
Abstract
Objective
In healthy volunteers cerebral blood flow starts to recover towards baseline within a few minutes of continued hyperventilation due to normalisation of perivascular pH. We investigated the time-course of changes in middle cerebral artery mean flow velocity (FVm) and intracranial pressure (ICP) in head-injured patients during sustained moderate reductions in arterial partial pressure of CO2 (PaCO2).
Design
Observational study.
Patients
Twenty-seven sedated, mechanically ventilated patients with severe head injury.
Interventions
Measurements were made during and after routine determination of CO2-reactivity: an acute 20% increase in respiratory minute volume was followed by a 10-min stabilisation period and 50 min of continued moderate hyperventilation at a constant PaCO2 (>3.5 kPa).
Measurements and results
FVm was monitored with transcranial Doppler, ICP was monitored with intraparenchymal probes. During the 50-min period with stable PaCO2 FVm increased in 36% of patients. All other patients showed a decline in FVm over the same time period. Overall FVm recovery was −0.03±0.14%·min-1. The time-course of ICP changes was significantly different from that of FVm, with ICP reaching its lowest value earlier than FVm (23±12 vs 37±20 min; P = 0.001) and returning more rapidly towards baseline than FVm (0.23±0.23 vs −0.03±0.14%·min-1; P< 0.0001).
Conclusions
Head-injured patients may adapt differently to hyperventilation than healthy volunteers. Potentially harmful reductions in cerebral blood flow may persist beyond the duration of useful ICP reduction.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
When healthy volunteers hyperventilate for a prolonged time-interval their cerebral blood flow (CBF) shows a relatively rapid adaptation starting almost immediately after onset of hyperventilation [1, 2], with recovery of CBF to approximately 90% of baseline values within 4 h of sustained hypocapnia [3]. This recovery is thought to be due to the return of cerebral vascular calibre towards baseline, following the normalisation of intracellular, and perivascular pH [4].
Hyperventilation has been used for several decades to reduce intracranial pressure (ICP) in head-injured patients [5, 6]. However, hyperventilation may have only relatively short-lived effects on ICP, and initial ICP reductions are often followed by recovery towards baseline, with restoration of pre-hyperventilation values within 60 min in some patients [7]. It is assumed that the short duration of the ICP reduction is due to the transient nature of the underlying vascular changes [8]. However, CBF responses in head-injured patients may differ from healthy subjects, with no recovery of CBF after 48 h of continuous hyperventilation in some patients [9].
Most patient studies of CBF adaptation to hyperventilation have focused on intervals of hypocapnia ≥24 h [10, 11]. More recently, long-term hyperventilation is less commonly used, and hypocapnia tends to be used for short periods to treat critical elevations in ICP [12, 13]. However, there are only sparse data on the short term responses of cerebral haemodynamics to moderate reductions in arterial partial pressure of CO2 (PaCO2). We have used transcranial Doppler (TCD) data acquired during and after routine CO2-reactivity testing to characterise the time-course of CBF changes during the first 60 min after a step decrease in PaCO2. We also tested the hypothesis that the time-course of changes observed in ICP is directly related to that seen in FVm. These data have previously been presented in abstract form [14].
Methods and materials
Data from adult patients (age >16 years) admitted to our Neurosciences Critical Care Unit (NCCU) with severe or moderate traumatic brain injury are presented. At the time of the study all patients had deteriorated to a Glasgow Coma Score (GCS) ≤8 and needed continuous ICP monitoring and mechanical ventilation. As part of the clinical protocol on the Unit, all patients underwent routine testing of CO2-reactivity to aid prognostic stratification. Data collected from these CO2-reactivity studies were used for this paper. The collection of these data was prospectively considered by the multidisciplinary NCCU Users Group, and it was agreed that because assessment of CO2-reactivity was part of normal clinical management, and since no patient confidentiality issues were involved, formal informed consent was not required. Exclusion criteria included respiratory failure, a baseline PaCO2 <4.30 kPa, and failure to obtain satisfactory bilateral TCD signals. All patients were treated according to a cerebral perfusion pressure (CPP) orientated protocol aiming to keep CPP above 70 mmHg, ICP below 25 mmHg, and jugular bulb venous oxygen saturation (SjO2) above 50% [15]. During the studies all physiological parameters were maintained within the limits specified in the treatment guidelines of our unit. Outcome was assessed 6 months after injury using the Glasgow Outcome Scale (GOS) [16].
Mean arterial blood pressure (MAP) was monitored via the radial artery (Edwards Lifesciences, Irvine, Calif., USA). ICP was monitored using an intraparenchymal probe (Codman MicroSensors ICP Transducer, Codman & Shurtleff, Raynham, Mass., USA). End-tidal CO2 (ETCO2) was measured using mainstream capnography (Marquette Solar 8000 M, GE Medical Systems, UK). Bilateral TCD mean flow velocity (FVm) measurements from the middle cerebral artery (MCA) were used to assess changes in CBF (Multi Dop X4, DWL Elektronische Systeme, Sipplingen, Germany). The two 2-MHz probes were held in place with a Lam head rack [17].
During the study all patients were sedated with propofol, (2–5 mg·kg-1·h-1) and fentanyl (1–2 μg·kg-1·h-1), and paralysed (atracurium). Infusion rates of sedative and vasoactive drugs were not changed and body temperature was kept constant throughout the study period. After recording baseline data for 20 min and determination of baseline PaCO2, the minute volume of the respirator was increased by 15–20%. If this intervention resulted in physiological parameters outside treatment guidelines (PaCO2 <3.5 kPa and/or SJO2 <55%), the protocol was abandoned. After an initial “stabilisation period” of 10 min the new ETCO2 was maintained for at least 50 min at this level (“stable hyperventilation period”). To detect a possible protracted equilibration between PaCO2 and ETCO2, PaCO2 was determined at the beginning and at the end of the stable hyperventilation period. Patients’ whose PaCO2 changed by more than one standard deviation of the total precision of the used blood gas analyser (AVL Omni, AVL Graz, A-8020 Austria: total precision of measurements according NCCLS document EP5-T: SD <0.27 kPa; personal communication, J. Riegebauer, Roche Diagnostics, Graz, Austria) during the stable hyperventilation period were excluded from analysis.
Data were recorded continuously using the analogue output of the monitors, analogue-digital conversion, and waveform time integration [18], and stored on a portable computer for offline analysis. For analysis 1-min mean values were calculated for all parameters. FVm values from both hemispheres were averaged. Averaged data of the 20-min pre-hyperventilation baseline period and the data from the first minute following the 10-min stabilisation period were used to calculate the relative CO2- and ICP-reactivity as the percentage change from baseline in FVm and ICP, respectively, per kPa change in PaCO2. Despite constant PaCO2 the lowest values of FVm were rarely reached after exactly 10 min of hyperventilation; we therefore also calculated the maximal CO2-reactivity, based on the lowest value for FVm that was reached.
The speed of onset of changes in FVm and ICP were characterised by the time-constant (τ) of the time taken (tmin) to achieve the lowest values of FVm (FVmmin) and ICP. Recovery was characterised by the slopes of linear functions that were fitted to the FVm and ICP data throughout the stable hyperventilation period (slopetot). However, using this approach it is possible to miss FVm recovery occurring relatively late during the stable hyperventilation period in patients with a negative slopetot. Therefore, in these patients we also calculated slopes for FVm between tmin and the end of the protocol (slopelate), if at least five data points (i.e., 5 min) were available to fit the linear regression function. The protocol and analysis are illustrated in Fig. 1. For comparisons of recovery of FVm in patients with that of healthy volunteers, previously published data were used [19]. These data were acquired using passive hyperventilation.

Schematic illustration of protocol and analysis. (FVm MCA mean flow velocity middle cerebral artery, τ time-constant of onset of hyperventilation induced changes, t min time interval between start of hyperventilation and lowest observed value for FVm [FVmmin]). FVmmin was used to calculate maximal CO2-reactivity, whereas “standard” CO2-reactivity was calculated from the first 1-min mean value of FVm of the stable hyperventilation period.
All data are presented as mean±standard deviation unless specified otherwise. Data were compared using paired t-tests with Bonferroni corrections where appropriate. For analysis of the interaction between time and the changes of FVm and ICP in patients and volunteers, two-way analysis of variance was used. Calculations were performed using SPSS 11.0 for Windows (SPSS, Chicago, Ill., USA).
Results
We investigated 30 head-injured patients. In one patient the study had to be abandoned because SJO2 fell below 55%. Two studies were excluded due to insufficient stability of PaCO2. Our analysis therefore includes data from 27 patients (four women, 23 men, mean age: 39 years, range 17–70 years, median GCS at admission: 5.5, range 3–12). Studies were carried out 3.7±2.7 days after injury. Clinical data and CT findings are summarised in Table 1. Haemodynamic and PaCO2 data at four time-points during the study are presented in Fig. 2. The increase in minute ventilation reduced PaCO2 from 5.10±0.35 kPa to 4.40±0.34 kPa (−0.70±0.27 kPa). CO2-reactivity was 26.5±16.3%·kPa-1. We found no significant correlations between the day of the study, GCS or GOS and CO2-reactivity. FVmmin was reached after 37±20 min. The mean time-constant for onset of FVm reduction due to hyperventilation was 8.9 min (range 2–48 min). Maximal CO2-reactivity calculated from FVmmin was significantly higher than that determined at the beginning of the stable hyperventilation period (35.4±19.4 vs 26.5±16.3%·kPa-1, P = 0.001).

Haemodynamic data at four time-points during the protocol. Data are shown as mean±SD. Dashed lines represent±1 SD of precision of blood gas measurements. [PaCO 2 arterial partial pressure of CO2, FVm mean flow velocity in the middle cerebral artery (mean of both hemispheres), MAP mean arterial blood pressure, ICP intracranial pressure, CPP cerebral perfusion pressure,† ICP increased significantly (P <0.01) between start and end of the stable hyperventilation period (paired t-tests with Bonferroni correction)]. *P< 0.01 between baseline and start stable hyperventilation.
FVm recovery
Overall slopetot was −0.03±0.14%·min-1. A negative slopetot was observed in 17 (63%) patients, i.e., FVm at the end of the stable hyperventilation period was lower than at the beginning of this period. In these 17 patients we attempted to calculate slopelate. Values ranging from –0.05%·min-1 to 0.95%·min-1 (0.18±0.24%·min-1) were found and seven patients showed evidence of late recovery (slopelate >0). There were no significant correlations between slopetot or slopelate and the extent of the change in PaCO2. Raw data of three patients are presented in Fig. 3. There were no significant correlations between slopetot and GCS, GOS, or age. Assuming linear recovery, we estimated the time to complete recovery of FVm in patients with positive slopetot or slopelate (n = 17). Values ranged from 81 min to 40 h (mean: 467 min). Using a general linear model we also addressed the question whether hemispheric recovery in the presence of a lesion within the MCA territory on CT is related to FVm recovery. However, the presence of a lesion did not significantly influence recovery. Comparison with data from healthy volunteers showed a significantly different time-course (Fig. 4, two-way ANOVA, significant interaction (P <0.05) between time and group).

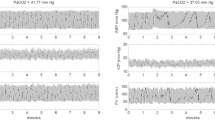
Examples of time-course of flow velocity in three patients. (FVm mean flow velocity, ICP intracranial pressure, MCA middle cerebral artery, PaCO 2 arterial partial pressure of CO2).

Comparison of recovery of FVm in head-injured patients and healthy volunteers. Pooled data from 27 patients and nine healthy volunteers (mean±SEM), showing more rapid recovery of middle cerebral artery mean flow velocity (average value of both hemispheres) in volunteers. Two-way ANOVA, significant interaction (P <0.05) between time and group.
Time-course of ICP changes and comparison with FVm
ICP-reactivity ranged from 2.0%·kPa-1 to 85.7%·kPa-1 (38.8±24.9%·kPa-1). There was a significant correlation between ICP-reactivity and CO2-reactivity (r2 = 0.39, P< 0.001) but no significant correlation between baseline ICP and ICP-reactivity. The time-constant for onset of the initial ICP reduction (mean 6 min, range 1–19 min) was comparable to the one for FVm (P >0.05); however, the lowest values for ICP were reached significantly earlier (23±12 min) for ICP than for FVm (P = 0.001). Slopetot for ICP-recovery was 0.23±0.23%·min-1 (range: -0.07–0.70%·min-1). In eight patients (30%) ICP had reached or exceeded baseline ICP by the end of the study. Only five patients had a negative slopetot for ICP-recovery, but in all these patients there was evidence of late-onset recovery with positive values for slopelate. During the stable hyperventilation period, ICP changes did not follow those of FVm. Overall the time-courses of FVm and ICP were significantly different (significant interaction between time and ICP and FVm, P< 0.0001, Fig. 5). Looking at individual patients, of the ten patients with positive slopetot three showed recovery of FVm that was faster than ICP-recovery and in six (including two women) ICP recovered faster than FVm. In a single patient FVm and ICP recovered at approximately equivalent rates. In the 17 patients with negative slopetot one had a similar time-course for FVm and ICP, in all others (including two women) ICP recovered more rapidly than FVm. We found no predictor for the response in FVm and ICP.

Time-course of changes in mean flow velocity, intracranial pressure and cerebral perfusion pressure in patients. Pooled data from all patients (mean±SEM), showing more rapid recovery of intracranial pressure than middle cerebral artery mean flow velocity (average value of both hemispheres).
Discussion
This study has found a persistent reduction of FVm over the first hour of moderate hyperventilation in a majority of patients and a discordance in the time-course of recovery of FVm and ICP during sustained moderate hyperventilation in head-injured patients.
Crucial to the interpretation of our data is the stability of PaCO2 during the stable hyperventilation phase. The precision of blood gas measurements is limited and the range of PaCO2 values that we accepted as stable is considerable. In 14 patients we determined PaCO2 at an additional time-point, and all these values were clearly within the precision limits specified in the methods section (Fig. 2). While smaller PaCO2 variations within the precision limits would be difficult to quantify but could affect determination of tmin, they would affect both FVm and ICP similarly, and would not confound comparisons between these two variables.
CO2-reactivity in women has been shown to be greater than in men [20]. To the best of our knowledge there are no published data concerning gender and adaptation of CBF to prolonged changes in PaCO2. In our small series of patients there are only four (premenopausal) women and our data do not allow us to assess the impact of gender on the response of CBF to sustained hypocapnia after head-injury.
Is the decrease in FVm we observed during 1 h of sustained hyperventilation due to the injury or to methodological differences between studies in patients and volunteers? Volunteer studies have mostly been performed using active hyperventilation. It could be argued that hyperventilation leads to cerebral activation that is at least partly responsible for the observed recovery of CBF. However, we used passive hyperventilation in our volunteers [19] and the rate of recovery that we saw using this method is comparable to earlier studies using active hyperventilation [1, 2]. This makes it unlikely that the observed difference is due to this methodological aspect. Another aspect that makes comparisons between volunteer and patient studies difficult is the effect of treatment. Our patients were sedated with propofol and were commonly receiving vasoactive drugs such as dopamine and noradrenaline. These factors could theoretically interfere with CBF recovery. Whilst in the presence of propofol CO2-reactivity is preserved, albeit somewhat reduced [21, 22], the effects of propofol, noradrenaline, and dopamine on CBF adaptation to sustained hypocapnia have not been investigated. A further concern is the influence of arterial partial pressure of O2 (PaO2) on recovery of FVm. It has recently been shown that hyperoxia when compared to normoxia reduces FVm recovery in healthy volunteers [19]. However, we found no correlation between PaO2 and slopetot in our patients. Our data do not allow us to draw conclusions as to whether the observed pattern is due to therapy or to the injury per se. But the fact that there was no relation between the time-course of adaptation and admission GCS, presence of a lesion within the MCA territory on CT, or outcome suggests that our observations are not only related to the injury per se. Further studies are needed to address this question.
ICP reductions with hyperventilation are generally attributed to changes in cerebral blood volume (CBV), and the concordant reductions in FVm and ICP with the induction of hyperventilation suggests that an alteration in cerebral vascular calibre was responsible for both changes. However, the clear discordance in the recovery patterns of these two parameters with continued hyperventilation suggests that different mechanisms were operating for CBF and CBV recovery during this period. The discordance in CBF and ICP recovery patterns may be due to effects in separate microvascular compartments; however, our data do not allow us to test this hypothesis. ICP recovery may be caused by the transient effects of hypocapnia in a segment of the vascular tree that is not investigated by TCD, e.g., the venous compartment, while the effect of hypocapnia on arterial resistance vessels and hence CBF may be more persistent. However, changes in one or more of the other intracranial compartments, cerebrospinal fluid (CSF), and brain volume, also need to be considered. Data are available on changes in CSF volume with hyperventilation in the setting of intracranial hypertension. Artru [23] studied the effects of hypocapnia in dogs with a simulated intracranial mass, and found a reduction in ICP for the complete duration of a 4-h study, despite CBV re-expansion to baseline values during the studies. The mechanism responsible was a concomitant reduction in CSF volume, probably due to a hypocapnia-induced improvement in the access of intracranial CSF to spinal sites of CSF absorption. Production of CSF did not change during the study. While the literature provides no information regarding compartmental changes in brain tissue volume with hyperventilation, Artru’s study did not find an increase in total brain tissue water. By avoiding low SJO2 we attempted to avoid hyperventilation-induced ischaemia. However, we cannot exclude that the ICP increases may have been at least partly induced by developing cytotoxic oedema. While further studies are needed, we believe that the present data support the inference that the discordant changes in CBF and ICP recovery patterns are due to changes in CBV in a vascular compartment not accessible to investigation with TCD, possibly the venous circulation. However, the studies by Artru [23] were conducted in animals without head trauma, and we cannot exclude the possibility that CSF-related mechanisms may contribute to this process.
Since these data were acquired as a part of routine CO2-reactivity testing, we were constrained by the protocol used in our unit for this purpose. Consequently, we acquired data for a maximum of 60 min of hyperventilation, and discontinued hyperventilation if there was any evidence of jugular venous desaturation. This limits the applicability of our results to understanding the physiological effects of sustained hyperventilation beyond this time frame, as might be employed in some patients with intracranial hypertension.
Our data raise the question which is the best time-point for determination of CO2-reactivity after a change in PaCO2? Generally, for determination of CO2-reactivity methods similar to ours are used [24, 25, 26], and reactivities are calculated 10 min or 15 min after changing the ventilator settings. Such a time-point may not represent the maximal response in many patients, but rather an arbitrary value that may depend on the rate and extent of induction of hypocapnia. Techniques that depend on steady-state intracranial haemodynamics with data collection over several minutes, such as positron emission tomography, need to be interpreted in the light of the possible instability of CBF during measurements and of the time lag between the start of hyperventilation and the actual measurements. As continuing and progressive CBF reductions may occur for up to an hour after initiation of hyperventilation, assessments of physiology in the immediate post-hyperventilation period should not be extrapolated beyond the time frame of the study.
In summary, our results show that the adaptation to sustained reductions in PaCO2 in patients may be different from that observed in healthy volunteers. It is unclear whether this difference is due to treatment or the injury per se. During sustained hyperventilation in head-injured patients, adaptation of CBF has a different temporal profile than the changes in ICP and the beneficial effect of hypocapnia on ICP may be outlived by potentially harmful decreases in CBF.
References
Ellingsen I, Hauge A, Nicolaysen G, Thoresen M, Walloe L (1987) Changes in human cerebral blood flow due to step changes in PAO2 and PACO2. Acta Physiol Scand 129:157–163
Poulin MJ, Liang PJ, Robbins PA (1998) Fast and slow components of cerebral blood flow response to step decreases in end-tidal PCO2 in humans. J Appl Physiol 85:388–397
Raichle ME, Posner JB, Plum F (1970) Cerebral blood flow during and after hyperventilation. Arch Neurol 23:394–403
Maddock RJ (2001) The lactic acid response to alkalosis in panic disorder : an integrative review. J Neuropsychiatry Clin Neurosci 13:22–34
Shenkin HA, Bouzarth WF (1970) Clinical methods of reducing intracranial pressure. Role of the cerebral circulation. N Engl J Med 282:1465–1471
Shapiro HM (1975) Intracranial hypertension: therapeutic and anesthetic considerations. Anesthesiology 43:445–471
Ropper AH (1993) Treatment of intracranial hypertension. In: Ropper AH (ed) Neurological and Neurosurgical Intensive Care. Raven, New York, pp 29–52
Yundt KD, Diringer MN (1997) The use of hyperventilation and its impact on cerebral ischemia in the treatment of traumatic brain injury. Crit Care Clin 13:163–184
Obrist WD, Langfitt TW, Jaggi JL, Cruz J, Gennarelli TA (1984) Cerebral blood flow and metabolism in comatose patients with acute head injury. Relationship to intracranial hypertension. J Neurosurg 61:241–253
Cold GE, Jensen FT, Malmros R (1977) The cerebrovascular CO2 reactivity during the acute phase of brain injury. Acta Anaesthesiol Scand 21:222–231
Muizelaar JP, Marmarou A, Ward JD, Kontos HA, Choi SC, Becker DP, Gruemer H, Young HF (1991) Adverse effects of prolonged hyperventilation in patients with severe head injury: a randomized clinical trial. J Neurosurg 75:731–739
http://www2.braintrauma.org/guidelines/ (accessed 28th July 2004).
Czosnyka M, Smielewski P, Piechnik S, Schmidt EA, Al-Rawi PG, Kirkpatrick PJ, Pickard JD (1999) Hemodynamic characterization of intracranial pressure plateau waves in head-injury patients. J Neurosurg 91:11–19
Steiner LA, Balestreri M, Johnston AJ, Czosnyka M, Coles JP, Pickard JD, Menon DK (2003) Hyperventilation-induced reductions in cerebral blood flow velocity outlive the reduction in intracranial pressure in head-injured patients. Crit Care 7:P074
Patel HC, Menon DK, Tebbs S, Hawker R, Hutchinson PJ, Kirkpatrick PJ (2002) Specialist neurocritical care and outcome from head injury. Intensive Care Med 28:547–553
Jennett B, Bond M (1975) Assessment of outcome after severe brain damage. Lancet 1:480–484
Lam AM (1995) Intraoperative transcranial Doppler monitoring. Anesthesiology 82:1536–1537
Zablotny W, Czosnyka M, Smielewski P (1994) Portable software for intracranial pressure recording and waveform analysis. In: Nagai H, Kamiya K, Ishii S (eds) Intracranial Pressure IX. Springer, Tokyo, pp 439–440
Johnston AJ, Steiner LA, Balestreri M, Gupta AK, Menon DK (2003) Hyperoxia and the cerebral hemodynamic responses to moderate hyperventilation. Acta Anaesthesiol Scand 47:391–396
Kastrup A, Thomas C, Hartmann C, Schabet M (1997) Sex dependency of cerebrovascular CO2 reactivity in normal subjects. Stroke 28:2353–2356
Eng C, Lam AM, Mayberg TS, Lee C, Mathisen T (1992) The influence of propofol with and without nitrous oxide on cerebral blood flow velocity and CO2 reactivity in humans. Anesthesiology 77:872–879
Strebel S, Kaufmann M, Guardiola PM, Schaefer HG (1994) Cerebral vasomotor responsiveness to carbon dioxide is preserved during propofol and midazolam anesthesia in humans. Anesth Analg 78:884–888
Artru AA (1987) Reduction of cerebrospinal fluid pressure by hypocapnia: changes in cerebral blood volume, cerebrospinal fluid volume, and brain tissue water and electrolytes. J Cereb Blood Flow Metab 7:471–479
van Santbrink H, Maas AI, Avezaat CJ (1996) Continuous monitoring of partial pressure of brain tissue oxygen in patients with severe head injury. Neurosurgery 38:21–31
Carmona Suazo JA, Maas AI, van den Brink WA, van Santbrink H, Steyerberg EW, Avezaat CJ (2000) CO2 reactivity and brain oxygen pressure monitoring in severe head injury. Crit Care Med 28:3268–3274
Lee JH, Kelly DF, Oertel M, McArthur DL, Glenn TC, Vespa P, Boscardin WJ, Martin NA (2001) Carbon dioxide reactivity, pressure autoregulation, and metabolic suppression reactivity after head injury: a transcranial Doppler study. J Neurosurg 95:222–232
Acknowledgements
We thank Hugh Richards (Academic Neurosurgery, University of Cambridge, UK) and Raymond Salvador (Wolfson Brain Imaging Centre, University of Cambridge) for statistical advice. M. Czosnyka is on unpaid leave from the Warsaw University of Technology, Poland
Author information
Authors and Affiliations
Corresponding author
Additional information
Grant information: L. Steiner was supported by a grant from the Margarete und Walter Lichtenstein-Stiftung (Basel, Switzerland), a Myron B. Laver Grant (Department of Anaesthesia, University of Basel, Switzerland), by the Swiss National Science Foundation, and was recipient of an Overseas Research Student Award (Committee of Vice-Chancellors and Principals of the Universities of the United Kingdom). A. Johnston was recipient of a grant from Codman. J. Coles was a Wellcome Research Training Fellow. This work was further supported by a grant from the Medical Research Council (Grant No. G9439390, ID 56833).
Rights and permissions
About this article
Cite this article
Steiner, L.A., Balestreri, M., Johnston, A.J. et al. Sustained moderate reductions in arterial CO2 after brain trauma Time-course of cerebral blood flow velocity and intracranial pressure. Intensive Care Med 30, 2180–2187 (2004). https://doi.org/10.1007/s00134-004-2463-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00134-004-2463-6