse events such as hemorrhagic episodes; see also eTable 8 in ESM). Decrease in platelet count at D3 was more frequent in the PMX-treated than in the control groups (p = 0.002). At day 3, the catecholamine infusion rate (µg/kg/min) was identical in both groups [0.31 (0–4.00) in PMX-HP and 0.27 (0–9.09) in the control group] (p = 0.71). The number of days without catecholamine treatment during the first 7 days was 2.0 (0.0–7.0) in PMX-HP and 3.0 (0.0–0.7) in the control group (p = 0.0710). The incidence of surgical revision was identical both groups (29 in PMX-HP vs. 26 in the control group).

Cumulative incidence of death overtime in the two arms: HP-PMX (continuous line) and standard treatment (hashed line). No significant difference was observed (p = 0.1067)
Hemoperfusion sessions
Among the 119 patients of the PMX-HP arm, 3 had no session completed for a technical problem (n = 1), severe hemodynamic dysfunction and early death (n = 2). Twelve patients could not have the second session because of technical problems (n = 2), death or major hemodynamic instability (n = 10). Among the 220 sessions performed, premature interruption was observed in 25 cases (11 %) mainly during the first session and especially due to circuit clotting (eTable 7 in ESM). In total, 2 PMX sessions were completed in only 81 of 119 patients (69.8 %).
Post hoc analysis
After adjusting on the McCabe score, age or SOFA score, mortality on day 28 and 90 remained similar between the two groups (p = 0.34 and 0.23, respectively) (see ESM). Comparison between the sub-group having completed the two sessions (n = 81) and controls alive at day 2 (n = 111) did not show any difference in mortalities at day 28 (n = 15; 18.5 % in the PMX-HP group vs. n = 20; 18.0 % in the control group (p = 0.93). Also, no differences were observed at day 90 (25.9 vs. 22.5 %, respectively; p = 0.59).
SOFA scores at day 7 between the two arms [5 (3–15) vs. 5 (0–17)] and at day 14 [3 (0–11) vs. 3 (0–12)] were identical (p = 0.80 and p = 0.61, respectively), as were the variation in SOFA from baseline to day 7 [−5(−11–6) in PMX-HP vs. −5(−11–9) in control] (p = 0.78). RRT in the PMX-HP group was more frequent than in control group [n = 63 (53 %) in PMX-HP vs. n = 42 (37 %), respectively], (p = 0.016). Durations of mechanical ventilation in PMX-HP and control groups were 6 (1–104) and 6.5 (1–53) days, respectively (p = 0.53), and lengths of ICU stay in the PMX-HP and control groups were 11 (1–104) and 10 (1–73) days, respectively (p = 0.49). RRT in the PMX-HP group was more frequent than in the control group [n = 63 (53 %) in PMX-HP vs. n = 42 (37 %), respectively] (p = 0.02). Additionally, analysis based on adequacy of surgery or plasma IL-6 levels showed no difference in mortality rates (see ESM). Interestingly, mortality of the sub-group of patients with moderate IL-6 level was higher in PMX-HP-treated patients (eFigure 2 in ESM).
Discussion
This randomized controlled study is the largest testing of PMX-HP hemoperfusion in peritonitis-induced SS. No significant differences in mortality at day 28 were observed between the PMX hemoperfusion arm and the conventional arm. No differences in hemodynamic patterns and organ failure evolution were observed. The latter effect differs from previous reports, which reported a benefit with the PMX-HP technique to improve systemic hemodynamic or oxygenation parameters and/or mortality [10, 16, 17]. Particularly, in 2009, a RCT on abdominal sepsis with SS showed a benefit of PMX-HP on hemodynamic parameters [17], with a reduction in mortality in a post hoc analysis from 53 % in the control group to 32 % in the PMX group. After observation of this result after an interim analysis, the Ethical Committee decided to stop the trial [17]. Some aspects of this study should be highlighted. First, no information was given on the screened population over 3 years and on the criteria used to select the enrolled patients. Second, the primary endpoint was just mean arterial pressure and vasopressor index, and the study was not powered to show a reduction in mortality. The observed reduction in 28-day mortality was observed only after adjustment of the SOFA score. Third, the small number of enrolled patients (n = 64) with an unusual high mortality rate in the control group (53 %) may limit the generalization of the conclusion of this study. In our study, similar mortality rates in ITT and on per protocol in both arms at day 28 were largely lower than the one observed in the control arm in the EUPHAS study [17], an aspect that may explain the different results compared to the present trial. The relatively high proportion of session failure (11 %) in our study was exactly the same as the one reported in the EUPHAS study (11.7 %). Although not tested in the EUPHAS study, we did not find any differences between the two arms using the sub-group of patients having completed the two PMX sessions. This negative result was recently supported by a large retrospective study [21] collecting data of Japanese patients having abdominal surgery for peritonitis with shock. Patients treated by one or two PMX-HP sessions had a similar mortality at day 28 (17 %) compared to propensity-matched patients not treated by PMX-HP (16.3 %).
The post hoc analysis in our study testing the impact of the important confounding factors (comorbidities, adequacy of surgery, and intensity of inflammation) confirmed the absence of positive signals when PMX-HP was used. Surprisingly, the HP-PMX group of patients with a moderate inflammation (plasma IL-6 level < the median value) had a significantly lower survival rate at day 28 compared to the conventional arm. Although not easy to explain, this observation could match with the results we observed in a RCT testing continuous hemofiltration at the early phase of severe sepsis which showed a worse outcome in the hemofiltrated group [9]. Together, these observations suggest a cautious use of extracorporeal techniques during the early phase of SS. Although not significant, there was an 8.2 % increase of mortality at day 28 in the PMX-HP group. The circuit, the device itself, might stimulate mechanisms deteriorating organ function and/or prognosis as suggested by the significant reduction in platelet number and the higher incidence of RRT in the HP-PMX treated group.
Limitation of our study
Our study has several limitations. First, the chosen 37 % mortality rate in the control group was finally too high regarding the 23 % observed in our study. This mortality rate was chosen for the protocol because it fitted well with previously reported death rates [3, 10, 21]. The most recent trials on SS published after the initiation of our trial reported mortality rates below 30 % in the control arm [4, 22]. Second, a 20 % reduction in mortality appeared too optimistic for the HP-PMX treated arm. Such a reduction was previously reported with PMX-HP [17] and we considered that such a high cost therapy should have a clear prognostic effect before generalization. Third, the relatively high incidence of incomplete treatment with PMX sessions (38 %) appears higher than those previously reported [10, 17]. Most of these interruptions resulted from early death or from very unstable hemodynamic conditions. The proportion of interrupted sessions due to clotting was only 11 %, an incidence previously reported [10, 17]. Occurring mainly during the first session, this suggests a procoagulant activity in the early phase of shock as previously shown [22]. The absence of any positive signal when only the patients having two sessions were analyzed attenuates the potential impact of an incomplete treatment. Fourth, the endotoxin circulating level was not measured, making enrollment of patients with low ET levels possible with a theoretical limited effect of PMX. The type of patients with peritonitis and the practical decisions made for therapy support the idea to use such an ET removal method in cases of high incidence of Gram-negative bacteria. ET levels were also not measured in the EUPHAS trial [17], as in the Japanese negative propensity-matched analysis [21].
In conclusion, despite the underpowered size of the study, the absence of any positive signal in this randomized control trial pleads against the use of PMX membranes to treat peritonitis-induced SS after surgery.
References
Liu V, Escobar GJ, Greene JD, Soule J, Whippy A, Angus DC, Iwashyna TJ (2014) Hospital deaths in patients with sepsis from 2 independent cohorts. JAMA 312:90–92
Koperna T, Schulz F (1996) Prognosis and treatment of peritonitis. Do we need new scoring systems? Arch Surg 131:180–186
Riche FC, Dray X, Laisne MJ, Mateo J, Raskine L, Sanson-Le Pors MJ, Payen D, Valleur P, Cholley BP (2009) Factors associated with septic shock and mortality in generalized peritonitis: comparison between community-acquired and postoperative peritonitis. Crit Care 13:R99
Opal SM, Laterre PF, Francois B, LaRosa SP, Angus DC, Mira JP, Wittebole X, Dugernier T, Perrotin D, Tidswell M, Jauregui L, Krell K, Pachl J, Takahashi T, Peckelsen C, Cordasco E, Chang CS, Oeyen S, Aikawa N, Maruyama T, Schein R, Kalil AC, Van Nuffelen M, Lynn M, Rossignol DP, Gogate J, Roberts MB, Wheeler JL, Vincent JL (2013) Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA 309:1154–1162
Losser MR, Bernard C, Beaudeux JL, Pison C, Payen D (1997) Glucose modulates hemodynamic, metabolic, and inflammatory responses to lipopolysaccharide in rabbits. J Appl Physiol 83:1566–1574
Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, Chen RO, Brownstein BH, Cobb JP, Tschoeke SK, Miller-Graziano C, Moldawer LL, Mindrinos MN, Davis RW, Tompkins RG, Lowry SF (2005) A network-based analysis of systemic inflammation in humans. Nature 437:1032–1037
McCloskey RV, Straube RC, Sanders C, Smith SM, Smith CR (1994) Treatment of septic shock with human monoclonal antibody HA-1A. A randomized, double-blind, placebo-controlled trial. CHESS Trial Study Group. Ann Intern Med 121:1–5
Joannes-Boyau O, Honore PM, Perez P, Bagshaw SM, Grand H, Canivet JL, Dewitte A, Flamens C, Pujol W, Grandoulier AS, Fleureau C, Jacobs R, Broux C, Floch H, Branchard O, Franck S, Roze H, Collin V, Boer W, Calderon J, Gauche B, Spapen HD, Janvier G, Ouattara A (2013) High-volume versus standard-volume haemofiltration for septic shock patients with acute kidney injury (IVOIRE study): a multicentre randomized controlled trial. Intensive Care Med 39:1535–1546
Payen D, Mateo J, Cavaillon JM, Fraisse F, Floriot C, Vicaut E, Hemofiltration, Sepsis Group of the College National de Reanimation et de Medecine d’Urgence des Hopitaux e-U (2009) Impact of continuous venovenous hemofiltration on organ failure during the early phase of severe sepsis: a randomized controlled trial. Crit Care Med 37:803–810
Vincent JL, Laterre PF, Cohen J, Burchardi H, Bruining H, Lerma FA, Wittebole X, De Backer D, Brett S, Marzo D, Nakamura H, John S (2005) A pilot-controlled study of a polymyxin B-immobilized hemoperfusion cartridge in patients with severe sepsis secondary to intra-abdominal infection. Shock 23:400–405
Nakamura T, Ebihara I, Shoji H, Ushiyama C, Suzuki S, Koide H (1999) Treatment with polymyxin B-immobilized fiber reduces platelet activation in septic shock patients: decrease in plasma levels of soluble P-selectin, platelet factor 4 and beta-thromboglobulin. Inflamm Res 48:171–175
Nakamura T, Kawagoe Y, Suzuki T, Shoji H, Ueda Y, Koide H (2007) Polymyxin B-immobilized fiber hemoperfusion with the PMX-05R column in elderly patients suffering from septic shock. Am J Med Sci 334:244–247
Nemoto H, Nakamoto H, Okada H, Sugahara S, Moriwaki K, Arai M, Kanno Y, Suzuki H (2001) Newly developed immobilized polymyxin B fibers improve the survival of patients with sepsis. Blood Purif 19:361–368 (discussion 368–369)
Shoji H (2003) Extracorporeal endotoxin removal for the treatment of sepsis: endotoxin adsorption cartridge (Toraymyxin). Ther Apher Dial 7:108–114
Suzuki H, Nemoto H, Nakamoto H, Okada H, Sugahara S, Kanno Y, Moriwaki K (2002) Continuous hemodiafiltration with polymyxin-B immobilized fiber is effective in patients with sepsis syndrome and acute renal failure. Ther Apher 6:234–240
Cruz DN, Perazella MA, Bellomo R, de Cal M, Polanco N, Corradi V, Lentini P, Nalesso F, Ueno T, Ranieri VM, Ronco C (2007) Effectiveness of polymyxin B-immobilized fiber column in sepsis: a systematic review. Crit Care 11:R47
Cruz DN, Antonelli M, Fumagalli R, Foltran F, Brienza N, Donati A, Malcangi V, Petrini F, Volta G, Bobbio Pallavicini FM, Rottoli F, Giunta F, Ronco C (2009) Early use of polymyxin B hemoperfusion in abdominal septic shock: the EUPHAS randomized controlled trial. JAMA 301:2445–2452
Levy MM, Fink MP, Marshall JC, Abraham E, Angus D, Cook D, Cohen J, Opal SM, Vincent JL, Ramsay G, International Sepsis Definitions C (2003) 2001 SCCM/ESICM/ACCP/ATS/SIS international sepsis definitions conference. Intensive Care Med 29:530–538
Scheller J, Garbers C, Rose-John S (2014) Interleukin-6: from basic biology to selective blockade of pro-inflammatory activities. Semin Immunol 26:2–12
Wacha H, Hau T, Dittmer R, Ohmann C (1999) Risk factors associated with intraabdominal infections: a prospective multicenter study. Peritonitis Study Group. Langenbecks Arch Surg 384:24–32
Iwagami M, Yasunaga H, Doi K, Horiguchi H, Fushimi K, Matsubara T, Yahagi N, Noiri E (2014) Postoperative polymyxin B hemoperfusion and mortality in patients with abdominal septic shock: a propensity-matched analysis. Crit Care Med 42:1187–1193
Ranieri VM, Thompson BT, Barie PS, Dhainaut JF, Douglas IS, Finfer S, Gardlund B, Marshall JC, Rhodes A, Artigas A, Payen D, Tenhunen J, Al-Khalidi HR, Thompson V, Janes J, Macias WL, Vangerow B, Williams MD, Group P-SS (2012) Drotrecogin alfa (activated) in adults with septic shock. N Engl J Med 366:2055–2064
Acknowledgments
The investigators thank all the intensivists participating in screening and enrollment and the nursing teams taking care of the patients. We thank Carole Guignon and Houda Haloui for their outstanding efforts in the monitoring of the study and their help for data collection, database and quality control. The study was conducted with autonomous funding from each of the participating centers. Polymyxin B cartridges for hemoperfusion (Toraymyxin) were provided free of charge by Toray Industries, Tokyo, Japan; Meditor SAS, Hoenheim, France, participated in logistic aspects of conducting the trial. We thank Paul Kirchner (San Francisco) for assistance in preparing and reviewing the manuscript, who was compensated. Study sponsored by the Torey Medical (Japan) and Meditor SA (France). The sponsors provided the circuits and the PMX membranes to perform the trial
Conflicts of interest
None.
Author information
Authors and Affiliations
Consortia
Corresponding authors
Additional information
Take-home message: The benefit of post-operative endotoxin adsorption by hemoperfusion in septic shock related to peritonitis tested in a randomized controlled trial did not show a reduction in 28- and 90-day mortality when compared to conventional group. Early polymyxin B hemoperfusion cannot be recommended in septic shock patients secondary to peritonitis.
Trial Registration: Clinical trials.gov Identifier: NCT01222663.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Participating centers and investigators
Participating centers and investigators
Department of Anesthesiology and Critical Care & SAMU, Lariboisière University Hospital AP-HP & Unité INSERM 1160 University Paris 7, Paris, France, D. Payen MD, PhD (Principal Investigator), A.C. Lukaszewicz MD, PhD; Medical Intensive Care, University Hospital Poitiers, France (coordinating center), R. Robert MD, PhD, R. Coudroy MD; Department of Anesthesia and Critical Care, Pontchaillou University Hospital, Rennes, France, Y. Launey MD, N. Nesseler MD, Mallédant YMD, PhD, P. Seguin MD, PhD; Intensive Care, Hospital of Roanne, France, M. Kaaki, P. Beuret MD; Department of Anesthesia and Critical Care, University Charles Nicolle Hospital, Rouen, France, B. Veber MD, PhD, P. Gouin MD, PG. Guitard MD, A. Lamacz MD; Department of Anesthesia and Critical Care, University Hospital Civil of Strasbourg, Strasbourg, France, J. Pottecher MD, PhD; Department of Anesthesia and Critical Care, Haut-Lévêque University Hospital, Bordeaux, France, O. Joannes-Boyau MD, A. Dewitte MD, C. Fleureau MD; Medical-Surgical Intensive Care Unit, District Hospital Center, La Roche sur-Yon, France, L. Martin-Lefevre MD, J. Reignier MD, PhD; Department of Anesthesia and Critical Care, University Hospital, Clermont-Ferrand, France, J.M. Constantin MD, PhD, M. Jabaudon MD; Surgical Intensive Care Unit, University Hospital, Poitiers, France, O. Mimoz MD, PhD, H. Nanadoumagar MD; Department of Anesthesia and Critical Care, Trousseau University Hospital, Tours, France, M. Ferrandière MD, A.C. Tellier MD; Department of Anesthesia and Critical Care, University Hospital, Lille, France, E. Kipnis MD, PhD, M. Boyer-Beyssère MD; Department of Intensive Care, Saint-Jean Hospital, Perpignan, France, C. Vela MD, O. Dematteis MD; Medical-Surgical Intensive Care, District Hospital, Saint-Malo, France, S. Chevallier MD, J.P. Gouello MD; Department of Intensive Care, District Hospital, Lens, France, J. Mallat MD, D. Thévenin MD; Medical-Surgical Intensive Care, District Hospital, Orléans, France, T. Boulain MD; Intensive Care, Dupuytren University Hospital, Limoges, France, A.P. Dugard MD, B. François MD, Department of Anesthesiology and Critical Care, University Hospital Saint-Louis, Paris, France, C. Le Gall MD; Medical-Surgical Intensive Care, District Hospital, Dieppe, France, J.P. Eraldi MD, P. Rigaud MD.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Payen, D.M., Guilhot, J., Launey, Y. et al. Early use of polymyxin B hemoperfusion in patients with septic shock due to peritonitis: a multicenter randomized control trial. Intensive Care Med 41, 975–984 (2015). https://doi.org/10.1007/s00134-015-3751-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00134-015-3751-z