
Abstract
Carbonate minerals are ubiquitous in most sediment-hosted mineral deposits. These deposits can contain a variety of carbonate types with complex paragenetic relationships. When normalized to chondritic values (CN), rare-earth elements and yttrium (REE+YCN) can be used to constrain fluid chemistry and fluid-rock interaction processes in both low- and high-temperature settings. Unlike other phases (e.g., pyrite), the application of in situ laser ablation-inductively coupled plasma-mass spectroscopy (LA-ICP-MS) data to the differentiation of pre-ore and hydrothermal carbonates remains relatively untested. To assess the potential applicability of carbonate in situ REE+Y data, we combined transmitted light and cathodoluminescence (CL) petrography with LA-ICP-MS analysis of carbonate mineral phases from (1) the Proterozoic George Fisher clastic dominated (CD-type) massive sulfide deposit and from (2) correlative, barren host rock lithologies (Urquhart Shale Formation). The REE+YCN composition of pre-ore calcite suggests it formed during diagenesis from diagenetic pore fluids derived from ferruginous, anoxic seawater. Hydrothermal and hydrothermally altered calcite and dolomite from George Fisher is generally more LREE depleted than the pre-ore calcite, whole-rock REE concentrations, and shale reference values. We suggest this is the result of hydrothermal alteration by saline Cl--rich mineralizing fluids. Furthermore, the presence of both positive and negative Eu/Eu* values in calcite and dolomite indicates that the mineralizing fluids were relatively hot (>250°C) and cooled below 200–250°C during ore formation. This study confirms the hypothesis that in situ REE+Y data can be used to differentiate between pre-ore and hydrothermal carbonate and provide important constraints on the conditions of ore formation.
Similar content being viewed by others

Introduction
In many mineral systems, carbonate minerals constitute a significant proportion of the host rock and gangue mineralogy. Complex carbonate parageneses are often found in ore deposits hosted in sedimentary rocks (Cline et al. 2005; Hitzman et al. 2005; Leach et al. 2005), and can derive from detrital, diagenetic, hydrothermal, and tectonic or metamorphic processes. Such carbonate-rich ore systems also typically lack an extensive alteration footprint due to the effective buffering of acidic hydrothermal fluids by carbonate-rich lithologies. The complex carbonate paragenesis and the limited alteration footprint are major challenges when investigating, and exploring for, ore systems in carbonate-rich rocks.
Recent analytical developments are providing exploration geoscientists with new techniques for confronting these challenges. For example, new techniques that enable the generation of large C and O isotope datasets have shown how alteration halos in carbonate-hosted mineral systems may extend beyond bulk-rock lithogeochemical anomalies (e.g., off-axis integrated cavity output spectroscopy; Barker et al. 2013). As a result of being generated using bulk-rock techniques, such isotopic datasets may need to be supplemented by in situ techniques in order to constrain hydrothermal and background processes in samples with a complex paragenetic history.
In low-temperature (e.g., marine or diagenetic) environments, in situ trace element analysis of carbonate minerals (e.g., laser ablation ICP-MS) has attracted increasing usage (e.g., Webb et al. 2009; Himmler et al. 2010). As the partitioning of rare-earth elements and yttrium (REE+Y) between fluids and carbonate minerals is well constrained (e.g., Morgan and Wandless 1980; Tanaka and Kawabe 2006; Perry and Gysi 2020) the fractionation of REE+Y can provide important constraints on physicochemical conditions (pH, temperature, ligand type, or fluid-rock interaction; e.g., Bau 1991; Bau and Möller 1992; Barker et al. 2006). The REE+Y composition of carbonate can also vary significantly due to intrinsic processes such as closed-system effects and the carbonate growth rate (e.g., Kontak and Jackson 1995; Barker and Cox 2011). Altogether, there is potential for the REE+Y systematics of diagenetic and hydrothermal carbonates in constraining key aspects of fluid chemistry. Nevertheless, the direct application of in situ REE+Y analysis of carbonate minerals has remained relatively untested for hydrothermal mineral systems (e.g., Magnall et al. 2016; Vaughan et al. 2016), despite the wealth of information that is available from these types of analyses (Debruyne et al. 2016; Smrzka et al. 2019).
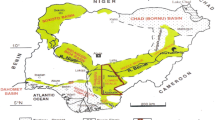
The need to differentiate between pre-ore and hydrothermal carbonate signatures is particularly relevant in the Carpentaria province (Fig. 1), where several of the world’s largest clastic-dominated (CD-type) Zn-Pb massive sulfide deposits are hosted in sedimentary rocks with multiple types of carbonate (e.g., Perkins and Bell 1998; Large et al. 2005; McGoldrick et al. 2010). Previous bulk-rock lithogeochemical studies have found large-scale (several km) halos of carbonate-associated elements (Mn and Fe) related to these hydrothermal systems (Large and McGoldrick 1998; Large et al. 2000). However, both Fe and Mn can also be enriched in carbonate minerals by background processes in marine or diagenetic environments (e.g., Brand and Veizer 1980; Kah 2000; Wittkop et al. 2020). As a result, bulk-rock and conventional in situ major-element analyses (i.e., electron probe microanalysis) alone may not be sufficient to differentiate between hydrothermal and background carbonate signatures.

a Map showing the Carpentaria province. Major Zn-Pb deposits in the Mount Isa Inlier and in the McArthur basin are denoted by stars. b Simplified superbasin stratigraphy for the Mount Isa Inlier (after Southgate et al. 2000). Stars denote approximate locations of the George Fisher (GF), Hilton (HI), Mount Isa (MI), Lady Loretta (LL), McArthur River (HYC), and Century (CE) clastic-dominated Zn-Pb massive sulfide deposits. c Map of the Mount Isa area showing simplified bedrock geology (after Gibson et al. 2017). The locations of the George Fisher, Hilton, and Mount Isa deposits and the unmineralized drill-hole (Shovel Flats) are denoted by stars and a circle respectively. d Stratigraphy of the Mount Isa Group (approximate formation thicknesses adapted from Neudert 1983)
In this study, we test the hypothesis that in situ REE+Y data can be used to differentiate diagenetic and hydrothermal carbonate mineral phases. We combine transmitted-light and cathodoluminescence (CL) petrography with LA-ICP-MS trace-element analyses of carbonates from drill-core samples through the main ore bodies at the southern Carpentaria George Fisher deposit (165 Mt at 9.1% Zn, 3.4% Pb, and 55 g/t Ag; Glencore 2019) and from a drill hole that intersected correlative, unmineralized host-rock lithologies (Urquhart Shale Formation). The in situ data are then compared to REE+Y compositions from whole-rock data from the same samples.
Rare-earth elements and yttrium (REE+Y)
The REE+Y are hard cations (high charge/radius ratio) and all have broadly similar chemical behavior, although subtle differences in solubility occur due to decreasing ionic radii from the light to the heavy REE+Y (e.g., Bau 1991; Migdisov et al. 2009; Williams-Jones et al. 2012). As a consequence of the Oddo-Harkins effect, elements with even atomic numbers are more abundant than those with uneven atomic numbers; therefore, the REE+Y are typically normalized to reference values (e.g., post-Archean Australian shale - PAAS, McLennan 1989; chondrite, McDonough and Sun 1995), which allows for better evaluation of the relative fractionation from different reservoirs and among individual REE+Y subgroups (light, middle, and heavy rare earth elements; LREE, MREE, and HREE). This fractionation can then be used to trace biogeochemical processes and to constrain fluid chemistry in low- and high- temperature settings (for reviews see Debruyne et al. 2016 and Smrzka et al. 2019).
The REE+Y are mostly trivalent (3+), apart from Ce (3+, 4+), Eu (2+, 3+), and Yb (2+, 3+), which are redox sensitive. The redox sensitivity of Yb is only important at high temperatures (> 420°C), whereas Ce and Eu can undergo valence changes at ambient and lower hydrothermal temperatures under geochemically reasonable fO2 conditions (for example at pH = 3; Fig. 2b). The fractionation of Ce and Eu relative to their neighbouring REE can, therefore, be used to constrain fluid redox conditions (e.g., De Baar et al. 1983; Sverjensky 1984; Bau and Möller 1992). This contrasting behavior is expressed as empirical anomalies relative to reference values, which can either be determined linearly or geometrically (e.g., Lawrence et al. 2006). The Ce anomalies (Ce/Ce*N) are typically shale-normalized (e.g., to PAAS; Ce/Ce*SN) in order to evaluate seawater or pore water redox, whereas Eu anomalies are commonly chondrite-normalized (Eu/Eu*CN) to avoid overestimation of Eu/Eu* values due to the negative Eu anomaly of PAAS (Fig. 2a).

a Chondrite normalized (McDonough and Sun 1995) rare earth element and Y profiles for: the post-Archean Australian shale reference material (black; PAAS, McLennan 1989), the Mount Isa Group (grey; Breakaway Shale, Native Bee Siltstone, Spear/Kennedy Siltstone, Magazine Shale; Nance and Taylor 1976), hydrothermal fluid literature data (red; Michard and Albarède 1986; Michard 1989; James et al. 1995; James and Elderfield 1996; Bau and Dulski 1999), modern oxygenated seawater data (light blue; James et al. 1995; Alibo and Nozaki 1999; Haley et al. 2004; Deng et al. 2017), modern anoxic seawater data (dark blue; Schijf et al. 1995; Bau et al. 1997), and modern sediment pore water data (light purple; Haley et al. 2004; Soyol-Erdene and Huh 2013; Deng et al. 2017). b Redox equilibria for Eu3+/Eu2+, Yb3+/Yb2+, Fe3+/Fe2+, and SO42-/H2S as a function of temperature and oxygen fugacity (after Bau and Möller 1992)
Yttrium and Ho exhibit similar geochemical behavior, although Ho is preferentially complexed and removed from fluids by Fe-oxides and organic particles; consequently, seawater and seawater-derived fluids preserve super-chondritic (>28) Y/Ho values, whereas crustal fluids have chondritic Y/Ho values (Zhang et al. 1994; Nozaki et al. 1997; Bau and Dulski 1999). Similarly, there are subtle differences in the adsorption of LREE, MREE, HREE, and Ce onto organic particles, oxide phases, or mineral phases (e.g., German and Elderfield 1989; Sholkovitz et al. 1994; Alibo and Nozaki 1999). These differences affect the absolute and relative abundances of REE+Y in seawater as a function of redox state and water depth; further modification can then occur due to mineral formation or redox reactions in early diagenetic pore waters (e.g., Haley et al. 2004; Abbott et al. 2019; for REE+Y reference profiles see Fig. 2a and for a recent review on REE+Y fractionation see Smrzka et al. 2019).
The REE+Y partition strongly into carbonate minerals (e.g., Tanaka and Kawabe 2006; Voigt et al. 2017; Perry and Gysi 2020), are enriched in crustal rocks (>chondrite), and occur only as trace components in fluids (<chondrite; Fig. 2a). As a result, high fluid-rock ratios are necessary to alter whole-rock REE+Y budgets, whereas fluid REE+Y compositions can be strongly affected even at low fluid-rock ratios (Michard and Albarède 1986; Bau 1991). Therefore, REE+Y signatures of marine and diagenetic carbonate mineral phases are good archives of fluid chemistry and generally unaffected by later diagenesis (e.g., Webb et al. 2009; Liu et al. 2019).
Ligand concentration, pH, and temperature can have strong effects on REE+Y solubility in aqueous solutions (e.g., Michard 1989; Craddock et al. 2010; Williams-Jones et al. 2012) and the REE+Y composition of hydrothermal fluids can be extremely variable (Fig. 2a). Fluid-mineral interactions under hydrothermal conditions can also modify the REE+Y signatures of carbonate mineral phases (Vaughan et al. 2016). Commonly, hydrothermal fluids have positive Eu/Eu* and REE+YCN profiles that are LREE enriched, which has been interpreted to directly result from dissolution of plagioclase during fluid-rock interaction (e.g., Klinkhammer et al. 1994; Douville et al. 1999). Empirical and experimental studies suggest that both the LREE and Eu2+ are more soluble in high temperature Cl--rich fluids (Migdisov et al. 2009; Craddock et al. 2010; Williams-Jones et al. 2012). Because of the higher solubility of Eu2+ relative to Eu3+, the temperature-dependent redox sensitivity of Eu has a strong effect on Eu/Eu*; for example, under geochemically reasonable fO2 and acidic conditions the more soluble Eu2+ is only dominant over Eu3+ at temperatures above 200–250°C (Fig. 2b; Sverjensky 1984; Bau 1991; Bilal 1991). As a consequence, positive Eu/Eu* can only develop in hot (>200–250°C) hydrothermal fluids or in extremely reducing environments. Divalent Eu, however, is not readily incorporated into the carbonate crystal lattice, which is why positive Eu/Eu* can only develop in carbonate mineral phases that formed from hot fluids that cooled below 200–250°C in order to stabilize Eu3+ (Bau and Möller 1992). Many hydrothermal carbonates also preserve LREE depletion relative to PAAS (e.g., Roberts et al. 2009; Debruyne et al. 2013; Magnall et al. 2016). Such LREE-depleted REE+Y signatures in hydrothermal precipitates have been interpreted to result from co-precipitation with LREE-enriched phases (Roberts et al. 2009), LREE scavenging by fluid-mineral interaction along the fluid pathway (e.g., by monazite; Debruyne et al. 2016), inherited REE+Y signatures from fluid-rock interaction (Lüders et al. 1993; Hecht et al. 1999), or LREE retention in the fluid by Cl--complexes (Craddock et al. 2010; Magnall et al. 2016; Perry and Gysi 2018).
Geological background
The Carpentaria Province comprises the Mount Isa Inlier and the McArthur Basin, which formed in an intracontinental setting during the Paleo- to Mesoproterozoic (Betts et al. 2002, 2016; Giles et al. 2002). During this time, three unconformity-bound superbasins formed due to episodic rifting, sag phase, and basin inversion; those superbasins can be further subdivided into twelve supersequences (Fig. 1; Jackson et al. 2000; Southgate et al. 2000, 2013). The George Fisher, Hilton, and Mount Isa massive sulfide deposits are hosted by the Urquhart Shale Formation, which is part of the Mount Isa Group of the Isa Superbasin (Southgate et al. 2000, 2013). Closure of the superbasins was initiated by the onset of the Isan orogeny (ca. 1600 Ma), during which the rocks of the Mount Isa Inlier underwent multiple phases of deformation in at least four separate north-south or east-west directed deformation events (Page and Bell 1986; Connors and Page 1995; Bell and Hickey 1998).
The Urquhart Shale Formation was deposited at ca. 1652 ± 7 Ma and comprises mainly laminated to bedded mudstones and siltstones, which are variably calcareous, dolomitic, carbonaceous, and pyritic (Page and Sweet 1998; Painter et al. 1999; Page et al. 2000). The depositional environment of the Urquhart Shale Formation was interpreted as either a carbonate slope (Neudert 1983; Painter, 2003) or deeper-water environment (Domagala et al. 2000). Moderate concentrations of organic carbon (< 2 wt.%) and Mo (< 30 ppm) are consistent with anoxic and ferruginous seawater, which is further supported by sulfur isotope values of diagenetic pyrite that do not preserve evidence of extreme sulfate limitation under euxinic conditions (Rieger et al. 2020a, 2021a).
Carbonates are major rock-forming mineral phases in the Urquhart Shale. There are several types of detrital, diagenetic, and hydrothermal calcite and dolomite. Zoned, porous, silt-sized detrital clasts of calcite and dolomite were deposited together with other clastic mineral grains (Painter et al. 1999) and may have been entirely replaced by diagenetic carbonate (Chapman 1999). Diagenetic carbonates comprise calcareous and dolomitic cements, which have been interpreted as supratidal crusts (e.g., Neudert 1983; Painter et al. 1999), seafloor cement (Domagala et al. 2000), or as carbonate alteration (Chapman 1999). Furthermore, nodular carbonates have been interpreted as diagenetic precipitates (e.g., Chapman 1999, 2004; Domagala et al. 2000; Painter, 2003), as diagenetic pseudomorphs after sulfate minerals (e.g., van den Heuvel 1969; McClay and Carlile 1978; Painter et al. 1999), or as hydrothermal replacement textures (Perkins 1997; Perkins and Bell 1998).
In and around the George Fisher, Hilton, and Mount Isa deposits there are several types of calcite, dolomite, and siderite veins, infill, and breccias associated with the Zn-Pb and Cu ore systems (e.g., Waring 1990; Valenta 1994; Chapman 1999). Generally, carbonate mineral phases are more dolomitic and Fe- and Mn-bearing at the Mount Isa and George Fisher deposits relative to the un-mineralized Urquhart Shale (Painter, 2003; Rieger et al. 2021a). Furthermore, the ore forming systems have likely produced 18O-depleted carbonate haloes around the mineral deposits in the Urquhart Shale Formation beyond visible alteration (Waring 1990; Chapman 1999). Besides differences in carbonate mineralogy and isotopic composition, the footprints of these CD-type massive sulfide deposits are evident from element enrichment (e.g., Tl, Mn, Ag) and depletion (e.g., Na, Sr) in whole-rock data (Painter, 2003; Rieger et al. 2021a). The main sulfide minerals at George Fisher are formed in multiple paragenetic stages and comprise pyrite, sphalerite, galena, and pyrrhotite, with minor quantities of chalcopyrite (Chapman 1999, 2004; Murphy 2004; Rieger et al. 2020a). Recently, Rieger et al. (2020a) described pre-ore diagenetic fine-grained pyrite followed by three ore stages: (1) stratabound sphalerite + pyrite ± galena, (2) breccia-hosted galena + sphalerite + pyrite + pyrrhotite, and (3) vein and breccia-hosted pyrite + pyrrhotite + chalcopyrite ± galena and sphalerite.
A whole-rock lithogeochemistry study on Urquhart Shale samples from an unmineralized drill-core and from the George Fisher deposit has shown that REE+Y, Si, Al, Ti, Sc, Nb, and Th were immobile during hydrothermal alteration, and that mineralization at George Fisher resulted in dilution of these immobile elements (Rieger et al. 2021a). The nature of any REE+Y fractionation was not discussed in Rieger et al. (2021a) and the data are presented here in Fig. 3 and are also available in Rieger et al. (2020b), and fully discussed below. The REE+YCN profiles for Urquhart Shale and massive sulfide samples are LREE enriched relative to chondrite, and have relatively flat MREE and HREE profiles, as well as negative Eu/Eu* and chondritic Y/Ho values (Fig. 3). These REE+YCN profiles are generally very similar to PAAS and to other formations from the Mount Isa Group, yet, are offset to lower concentrations (Fig. 3; Nance and Taylor 1976; McLennan 1989).

Chondrite normalized (McDonough and Sun 1995) rare earth element and Y reference profiles for: PAAS (black; McLennan 1989), the Mount Isa Group (green; Breakaway Shale, Native Bee Siltstone, Spear/Kennedy Siltstone, Magazine Shale; Nance and Taylor 1976), whole-rock data from unmineralized Urquhart Shale (blue; Shovel Flats; Rieger et al. 2020b, 2021a), whole-rock data from mineralized Urquhart Shale (orange; George Fisher; Rieger et al. 2020b, 2021a), and whole-rock data from massive sulfides (dark red; George Fisher; Rieger et al. 2020b, 2021a)
Methods
Sampling and petrography
Representative samples were selected from a drill-core that intersected unmineralized Urquhart Shale (Shovel Flats drill-hole; n = 91) and from 4 drill holes that intersected mineralized Urquhart Shale at the George Fisher deposit (drill hole 8C K751, n = 61; 10C K795, n = 77; 10C K798, n = 57; 12C I797, n = 30). At George Fisher, representative samples were taken from the main ore bodies, weakly mineralized sections, barren siltstones or mudstones, and from sections of stratigraphically continuous hanging-wall stratigraphy.
Drill-core samples were examined using a binocular microscope and key samples were selected for polished thin-section preparation (n = 95). Thin sections were further examined using transmitted- and reflected-light microscopy and representative carbonate samples were selected for cathodoluminescence (CL) petrography (n = 30). The hot-cathode optical CL system was operated at 14 keV and 0.15-0.20 mA. To capture zonation and differences in luminescence, exposure times were varied and quartz grains were used as reference for luminescence intensity, because quartz is characterized by very low CL relative to carbonates or feldspars (Marshall 1988).
Electron probe micro-analyzer (EPMA)
Quantitative wavelength-dispersive spectrometry (WDS) was performed on a JEOL Superprobe JXA 8230, Hyperprobe JXA 8500F and Hyperprobe JXA-8530Fplus. The samples were coated with a 20-nm-thick carbon film and quantified for Mg, Ca, Fe, Mn, and Sr (and additionally for Ba and Zn in some samples) in carbonates using an acceleration voltage of 15 kV, a beam current of 10 nA, a probe size of 5–40 μm depending on the grain size and relatively short measurement times between 10 to 50 s (for both backgrounds (±) and peak). Those analytical parameters are reported as optimal for precise electron probe micro analyses of carbonates (Zhang et al. 2019). The φ(ρZ) matrix correction scheme (CITZAF; Armstrong 1995) was applied and natural standards were analysed during the course of the analytical sessions to ensure the quality of measured data. Standards were calcite (Ca), dolomite (Ca, Mg), siderite (Fe), strontianite (Sr), rhodonite (Mn), BaSi2O5 for Ba and ZnS for Zn. Under these conditions, analytical errors (2δ) and detection limits were 1.5% and 300 ppm for Ca, 0.4% and 300 ppm for Mg, Mn, and Fe, 0.1% and 200 ppm for Zn and Sr, and 0.1% and 350 ppm for Ba.
Mass spectroscopy (LA-ICP-MS)
Laser ablation ICP-MS of 19 polished thin sections was carried out using the Analyte Excite 193 nm ArF* excimer-based laser ablation (LA) system (Teledyne Photon Machines, Bozeman, MT, USA), coupled to the quadrupole-ICP-MS iCAP from Thermo Scientific. The LA-system is equipped with a HelEx II 2-volume ablation cell. Helium was used as a carrier gas for aerosol transport from the sample surface to the ICP and was mixed downstream with Ar as a make-up gas before entering the plasma. Operational parameters of the ICP-MS instrument and LA-unit were tuned for maximum sensitivity, low oxide formation based on the 232Th16O/ 232Th ratio and low laser-induced elemental fractionation based on the 232U/ 232Th ratio using NIST SRM 610. We used 43Ca as internal standard and the certified reference material NIST610 for calibration for all elements. Samples were ablated with a spot size of 50 μm, for 30 s with a repetition rate of 10Hz and an energy density of 2–3 J/cm2. Time intervals for data reduction were selected by visual inspection of each spectrum using Iolite™ (Paton et al. 2011) and the data reduction scheme X_trace_elemets_IS (Woodhead et al. 2007). Uncertainty estimates for the elements measured are based on repeated measurement of the reference material MACS-3 and ECRM-752 and are in general below 10 % (relative percent). Data were then screened for contamination by other mineral phases than carbonates (e.g., silicates, phosphates, sulfides) by using the elements Al, K, P, S, Si, and Ti. Data with high concentrations of these elements (Al + K + P + S + Si + Ti > 1000 ppm) were then rejected.
The Ce/Ce* and Eu/Eu* values were calculated geometrically (cf., Lawrence et al. 2006):
\( \mathrm{Eu}/{\mathrm{Eu}}_{CN}^{\ast }={\left(\frac{Eu}{{\left(S{m}^2\ast Tb\right)}^{\frac{1}{3}}}\right)}_{CN} \) and \( \mathrm{Ce}/{\mathrm{Ce}}_{SN}^{\ast }={\left(\frac{Ce}{\Pr \ast \left(\frac{\Pr }{Nd}\right)}\right)}_{SN} \)
Lanthanum was not used to calculate Ce* in order to avoid the influence of La anomalies on Ce/Ce* values (cf., Bau and Dulski 1996).
Results
Carbonate in the Urquhart Shale Formation
Detrital carbonate grains are most abundant in siltstone lithologies and preserve similar grain size (≤ 20 μm) and morphology (sub-rounded to angular) to detrital feldspar and quartz grains. Micritic calcite and dolomite is common throughout all lithologies and is very fine-grained (< 5 μm). These detrital and micritic carbonates were too fine grained for LA-ICP-MS analyses and will not be discussed further. Carbonate nodules are interbedded with laminated, pyritic, carbonaceous siltstones. Nodules are, typically, several cm long and wide in the lateral and vertical direction (Fig. 4a, b). Calcite is the dominant carbonate phase in the carbonate nodules and calcite grains are anhedral to subhedral and >10 μm. Coarse-grained (>100 μm) calcite veins cross-cut all lithologies and often contain euhedral pyrite. Where the veins cross-cut carbonate nodules, euhedral pyrite can occur in the nodules.

a-e Unmineralized nodular carbonate sample from the background drill-core. a Hand sample photograph. b Thin section photograph. c Transmitted light microphotograph; the arrows indicate respective mineral phases (blue = calcite; white = quartz; yellow = pyrite). d Cathodoluminescence microphotograph of the area shown in c, white circles indicate LA-ICP-MS analysis spots in calcite and the blue arrow indicates a K-feldspar grain with blue luminescence. e Chondrite-normalized REE+Y profiles of calcite from this sample (PR832SF080). f–i Mineralized nodular carbonate sample from George Fisher. f Hand sample photograph. g Thin section photograph. h Transmitted light microphotograph; the arrows indicate respective mineral phases (purple = dolomite and calcite; white = quartz; yellow = pyrite; light orange = sphalerite). i Cathodoluminescence microphotograph of area shown in h; the white circle indicates a LA-ICP-MS analysis spot. Note the strong difference in the luminescence signal between bright, yellow calcite and dull, red dolomite. j Chondrite-normalized REE+Y profiles of calcite and dolomite from this sample (PRK751017)
Carbonate at the George Fisher deposit
Both calcite and dolomite have been observed in the paragenesis at the George Fisher deposit. When calcite and dolomite occur together, calcite can be differentiated by a brighter luminescence, whereas dolomite commonly has a red, low-intensity luminescence signal (Fig. 4i). Nodular carbonates at George Fisher have similar morphologies to those from Shovel Flats samples, but comprise mostly anhedral to subhedral finer-grained dolomite and minor amounts of coarser-grained calcite. The edges of carbonate nodules are also commonly replaced by sphalerite (Fig. 4h).
In the sulfide paragenesis described by Rieger et al. (2020a, b); i.e., ore stages 1, 2, and 3), there are multiple phases of hydrothermal carbonate. The most abundant type of this carbonate is anhedral, coarse-grained (>100 μm), and Fe-rich dolomite; it is generally observed with anhedral quartz, and is in textural disequilibrium with sphalerite and galena, which is indicated by irregular grain boundaries and partial replacement of the dolomite by the sulfides (e.g., Fig. 5h, i). Dolomite, therefore, likely formed before sulfide precipitation of the individual ore stages.

a-e Carbonate in a stratabound massive sulfide sample from ore stage 1. a Hand sample photograph. b Thin section photograph. c Transmitted light microphotograph, the arrows indicate respective mineral phases (orange = dolomite; white = quartz; yellow = pyrite; light orange = sphalerite). d Cathodoluminescence microphotograph of the area shown in c; the white circles indicate LA-ICP-MS analysis spots in dolomite. e Chondrite-normalized REE+Y profiles' of dolomite from this sample (PRK798C014). f-i Carbonate in a massive sulfide breccia sample from ore stage 2. f Hand sample photograph. g Thin section photograph. h Transmitted light microphotograph; the arrows indicate respective mineral phases (orange = dolomite; white = quartz; light orange = sphalerite). i Cathodoluminescence microphotograph of the area shown in h; the white circles indicate LA-ICP-MS analysis spots in dolomite. j Chondrite-normalized REE+Y profiles' of dolomite from this sample (PRK751024)
In ore stage 1, this Fe-rich dolomite occurs in stratabound veins together with massive sulfide (e.g., Fig. 5c, d). In the later ore stages 2 and 3, this dolomite occurs as irregular clasts in massive sulfide breccias (e.g., Fig. 5h, i). The other, less abundant, types of carbonate that are associated with massive sulfides are calcites and dolomites that have planar crystal faces and are in textural equilibrium with quartz and sphalerite (Fig. 6d, e). In the samples studied here, these calcite and dolomite variants have only been found associated with ore stage 2. They are coarse-grained (> 100 μm) and occur in cross-cutting, mineralized carbonate-quartz veins or in carbonate-quartz infill adjacent to massive sulfide (Fig. 6). To simplify the later discussion, the carbonate variants that are associated with the massive sulfides will be grouped into two types: hydrothermal dolomite type A, which comprises dolomite in textural disequilibrium with sulfide minerals; and hydrothermal calcite and dolomite type B, which comprises carbonate in textural equilibrium with sulfide minerals. In the ore stage samples no phosphate-bearing mineral phases were observed.

a-i Carbonate veins and infill in a massive sulfide breccia sample from ore stage 2 (sample PRK7952037). a Hand sample photograph. b Thin section photograph. c Thin section photograph. d Transmitted light microphotograph of the area indicated in b; the arrows indicate respective mineral phases (dark red = dolomite; pink = calcite; white = quartz; light orange = sphalerite). Textural equilibrium of carbonate and quartz phases with ore-stage sphalerite is indicated by planar crystal faces orientated at 120°. e Cathodoluminescence microphotograph of the area shown in d; the white circles indicate LA-ICP-MS analysis spots in dolomite and calcite. f REE+Y profiles of dolomite from this sample. g Transmitted light microphotograph of the area indicated in c; the arrows indicate respective mineral phases (dark red = dolomite; white = quartz; light orange = sphalerite). h Cathodoluminescence microphotograph of the area shown in g; the white circles indicate LA-ICP-MS analysis spots in dolomite. i REE+Y profiles of calcite from this sample
There are also barren veins with coarse-grained calcite (> 100 μm) at George Fisher that cross-cut massive sphalerite of ore stage 1. In the samples studied here, the paragenetic relationship of these barren calcite veins with ore stage 2 and 3 could not be observed.
Major-element data—electron probe micro-analyzer (EPMA)
Major-element data for carbonates plot between the endmember compositions of calcite, dolomite and ankerite (Fig. 7). Calcite grains plot near the modal composition of calcite with MgCO3, FeCO3, and MnCO3 concentrations generally below 1.6 wt.%, 1.5 wt.%, and 1.1 wt.%, respectively. Dolomite and ankerite grains plot between the endmember modal compositions of the two minerals and preserve a range of MgCO3, FeCO3, and MnCO3 concentrations between 9–46 wt.%, 2–37 wt.%, and 0.2–9 wt.%, respectively. Overall, the carbonate major element data from this study are consistent with data for the George Fisher and Hilton deposits from other studies (Fig. 7; Valenta 1988; Chapman 1999; Murphy 2004).

CaCO3-MgCO3-(Fe,Mn)CO3 ternary diagram of carbonates from the George Fisher deposit and the Shovel Flats drill core. The colored fields denote EPMA carbonate data from Murphy (2004), Chapman (1999), and Valenta (1988). Endmember compositions of calcite, dolomite, ankerite, and siderite are shown in grey
Rare-earth elements and yttrium (REE+Y)—LA-ICP-MS
The LA-ICP-MS carbonate data of all samples is presented in Rieger et al. (2021b). Total rare-earth element concentrations (ƩREE) of carbonates are generally within one order of magnitude of the post-Archean Australian shale (PAAS; Fig. 8, Fig. 9c), and several times higher than REE concentrations in modern seawater, diagenetic pore fluids, and hydrothermal fluids (cf. Fig. 2).

a–g Chondrite normalized (McDonough and Sun 1995) REE+Y profiles for different carbonate sub-groups from the George Fisher deposit and the Shovel Flats drill-core. a Nodular carbonate (calcite) from the Shovel Flats drill core. b Late calcite veins from the Shovel Flats drill-core. c Nodular carbonate (calcite and dolomite) from George Fisher. d Hydrothermal type A dolomite from George Fisher. e Hydrothermal type B calcite from George Fisher. f Hydrothermal type Β dolomite from George Fisher. g Late calcite veins from George Fisher. The colored fields in a-g denote the 25-75 percentile for the respective sub-groups, thin black lines are the REE+Y profiles of individual analyses, thick black lines are PAAS (McLennan 1989), and grey lines are median values of REE+Y whole-rock data of the respective samples that in situ data was generated from (whole-rock data from Rieger et al. 2020b, 2021a)

a Mn and Fe concentrations of calcite and dolomite from the Shovel Flats drill core and the George Fisher deposit. Dashed lines denote expected cathodoluminescence signal of carbonate phases as a function of Fe and Mn concentration (fields after Pierson 1981; Machel and Burton 1991). b Box and whisker plot of Ce/CeSN for individual carbonate sub-groups. Boxes are 50 % of the data (Q1 to Q3), black lines and balls indicate median and mean values respectively, whiskers denote extreme values, and circles and triangles are outliers with >1.5*Q3-Q1 and >3*Q3-Q1 respectively. c Box and whisker plot of REE+Y concentrations for individual carbonate sub-groups. d Y/Ho and Pr/YbSN for calcite and dolomite from the Shovel Flats drill core and the George Fisher deposit. e Y/Ho and Eu/Eu*CN values of calcite and dolomite from the Shovel Flats drill core and the George Fisher deposit. b–e also show reference values of chondrite (1; McDonough and Sun 1995), PAAS (2; McLennan 1989), oxygenated seawater (3; Alibo and Nozaki 1999), anoxic seawater (4; Bau et al. 1997), the median value of the Mount Isa Group (5; Nance and Taylor 1976), and the whole-rock median values of Urquhart Shale from the unmineralized Shovel Flats drill core (6), from the George Fisher deposit (7), and from massive sulfide samples from George Fisher (8; Rieger et al. 2020b, 2021a). Note that values of #3, and #3 and #4 are lower than the plotted x-axis for b and c
Generally, the nodular calcite samples from Shovel Flats have REE+YCN profiles similar to PAAS (Fig. 8a), whereas nodular calcite and dolomite at George Fisher have more variable REE+YCN profiles (Fig. 8c). Hydrothermal dolomite type A from ore stages 1, 2, and 3 has REE+YCN profiles that are LREE-depleted and preserves negative Eu/Eu* (Fig. 8d). Hydrothermal calcite and dolomite type B also have REE+YCN profiles that are depleted in LREE, but preserve positive Eu/Eu* (Fig. 8e, f). The highest Eu/Eu* values are preserved in hydrothermal carbonate type B (calcite and dolomite; Fig. 8e, f, Fig. 9e) and in calcite veins from the Shovel Flats drill hole (Fig. 8b, Fig. 9e). The Ce/Ce* values are distributed around unity and are slightly skewed to negative anomalies (Fig. 9b). The Y/Ho values are variable, but are generally distributed between 15 and 50, Pr/YbSN values are typically below 1 in George Fisher dolomite and calcite, and the greatest variability in both Y/Ho and Pr/YbSN values is preserved in nodular dolomite and calcite from George Fisher samples (Fig. 9d, e).
The in situ composition of nodular calcite from Shovel Flats samples preserves overall higher REE+Y concentrations than whole-rock samples, whereas carbonates at George Fisher are generally LREE-depleted (Fig. 10a). When the data are normalized to the whole-rock REE+Y value of the respective sample (Fig. 10b), the in situ carbonate data from George Fisher is LREE-depleted, whereas Shovel Flats nodular calcite REE+Y profiles are relatively flat. Based on whole-rock REE+Y concentrations (REE+Ywr in ppm) and total carbonate contents of respective samples (XRDcarb-wr in %; Rieger et al. 2021a), the mass balance of the REE+Y in carbonate (REE+Ycarb-wr in %) has been calculated from the in situ REE+Y concentrations (REE+YLA-ICP-MS in ppm):

a Chondrite normalized REE+Y profiles for samples from the Shovel Flats drill core and the George Fisher deposit. The lines denote median values for in situ carbonate data from the Shovel Flats drill core (dark blue) and from the George Fisher deposit (red), and for whole-rock REE+Y profiles from the Shovel Flats drill core (light blue; n = 41) and from the George Fisher deposit (orange; Urquhart Shale and massive sulfide; n = 69); the light purple (Shovel Flats) and light red (George Fisher) fields denote the 25–75 percentiles of the in situ carbonate REE+Y profiles. For more detailed whole-rock data see Fig. 3. b REE+Y profiles (medians and 25–75 percentiles) for individual carbonate sub-groups from the Shovel Flats drill core and the George Fisher deposit normalized to whole-rock REE+Y data of the respective sample that in situ data was generated from
Assuming that the analyzed carbonate is representative for the entire sample, carbonate contains 61 % (average value, n = 2 samples) and 49 % (average value, n = 8 samples) of total REE+Y concentrations in the samples from Shovel Flats and George Fisher respectively.
Discussion
Pre-ore carbonate
The unmineralized Paleoproterozoic Urquhart Shale Formation consists of carbonate-rich siltstones and mudstones that comprise detrital, micritic, and nodular carbonates (e.g., Neudert 1983; Chapman 1999; Painter et al. 1999). The detrital and micritic carbonate is fine grained (≤ 20 μm) and not amenable to laser ablation studies, but the nodular carbonates are coarser grained and occur both in the barren Urquhart Shale at Shovel Flats and in the George Fisher deposit. In the mineralized units the nodular carbonates pre-date the first phase of mineralization (Fig. 4; Chapman 2004; Rieger et al. 2021a), and therefore, provide constraints on carbonate formation under pre-ore conditions.
The nodular carbonates in the Urquhart Shale have been interpreted as pseudomorphs after sulfate evaporites (e.g., Painter et al. 1999) or as diagenetic precipitates (e.g., Domagala et al. 2000). Indicators for shallow-water, evaporitic conditions, such as halite casts and stromatolites, are preserved in other formations of the Mount Isa Group (McClay and Carlile 1978; Neudert and Russell 1981; Neudert 1983). However, sulfur isotope values of early diagenetic pyrite (cf., Painter et al. 1999; Rieger et al. 2020a) indicate that open-system conditions with respect to sulfate availability were dominant during deposition and early diagenesis of the Urquhart Shale. Such conditions are inconsistent with the closed-system conditions typical of evaporite formation and more typical of diagenetic carbonate precipitation (cf., Domagala et al. 2000; Chapman 2004). The composition of the nodular carbonates from the Urquhart Shale Formation is, therefore, considered representative of diagenetic pore fluids, which predated the hydrothermal system at George Fisher.
The REE+Y systematics in seawater and diagenetic pore fluids are typically controlled by redox processes (for a review see Smrzka et al. 2019). Unlike the modern oxygenated oceans, mid- and Paleoproterozoic oceans were mostly ferruginous (anoxic, non-sulfidic), with euxinic conditions spatially and temporally restricted to highly productive margins and with oxygenated conditions only developed in surface waters (e.g., Canfield 1998; Planavsky et al. 2011; Lyons et al. 2014). Indeed, the REE+Y compositions of Proterozoic carbonates are consistent with REE+Y systematics observed in modern anoxic basins (cf., Bau et al. 1997; Tang et al. 2016; Bellefroid et al. 2019). Such REE+Y systematics are characterized by both variable Ce/Ce*SN and variable Eu/Eu*CN values around unity, variable chondritic to super-chondritic Y/Ho values, and REE+YCN profiles that are similar to PAAS (e.g., Planavsky et al. 2010; Tang et al. 2016; Bellefroid et al. 2019). Compared to oxygenated seawater (Fig. 2a), these contrasting REE+Y signatures are likely the result of (1) the dissolution of LREE-, Ce-, and Ho-enriched phases, such as Mn-oxides, at the chemocline between oxidizing and reducing seawater, and (2) enhanced solubility of Eu2+ under reducing conditions (Sverjensky 1984; Planavsky et al. 2010).
Altogether, the nodular calcite preserves Ce/Ce*SN, Eu/Eu*CN, and Y/Ho values (Fig. 9b, e) that are consistent with pore waters derived from anoxic bottom waters during the deposition of the Urquhart Shale Formation. The existence of ferruginous conditions is supported by the sulfur isotope values of early diagenetic pyrite and low Mo concentrations from the Urquhart Shale Formation (Rieger et al. 2020a, 2021a) and in agreement with growing evidence for wide-spread ferruginous conditions in Proterozoic oceans (e.g., Poulton et al. 2010; Planavsky et al. 2011; Song et al. 2017). As such, the nodular carbonate chemistry from the unmineralized Urquhart Shale Formation preserves normal background marine and early-diagenetic signatures for the Proterozoic.
Carbonate at George Fisher
The George Fisher deposit was formed from a multi-stage mineralizing system (Chapman 2004; Rieger et al. 2020a), and it is possible that multiple processes contributed to the trace element composition of George Fisher carbonates. The deposit comprises a number of different carbonate generations (this study; Chapman 1999; Murphy 2004), including evidence of hydrothermal alteration that has resulted in dolomitization of pre-ore calcite (Rieger et al. 2021a). Hydrothermal dolomitization is clearly observed in the comparison between nodular carbonates from unmineralized (Fig. 4a–e; Shovel Flats) and mineralized (Fig. 4f–j; George Fisher) Urquhart Shale samples, whereby samples from the latter are partially replaced by sphalerite and have higher dolomite/calcite ratios (Fig. 4g, h; Rieger et al. 2021a).
Nodular carbonates from the unmineralized rocks also have homogenous CL signals (Fig. 4d), whereas those at George Fisher have irregular signals with relatively bright CL of calcite compared to dull and quenched CL of dolomite (Fig. 4i). Such dull and quenched luminescence is characteristic of carbonate minerals with high Fe and variable Mn concentrations (Pierson 1981; Machel and Burton 1991; Baele et al. 2019), which is also consistent with the major-element chemistry of George Fisher and Shovel Flats carbonates (Fig. 7, Fig. 9a). These irregular CL signals (Fig. 4i) and variable Fe and Mn concentrations (Fig. 9a), together with higher whole-rock dolomite/calcite ratios (Rieger et al. 2021a), are consistent with dolomitization of pre-ore calcite that was associated with the mineralization at George Fisher. Similar to calcite and dolomite in nodular carbonate samples at George Fisher, type A dolomite has dull to quenched luminescence (Fig. 5c, h). It is, therefore, likely that dolomitization was associated with all stages of mineralization at George Fisher.
This dolomitization and hydrothermal alteration has not resulted in fractionation of the whole-rock REE+YCN composition between Shovel Flats and George Fisher samples, apart from dilution of the REE+Y in the samples containing massive sulfide mineralization (Fig. 3; Rieger et al. 2021a). In contrast, the REE+YCN composition of hydrothermal carbonate from George Fisher is fractionated (LREE-depleted) relative to respective whole-rock and in situ pre-ore carbonate REE+YCN profiles (Fig. 10). One possibility is that REE+Y were fractionated during precipitation of another phase that formed in an assemblage with the hydrothermal carbonate. For example, phosphate minerals (e.g., monazite or apatite) have a high partition coefficient for the REE+Y and could have resulted in REE+Y-fractionated carbonate without changing the overall whole-rock composition (cf., Debruyne et al. 2016).
A chemical mass balance analysis of the whole-rock composition of samples from Shovel Flats and George Fisher indicates that P was relatively immobile and in fact was slightly depleted during hydrothermal alteration at George Fisher (Rieger et al. 2021a). Co-precipitation of a P-bearing phase would have only locally affected the REE+Y composition of carbonate at George Fisher (e.g., due to local dissolution and precipitation reactions of P-bearing phases). Nevertheless, there is no evidence of hydrothermal phosphate mineral phases in an assemblage with the ore stage sulfides at George Fisher, making this scenario unlikely. Rather, it is more likely that hydrothermal carbonate only accounts for a small fraction of the whole-rock REE+Y budget at George Fisher. For example, it is possible that the whole-rock REE+Y composition is dominated by pre-ore carbonate, which has a similar REE+YCN profile to the whole-rock (Figs. 4, 8, 10), and by phyllosilicate mineral phases, which are commonly important hosts for REE+Y in fine-grained sedimentary rocks (e.g., Condie 1991; Abbott et al. 2019). Based on these considerations, the following discussion will focus on hydrothermal carbonate at George Fisher and the implications for the hydrothermal system that can be derived from the carbonate REE+Y composition.
The REE+Y composition of hydrothermal dolomite should provide a record of fluid chemistry that includes both pre- and syn-sulfide hydrothermal events. For example, the sulfide replacement textures and irregular grain boundaries with sphalerite (Fig. 4g–i, Fig. 5c, d, h, i) are indicative of a pre-sphalerite timing for some dolomitization (i.e., type A) at the onset of mineralization (e.g., Chapman 1999). In contrast, the dolomite and calcite from type B formed in textural equilibrium with sphalerite (Fig. 6d, e). On the basis of these paragenetic relationships, the REE+Y chemistry of the pre-sphalerite dolomite (type A) should provide constraints on the hydrothermal fluids that caused the alteration of pre-ore calcite whereas type B carbonate should provide constraints on the hydrothermal fluids during sphalerite formation. In order to constrain the syn-sphalerite end-member of the hydrothermal fluid, the REE+Y composition of this type B carbonate is discussed first.
Type B calcite and dolomite are characterized by (1) LREE depletion relative to PAAS, respective whole-rock, and pre-ore calcite, (2) chondritic and sub-chondritic Y/Ho values, and (3) Eu/Eu* values >1 (Fig. 6f, i, Fig. 8e, f, Fig. 9d, e). Light-rare-earth-element fractionation is a common feature of hydrothermal fluids and there are a number of processes that could have produced the REE+YCN profiles in the type B carbonates (Fig. 6f, i, Fig. 8e, f). A mineralogical control on this LREE fractionation is unlikely because both calcite and dolomite are LREE-depleted relative to pre-ore calcite (Fig. 8a, e, f). This observation is consistent with other studies on hydrothermal gangue carbonates that indicate no significant REE+Y fractionation between calcite and dolomite (e.g., Hecht et al. 1999; Roberts et al. 2009). The depletion of LREEs in carbonate mineral phases can also be inherited from complexation-controlled REE+Y-leaching from crustal source rocks (e.g., Lüders et al. 1993) or from LREE-scavenging by mineral phases with high REE-partition coefficients (e.g., monazite) along the fluid pathways (e.g., Debruyne et al. 2013, 2016). The source rocks for the mineralization are unknown at George Fisher and whether the footwall units could contain significant amounts of phosphatic minerals is also unknown. However, if the REE+Y signatures were inherited from the metal source or fluid-rock interaction along the flow pathway, they would likely be more homogenous than the variability preserved in the REE+Y profiles of hydrothermal calcite and dolomite at George Fisher (Fig. 6f, i, Fig. 8e, f). Notably, the rocks of the Mount Isa Group and of the unconformably underlying Eastern Creek Volcanics have LREE-enriched REE+YCN profiles (Fig. 3; Nance and Taylor 1976; Hannan et al. 1993); as a consequence, a hydrothermal fluid buffered by these rocks would most likely not be LREE-depleted. Instead, the observed variability may result from closed-system Rayleigh fractionation during carbonate precipitation (e.g., Kontak and Jackson 1995), which is consistent with variable LREE concentrations in stage B calcites (e.g., Fig. 6i). Similar observations have been made from experimental data, where carbonate grown from static solutions preserved large variability in REE composition between individual growth zones (Barker and Cox 2011). Fluid composition and temperature also provide a major control on REE+Y fractionation (e.g., Michard 1989; Craddock et al. 2010; Williams-Jones et al. 2012). Unfortunately, there are no direct constraints on the composition of the mineralizing fluids at George Fisher (e.g., fluid inclusions). Based on the basin fill in the stratigraphy below the Carpentaria CD-type deposits, Cooke et al. (2000) suggested that the mineralizing fluids were likely oxygenated, neutral to mildly acidic, and saline brines (ca. 25 wt.% NaClequiv), which would have been highly effective in transporting Zn and Pb at moderate temperatures (150°C). Fluid inclusion data from the relatively undeformed Century deposit do indicate relatively low-temperature (<125°C) and high-salinity fluids (ca. 23 wt.% NaClequiv; Polito et al. 2006). Notably, salinity can have a strong effect on REE+Y solubility and fractionation. For example, experimental studies and numerical modelling of REE+Y partitioning and solubility in hydrothermal fluids suggest that chlorides are the dominant ligands for REE+Y-complexation at hydrothermal temperatures and that Cl--ligands form the strongest complexes with the REE+Y with the lowest charge/radius ratios (Eu2+ and LREE; e.g., Migdisov et al. 2009; Williams-Jones et al. 2012; Perry and Gysi 2018). If the mineralizing fluids for the George Fisher deposit were saline (cf., Cooke et al. 2000; Polito et al. 2006), Cl--complexes would have been the dominant ligands for REE+Y-complexation and could have resulted in stronger LREE-retention in the fluid during carbonate precipitation. Consequently, REE+YCN profiles of syn-ore carbonates should generally be LREE-depleted despite the effects of intrinsic processes.
A second common signature of type B carbonates at George Fisher are chondritic and sub-chondritic Y/Ho values ratios (Fig. 9d, e). Chondritic Y/Ho values are typical of hydrothermal fluids (Bau and Dulski 1999; Douville et al. 1999), whereas sub-chondritic Y/Ho values may indicate fractionation of Y relative to Ho. Experiments at ambient temperatures suggest that Ho partitions more strongly into calcite than Y (e.g., Tanaka and Kawabe 2006). Such differential partitioning is consistent with sub-chondritic Y/Ho values in type B calcite at George Fisher. There is, however, no experimental data available for Y/Ho fractionation during carbonate precipitation at hydrothermal temperatures. If future experiments show differential fractionation of Y and Ho into carbonate minerals at hydrothermal temperatures, sub-chondritic Y/Ho values may be a distinguishing criterium for hydrothermal carbonates.
A third dominant feature of type B carbonates at George Fisher are Eu/Eu* values >1 (Fig. 6f, i, Fig. 8e, f, Fig. 9b). The major control on Eu/Eu* is temperature (Fig. 2b; Sverjensky 1984; Bau 1991; Bilal 1991). In oxidized hydrothermal fluids, such as those suggested for the Carpentaria CD-type deposits (SO42- > H2S; Cooke et al. 2000), Eu/Eu* values >1 can only develop at high temperatures, because the more soluble Eu2+ is only stable at temperatures >200-250°C (Fig. 2b; Sverjensky 1984; Bau 1991; Bilal 1991). It is, therefore, unlikely that the George Fisher deposit formed from fluids that were initially cooler than 200-250°C. Divalent Eu is, however, not readily incorporated into the crystal lattice of carbonate minerals (Bau and Möller 1992); therefore, the hot, Eu2+-dominated hydrothermal fluids (>200-250°C) must have cooled in order to stabilize sufficient Eu3+ to produce Eu/Eu* values >1 in the type B carbonate at George Fisher. Such fluid cooling at George Fisher is consistent with thermodynamic models for sulfide formation (Rieger et al. 2020a), calcite twinning temperatures (Murphy 2004), and also with overall temperature constraints for the Urquhart Shale Formation (see summary for temperature constraints in Rieger et al. 2021a). Furthermore, this is consistent with Eu/Eu* values <1 preserved by type A dolomite from ore stage 2 (Fig. 5j). These Eu/Eu* values <1 in type A dolomites support the hypothesis that the hydrothermal fluids were hot at the onset of mineralization, and then cooled below 200-250°C before type B calcite and dolomite formation.
Dolomite that formed from dolomitization of pre-ore calcite at the onset of mineralization is preserved both in samples of mineralized nodular carbonate and in massive sulfide samples at George Fisher (Fig. 4f–i, Fig. 5a–i). Partially mineralized nodular carbonates preserve the highest textural variability, together with the highest variability in the REE+Y composition (Fig. 4e, j, Fig. 8c, Fig. 9b–d). Therefore, they likely comprise mixed REE+Y compositions from pre-ore diagenetic and hydrothermal end members, which is consistent with the large ranges of LREE-depletion relative to pre-ore calcite and the large variability of Y/Ho values (Fig. 8c, Fig. 9d). The REE+Y composition of type A dolomite at George Fisher is slightly more homogenous than that of mineralized nodular carbonate (Fig. 8d, Fig. 9d), and this may indicate that the REE+Y composition is more dominated by hydrothermal signatures as a result of progressive dolomitization. The REE+YCN profiles of type A dolomite are LREE-depleted relative to PAAS, pre-ore calcite, and to whole-rock REE+Y profiles (Fig. 4j, Fig. 5e, j, Fig. 8d, Fig. 10b). This LREE-depletion is similar to REE+YCN profiles of type B calcite and dolomite and consistent with Cl--complexation and LREE-retention in the fluid.
Chemical alteration and dissolution-reprecipitation of carbonate is not limited to the high-grade domains of ore deposits, and chemical and isotopic changes in carbonate can be traced beyond zones of visible alteration (e.g., Barker et al. 2013; Vaughan et al. 2016). In fact, isotopic studies indicate that large-scale 18O-depletion in carbonate is associated with the Mount Isa Cu-system (Waring 1990; Waring et al. 1998a), and similarly, 18O-depletion has also been reported for carbonate from the George Fisher Zn-Pb system (Chapman 1999). There is, therefore, the potential that the mineralizing fluids at George Fisher have also produced alteration of the REE+Y signatures beyond the highly mineralized zones. For example, sulfide formation would have resulted in the destabilization of chloride complexes and carbonate dissolution via the generation of acid (e.g., ZnCl2 + HS- → ZnS + H+ + 2Cl-). Such a fluid could have caused even higher solubilities of LREE as Cl--complexes, which could have resulted in LREE-depletion along the fluid migration pathway beyond visible alteration.
Late calcite veins postdate the stratabound Zn mineralization at George Fisher, and late calcite veins cross-cut all lithologies in the unmineralized Shovel Flats drill core. Cross-cutting calcite veins have also been described before at Mount Isa and George Fisher (e.g., Waring et al. 1998b; Murphy 2004). The calcite veins at George Fisher preserve REE+Y signatures similar to the pre-ore calcites and to PAAS (Fig. 8g, Fig. 9), which may indicate that they are rock-buffered. The late calcite veins from Shovel Flats preserve REE+Y signatures that are more similar to type B calcites (Fig. 8b, Fig. 9), which could represent a common hydrothermal affinity. The differences in REE+Y composition suggest that there are, at least, two generations of late calcite veins. These veins were, however, not the focus of this study and future work is needed to understand their significance and relationship to mineralization.
Overall, the Y/Ho values, Eu/Eu* values, and LREE-depleted REE+YCN profiles that are characteristic of George Fisher calcite and dolomite are consistent with interaction of a hot, saline (Cl--rich) hydrothermal fluid that subsequently cooled to temperatures below 200-250°C during fluid-rock interaction. Notably, such conditions correspond well with other CD-type systems, for which the thermal evolution of the hydrothermal fluids is well constrained (e.g., MacMillan Pass; Magnall et al. 2016).
Implications for the application of in situ REE+Y carbonate data
There are some important implications of the in situ REE+Y carbonate data from George Fisher, both for ore formation models and future exploration programs. The George Fisher deposit formed from a multi-stage system and all mineralization post-dated deposition of the host rock (Chapman 2004; Rieger et al. 2020a). The mineralizing fluids must have reacted with the host rock, therefore, in order to create sufficient porosity for fluid flow and base metal sulfide precipitation. The fractionation between whole-rock REE+Y profiles and carbonate in situ REE+Y profiles (Fig. 10a, b) indicates that (1) the whole-rock REE+Y composition is still dominated by diagenetic carbonate and other REE+Y-bearing phases after hydrothermal alteration, that (2) carbonate mineral phases were more sensitive to REE+Y alteration relative to the whole-rock, and that (3) fluid-rock ratios were not high enough to alter whole-rock REE+Y compositions (cf., Michard and Albarède 1986; Bau and Möller 1992). Altogether, this suggests that the hydrothermal fluids at George Fisher were highly selective in replacing and altering pre-ore carbonate mineral phases. Moreover, this means that REE+Y carbonate data could provide a more sensitive record of hydrothermal alteration relative to bulk-rock techniques. At this stage, however, the small number of samples analyzed at George Fisher is not sufficient to evaluate the 3D REE+Y footprint of the mineralizing system.
The paragenetic and geochemical data provide evidence of fluid-rock interaction that involved hot, acidic, saline, and base metal-bearing hydrothermal fluids. The resulting replacement and dolomitization of pre-ore calcite led to the development of secondary porosity, fluid cooling, and pH increase, which lowered base metal solubilities and caused sulfide precipitation. Such a diagenetic carbonate replacement model is consistent with (1) carbonate replacement textures (e.g., Fig. 4; Chapman 2004; Rieger et al. 2021a), with (2) fluid cooling indicated by Eu/Eu* <1 in type A dolomite opposed to Eu/Eu* >1 in type B dolomite and calcite (Fig. 8d–f, Fig. 9e), and with (3) Cl--rich hydrothermal fluids indicated by LREE-depleted REE+YCN profiles in George Fisher carbonates (Fig. 4j, Fig. 5e, j, Fig. 6f, i, Fig. 8c–f, Fig. 10). Notably, carbonate replacement was reported for a number of other CD-type massive sulfide systems in the Carpentaria province (Eldridge et al. 1993; Perkins and Bell 1998; Chapman 2004; Magnall et al. 2021; Spinks et al. 2021), which highlights the importance of the reactive carbonate-rich host rocks throughout the province.
More broadly, it is generally accepted that base-metal deposits in sedimentary basins form from saline hydrothermal brines (Bodnar et al. 2014; Heinrich and Candela 2014); and indeed, previous studies on Zn-Pb- and Cu-systems in the Carpentaria province propose that saline fluids were responsible for base-metal transport (Heinrich et al. 1989; Cooke et al. 2000; Polito et al. 2006). At George Fisher, the LREE-depleted REE+YCN profiles in calcite and dolomite (Fig. 4j, Fig. 5e, j, Fig. 6f, i, Fig. 8c–f, Fig. 10a, b) are consistent with LREE-retention in such Cl--rich saline hydrothermal fluids. Furthermore, there is evidence that LREE-depletion is a common feature of carbonate mineral phases from a number of mineral deposits that formed from saline fluids (Roberts et al. 2009; Debruyne et al. 2013; Genna et al. 2014; Magnall et al. 2016), whereas LREE depletion is not evident from carbonates that precipitated from low-salinity fluids that were dominated by HS-- or CO32--complexes (Maskenskaya et al. 2015; Vaughan et al. 2016). As a consequence, LREE depletion in calcites or dolomites relative to background REE+Y profiles may be an effective tracer for fluid-rock interaction involving saline hydrothermal fluids. So, if this hypothesis is supported by future studies, the analysis of REE+Y in carbonate mineral phases could be useful for future exploration programs in sedimentary basins in order to identify hydrothermally altered carbonate-rich lithologies.
In summary we show that LA-ICP-MS analyses of REE+Y in carbonate minerals can distinguish hydrothermal carbonate from pre-existing diagenetic carbonate, and this may be a useful exploration tool in combination with whole-rock and isotope geochemistry for mineral deposits hosted in sedimentary rocks. Future studies should test this hypothesis by systematically sampling the 3D grid around an (undeformed) deposit to assess the nature of the distal footprint on carbonate REE+Y systematics.
Data availability
The data is available via GFZ Data Service:
Rieger P, Magnall JM, Gleeson SA, Oelze M, Wilke FDH, Lilly R (2021) Differentiating between hydrothermal and diagenetic carbonates using rare earth element and yttrium (REE+Y) geochemistry: A case study from the Paleoproterozoic George Fisher massive sulphide Zn deposit. GFZ Data Services. https://doi.org/10.5880/GFZ.3.1.2020.005
References
Abbott AN, Löhr S, Trethewy M (2019) Are clay minerals the primary control on the oceanic rare earth element budget? Front Mar Sci 6:504
Alibo DS, Nozaki Y (1999) Rare earth elements in seawater: particle association, shale-normalization, and Ce oxidation. Geochim Cosmochim Acta 63:363–372
Armstrong JT (1995) Citzaf-a package of correction programs for the quantitative Electron Microbeam X-Ray-Analysis of thick polished materials, thin-films, and particles. Microbeam Anal 4:177–200
Baele J, Decrée S, Rusk B (2019) Cathodoluminescence applied to ore geology and exploration. Ore Depos Orig Explor Exploit 131–161
Barker SLL, Cox SF (2011) Oscillatory zoning and trace element incorporation in hydrothermal minerals: insights from calcite growth experiments. Geofluids 11:48–56
Barker SLL, Cox SF, Eggins SM, Gagan MK (2006) Microchemical evidence for episodic growth of antitaxial veins during fracture-controlled fluid flow. Earth Planet Sci Lett 250:331–344
Barker SLL, Dipple GM, Hickey KA, Lepore WA, Vaughan JR (2013) Applying stable isotopes to mineral exploration: Teaching an old dog new tricks. Econ Geol 108:1–9
Bau M (1991) Rare-earth element mobility during hydrothermal and metamorphic fluid-rock interaction and the significance of the oxidation state of europium. Chem Geol 93:219–230
Bau M, Dulski P (1996) Distribution of yttrium and rare-earth elements in the Penge and Kuruman iron-formations, Transvaal Supergroup, South Africa. Precambrian Res 79:37–55
Bau M, Dulski P (1999) Comparing yttrium and rare earths in hydrothermal fluids from the Mid-Atlantic Ridge: implications for Y and REE behaviour during near-vent mixing and for the Y/Ho ratio of Proterozoic seawater. Chem Geol 155:77–90
Bau M, Möller P (1992) Rare earth element fractionation in metamorphogenic hydrothermal calcite, magnesite and siderite. Mineral Petrol 45:231–246
Bau M, Möller P, Dulski P (1997) Yttrium and lanthanides in eastern Mediterranean seawater and their fractionation during redox-cycling. Mar Chem 56:123–131
Bell TH, Hickey KA (1998) Multiple deformations with successive subvertical and subhorizontal axial planes in the Mount Isa region; their impact on geometric development and significance for mineralization and exploration. Econ Geol 93:1369–1389
Bellefroid EJ, Planavsky NJ, Hood AVS, Halverson GP, Spokas K (2019) Shallow water redox conditions of the mid-Proterozoic Muskwa Assemblage, British Columbia, Canada. Am J Sci 319:122–157
Betts PG, Giles D, Lister GS, Frick LR (2002) Evolution of the Australian lithosphere. Aust J Earth Sci 49:661–695
Betts PG, Armit RJ, Stewart J, Aitken ARA, Ailleres L, Donchak P, Hutton L, Withnall I, Giles D (2016) Australia and nuna. Geol Soc London Spec Publ 424:47–81
Bilal BA (1991) Thermodynamic study of Eu3+/Eu2+ redox reaction in aqueous solutions at elevated temperatures and pressures by means of cyclic voltammetry. Zeitschrift für Naturforsch A 46:1108–1116
Bodnar RJ, Lecumberri-Sanchez P, Moncada D, Steele-MacInnis M (2014) 13.5—Fluid inclusions in hydrothermal ore deposits. Treatise Geochemistry, Second edn Elsevier, Oxford 119–142
Brand U, Veizer J (1980) Chemical diagenesis of a multicomponent carbonate system; 1, Trace elements. J Sediment Res 50:1219–1236
Canfield DE (1998) A new model for Proterozoic ocean chemistry. Nature 396:450–453
Chapman LH (1999) Geology and genesis of the George Fisher Zn-Pb-Ag deposit, Mount Isa, Australia. Unpubl. PhD Thesis, James Cook University 315
Chapman LH (2004) Geology and mineralization styles of the George Fisher Zn-Pb-Ag deposit, Mount Isa, Australia. Econ Geol 99:233–255
Cline JS, Hofstra AH, Muntean JL, Tosdal RM, Hickey KA (2005) Carlin-type gold deposits in Nevada: Critical geologic characteristics and viable models. Econ Geol 100th Anniv 451:484
Condie KC (1991) Another look at rare earth elements in shales. Geochim Cosmochim Acta 55:2527–2531
Connors KA, Page RW (1995) Relationships between magmatism, metamorphism and deformation in the western Mount Isa Inlier, Australia. Precambrian Res 71:131–153
Cooke DR, Bull SW, Large RR, McGoldrick PJ (2000) The importance of oxidized brines for the formation of Australian Proterozoic stratiform sediment-hosted Pb-Zn (Sedex) deposits. Econ Geol 95:1–18
Craddock PR, Bach W, Seewald JS, Rouxel OJ, Reeves E, Tivey MK (2010) Rare earth element abundances in hydrothermal fluids from the Manus Basin, Papua New Guinea: Indicators of sub-seafloor hydrothermal processes in back-arc basins. Geochim Cosmochim Acta 74:5494–5513
De Baar HJW, Bacon MP, Brewer PG (1983) Rare-earth distributions with a positive Ce anomaly in the Western North Atlantic Ocean. Nature 301:324–327
Debruyne D, Balcaen L, Vanhaecke F, Muchez P (2013) Rare earth element and yttrium characteristics of carbonates within the sediment-hosted Luiswishi and Kamoto Cu-Co deposits, Katanga Copperbelt (Democratic Republic of Congo-DRC). Geol Belg 16:76–83
Debruyne D, Hulsbosch N, Muchez P (2016) Unraveling rare earth element signatures in hydrothermal carbonate minerals using a source–sink system. Ore Geol Rev 72:232–252
Deng Y, Ren J, Guo Q, Cao J, Wang H, Liu C (2017) Rare earth element geochemistry characteristics of seawater and porewater from deep sea in western Pacific. Sci Rep 7:1–13
Domagala J, Southgate PN, McConachie BA, Pidgeon BA (2000) Evolution of the Palaeoproterozoic Prize, Gun and lower Loretta Supersequences of the Surprise Creek Formation and Mt Isa Group. Aust J Earth Sci 47:485–507
Douville E, Bienvenu P, Charlou JL, Donval JP, Fouquet Y, Appriou P, Gamo T (1999) Yttrium and rare earth elements in fluids from various deep-sea hydrothermal systems. Geochim Cosmochim Acta 63:627–643
Eldridge CS, Williams N, Walshe JL (1993) Sulfur isotope variability in sediment-hosted massive sulfide deposits as determined using the ion microprobe SHRIMP; II, A study of the HYC Deposit at McArthur River, Northern Territory, Australia. Econ Geol 88:1–26
Genna D, Gaboury D, Roy G (2014) Evolution of a volcanogenic hydrothermal system recorded by the behavior of LREE and Eu: Case study of the Key Tuffite at Bracemac–McLeod deposits, Matagami, Canada. Ore Geol Rev 63:160–177
German CR, Elderfield H (1989) Rare earth elements in Saanich Inlet, British Columbia, a seasonally anoxic basin. Geochim Cosmochim Acta 53:2561–2571
Gibson GM, Hutton LJ, Holzschuh J (2017) Basin inversion and supercontinent assembly as drivers of sediment-hosted Pb–Zn mineralization in the Mount Isa region, northern Australia. J Geol Soc Lond 174:773–786
Giles D, Betts P, Lister G (2002) Far-field continental backarc setting for the 1.80–1.67 Ga basins of northeastern Australia. Geology 30:823–826
Glencore (2019) Resources and reserves report: December 31 71. https://www.glencore.com/dam/jcr:0e7b6c0f-e670-49fe-9048-8582e7530dab/GLEN_2019_Resources_Reserves_Report%2D%2D.pdf
Haley BA, Klinkhammer GP, McManus J (2004) Rare earth elements in pore waters of marine sediments. Geochim Cosmochim Acta 68:1265–1279
Hannan KW, Golding SD, Herbert HK, Krouse HR (1993) Contrasting alteration assemblages in metabasites from Mount Isa, Queensland; implications for copper ore genesis. Econ Geol 88:1135–1175
Hecht L, Freiberger R, Gilg HA, Grundmann G, Kostitsyn YA (1999) Rare earth element and isotope (C, O, Sr) characteristics of hydrothermal carbonates: genetic implications for dolomite-hosted talc mineralization at Göpfersgrün (Fichtelgebirge, Germany). Chem Geol 155:115–130
Heinrich CA, Candela PA (2014) Fluids and ore formation in the Earth’s crust. In: Treatise on Geochemistry (Second Edition). Elsevier 1–28
Heinrich CA, Andrew AS, Wilkins RWT, Patterson DJ (1989) A fluid inclusion and stable isotope study of synmetamorphic copper ore formation at Mount Isa, Australia. Econ Geol 84:529–550
Himmler T, Bach W, Bohrmann G, Peckmann J (2010) Rare earth elements in authigenic methane-seep carbonates as tracers for fluid composition during early diagenesis. Chem Geol 277:126–136
Hitzman M, Kirkham R, Broughton D, Thorson J, Selley D (2005) The sediment-hosted stratiform copper ore system. Econ Geol 100:609–642
Jackson MJ, Scott DL, Rawlings DJ (2000) Stratigraphic framework for the Leichhardt and Calvert Superbasins: review and correlations of the pre-1700 Ma successions between Mt Isa and McArthur River. Aust J Earth Sci 47:381–403
James RH, Elderfield H (1996) Chemistry of ore-forming fluids and mineral formation rates in an active hydrothermal sulfide deposit on the Mid-Atlantic Ridge. Geology 24:1147–1150
James RH, Elderfield H, Palmer MR (1995) The chemistry of hydrothermal fluids from the Broken Spur site, 29 N Mid-Atlantic Ridge. Geochim Cosmochim Acta 59:651–659
Kah LC (2000) Depositional δ18O signatures in Proterozoic dolostones: constraints on seawater chemistry and early diagenesis. SEPM Spec Publ 67:345–360
Klinkhammer GP, Elderfield H, Edmond JM, Mitra A (1994) Geochemical implications of rare earth element patterns in hydrothermal fluids from mid-ocean ridges. Geochim Cosmochim Acta 58:5105–5113
Kontak DJ, Jackson S (1995) Laser-ablation ICP-MS micro-analysis of calcite cement from a Mississippi-Valley-Type Zn-Pb deposit, Nova Scotia: dramatic variability in REE content on macro- and micro-scales. Can Mineral 33:445–467
Large RR, McGoldrick PJ (1998) Lithogeochemical halos and geochemical vectors to stratiform sediment hosted Zn–Pb–Ag deposits, 1. Lady Loretta Deposit, Queensland. J Geochem Explor 63:37–56
Large RR, Bull SW, McGoldrick PJ (2000) Lithogeochemical halos and geochemical vectors to stratiform sediment hosted Zn–Pb–Ag deposits: part 2. HYC deposit, McArthur River, Northern Territory. J Geochem Explor 68:105–126
Large RR, Bull SW, McGoldrick PJ, Walters SG (2005) Stratiform and strata-bound Zn-Pb-Ag deposits in Proterozoic sedimentary basins, northern Australia. Econ Geol 100:931–963
Lawrence MG, Greig A, Collerson KD, Kamber BS (2006) Rare earth element and yttrium variability in South East Queensland waterways. Aquat Geochem 12:39–72
Leach DL, Sangster DF, Kelley KD, Large RR, Garven G, Allen CR, Gutzmer J, Walters S (2005) Sediment-hosted lead-zinc deposits: A global perspective. Econ Geol 100:561–607
Liu X-M, Hardisty DS, Lyons TW, Swart PK (2019) Evaluating the fidelity of the cerium paleoredox tracer during variable carbonate diagenesis on the Great Bahamas Bank. Geochim Cosmochim Acta 248:25–42
Lüders V, Moeller P, Dulski P (1993) REE fractionation in carbonates and fluorites. Formation of Hydrothermal Vein Deposits. A case study of the Pb-Zn-, barite and fluorite deposits of. Gebrüder Borntraeger, In, pp 133–150
Lyons TW, Reinhard CT, Planavsky NJ (2014) The rise of oxygen in Earth’s early ocean and atmosphere. Nature 506:307–315
Machel HG, Burton EA (1991) Factors governing cathodoluminescence in calcite and dolomite, and their implications for studies of carbonate diagenesis. SEPM Spec Publ 37–57
Magnall JM, Gleeson SA, Blamey NJF, Paradis S, Luo Y (2016) The thermal and chemical evolution of hydrothermal vent fluids in shale hosted massive sulphide (SHMS) systems from the MacMillan Pass district (Yukon, Canada). Geochim Cosmochim Acta 193:251–273
Magnall JM, Hayward N, Gleeson SA, Schleicher A, Dalrymple I, King R, Mahlstadt N (2021) The Teena Zn-Pb deposit (McArthur Basin, Australia). Part II: carbonate replacement sulfide mineralization during burial diagenesis - implications for mineral exploration. Econ Geol. https://doi.org/10.5382/econgeo.4845
Marshall DJ (1988) Cathodoluminescence of geological materials. Unwin Hyman
Maskenskaya OM, Drake H, Mathurin FA, Åström ME (2015) The role of carbonate complexes and crystal habit on rare earth element uptake in low-temperature calcite in fractured crystalline rock. Chem Geol 391:100–110
McClay KR, Carlile DG (1978) Mid-proterozoic sulphate evaporites at Mount Isa mine, Queensland, Australia. Nature 274:240–241
McDonough WF, Sun S-S (1995) The composition of the Earth. Chem Geol 120:223–253
McGoldrick P, Winefield P, Bull S, Selley D, Scott R (2010) Sequences, synsedimentary structures, and sub-basins: the where and when of SEDEX zinc systems in the southern McArthur Basin, Australia. Soc Econ Geol Spec Publ 15:1–23
McLennan SM (1989) Rare earth elements in sedimentary rocks: influence of provenance and sedimentary processes. Rev Mineral 21:169–200
Michard A (1989) Rare earth element systematics in hydrothermal fluids. Geochim Cosmochim Acta 53:745–750
Michard A, Albarède F (1986) The REE content of some hydrothermal fluids. Chem Geol 55:51–60
Migdisov AA, Williams-Jones AE, Wagner T (2009) An experimental study of the solubility and speciation of the Rare Earth Elements (III) in fluoride-and chloride-bearing aqueous solutions at temperatures up to 300°C. Geochim Cosmochim Acta 73:7087–7109
Morgan JW, Wandless GA (1980) Rare earth element distribution in some hydrothermal minerals: evidence for crystallographic control. Geochim Cosmochim Acta 44:973–980
Murphy TE (2004) Structural and stratigraphic controls on mineralization at the George Fisher Zn-Pb-Ag deposit, northwest Queensland, Australia. Unpubl. PhD Thesis, James Cook University 403
Nance WB, Taylor SR (1976) Rare earth element patterns and crustal evolution—I. Australian post-Archean sedimentary rocks. Geochim Cosmochim Acta 40:1539–1551
Neudert M (1983) A depositional model for the Upper Mount Isa Group and implications for ore formation. Unpubl. PhD Thesis, Australian National University 538
Neudert MK, Russell RE (1981) Shallow water and hypersaline features from the middle Proterozoic Mt Isa Sequence. Nature 293:284–286
Nozaki Y, Zhang J, Amakawa H (1997) The fractionation between Y and Ho in the marine environment. Earth Planet Sci Lett 148:329–340
Page RW, Bell TH (1986) Isotopic and structural responses of granite to successive deformation and metamorphism. J Geol 94:365–379
Page RW, Sweet IP (1998) Geochronology of basin phases in the western Mt Isa Inlier, and correlation with the McArthur Basin. Aust J Earth Sci 45:219–232
Page RW, Jackson MJ, Krassay AA (2000) Constraining sequence stratigraphy in north Australian basins: SHRIMP U–Pb zircon geochronology between Mt Isa and McArthur River. Aust J Earth Sci 47:431–459
Painter MGM (2003) The geochemical and mineralogical haloes around the Mt Isa base metal orebodies. Unpubl. PhD Thesis, University of Queensland 478
Painter MGM, Golding SD, Hannan KW, Neudert MK (1999) Sedimentologic, petrographic, and sulfur isotope constraints on fine-grained pyrite formation at Mount Isa Mine and environs, Northwest Queensland, Australia. Econ Geol 94:883–912
Paton C, Hellstrom J, Paul B, Woodhead J, Hergt J (2011) Iolite: Freeware for the visualisation and processing of mass spectrometric data. J Anal At Spectrom 26:2508–2518
Perkins WG (1997) Mount Isa lead-zinc orebodies: Replacement lodes in a zoned syndeformational copper-lead-zinc system? Ore Geol Rev 12:61–110
Perkins WG, Bell TH (1998) Stratiform replacement lead-zinc deposits; a comparison between Mount Isa, Hilton, and McArthur River. Econ Geol 93:1190–1212
Perry EP, Gysi AP (2018) Rare earth elements in mineral deposits: speciation in hydrothermal fluids and partitioning in calcite. Geofluids 2018:1–19
Perry E, Gysi AP (2020) Hydrothermal calcite-fluid REE partitioning experiments at 200 °C and saturated water vapor pressure. Geochim Cosmochim Acta 286:177–197
Pierson BJ (1981) The control of cathodoluminescence in dolomite by iron and manganese. Sedimentology 28:601–610
Planavsky N, Bekker A, Rouxel OJ, Kamber B, Hofmann A, Knudsen A, Lyons TW (2010) Rare earth element and yttrium compositions of Archean and Paleoproterozoic Fe formations revisited: new perspectives on the significance and mechanisms of deposition. Geochim Cosmochim Acta 74:6387–6405
Planavsky NJ, McGoldrick P, Scott CT, Li C, Reinhard CT, Kelly AE, Chu X, Bekker A, Love GD, Lyons TW (2011) Widespread iron-rich conditions in the mid-Proterozoic ocean. Nature 477:448–451
Polito PA, Kyser TK, Golding SD, Southgate PN (2006) Zinc deposits and related mineralization of the Burketown mineral field, including the world-class Century deposit, northern Australia: Fluid inclusion and stable isotope evidence for basin fluid sources. Econ Geol 101:1251–1273
Poulton SW, Fralick PW, Canfield DE (2010) Spatial variability in oceanic redox structure 1.8 billion years ago. Nat Geosci 3:486–490
Rieger P, Magnall JM, Gleeson SA, Lilly R, Rocholl A, Kusebauch C (2020a) Sulfur isotope constraints on the conditions of pyrite formation in the Paleoproterozoic Urquhart Shale Formation and George Fisher Zn-Pb-Ag deposit, Northern Australia. Econ Geol 115:1003–1020
Rieger P, Magnall JM, Gleeson SA, Schleicher AM, Bonitz M, Lilly R (2020b) The mineralogical and lithogeochemical footprint of the George Fisher Zn-Pb-Ag massive sulphide deposit in the Proterozoic Urquhart Shale Formation, Queensland, Australia. GFZ Data Services. https://doi.org/10.5880/GFZ.3.1.2020.003
Rieger P, Magnall JM, Gleeson SA, Schleicher AM, Bonitz M, Lilly R (2021a) The mineralogical and lithogeochemical footprint of the George Fisher Zn-Pb-Ag massive sulphide deposit in the Proterozoic Urquhart Shale Formation, Queensland, Australia. Chem Geol 560:119975
Rieger P, Magnall JM, Gleeson SA, Oelze M, Wilke FDH, Lilly R (2021b) Differentiating between hydrothermal and diagenetic carbonates using rare earth element and yttrium (REE+Y) geochemistry: A case study from the Paleoproterozoic George Fisher massive sulphide Zn deposit. GFZ Data Services. https://doi.org/10.5880/GFZ.3.1.2020.005
Roberts S, Palmer MR, Cooper MJ, Buchaus P, Sargent D (2009) REE and Sr isotope characteristics of carbonate within the Cu–Co mineralized sedimentary sequence of the Nchanga Mine, Zambian Copperbelt. Mineral Deposita 44:881–891
Schijf J, De Baar HJW, Millero FJ (1995) Vertical distributions and speciation of dissolved rare earth elements in the anoxic brines of Bannock Basin, eastern Mediterranean Sea. Geochim Cosmochim Acta 59:3285–3300
Sholkovitz ER, Landing WM, Lewis BL (1994) Ocean particle chemistry: the fractionation of rare earth elements between suspended particles and seawater. Geochim Cosmochim Acta 58:1567–1579
Smrzka D, Zwicker J, Bach W, Feng D, Himmler T, Chen D, Peckmann J (2019) The behavior of trace elements in seawater, sedimentary pore water, and their incorporation into carbonate minerals: a review. Facies 65:41
Song H, Jiang G, Poulton SW, Wignall PB, Tong J, Song H, An Z, Chu D, Tian L, She Z (2017) The onset of widespread marine red beds and the evolution of ferruginous oceans. Nat Commun 8:1–7
Southgate PN, Scott DL, Sami TT, Domagala J, Jackson MJ, James NP, Kyser TK (2000) Basin shape and sediment architecture in the Gun Supersequence: a strike-slip model for Pb–Zn–Ag ore genesis at Mt Isa. Aust J Earth Sci 47:509–531
Southgate PN, Neumann NL, Gibson GM (2013) Depositional systems in the Mt Isa Inlier from 1800 Ma to 1640 Ma: Implications for Zn–Pb–Ag mineralisation. Aust J Earth Sci 60:157–173
Soyol-Erdene T-O, Huh Y (2013) Rare earth element cycling in the pore waters of the Bering Sea Slope (IODP Exp. 323). Chem Geol 358:75–89
Spinks SC, Pearce MA, Liu W, Kunzmann M, Ryan CG, Moorhead GF, Kirkham R, Blaikie T, Sheldon HA, Schaubs PM, Rickard WDA (2021) Carbonate Replacement as the Principal Ore Formation Process in the Proterozoic McArthur River (HYC) Sediment-Hosted Zn-Pb Deposit, Australia. Econ Geol 116:693–718
Sverjensky DA (1984) Europium redox equilibria in aqueous solution. Earth Planet Sci Lett 67:70–78
Tanaka K, Kawabe I (2006) REE abundances in ancient seawater inferred from marine limestone and experimental REE partition coefficients between calcite and aqueous solution. Geochem J 40:425–435
Tang D, Shi X, Wang X, Jiang G (2016) Extremely low oxygen concentration in mid-Proterozoic shallow seawaters. Precambrian Res 276:145–157
Valenta RK (1988) Deformation, fluid flow and mineralization in the Hilton area, Mt Isa, Australia. Unpub. PhD Thesis, Monash University 284
Valenta R (1994) Deformation of host rocks and stratiform mineralization in the Hilton Mine area, Mt Isa. Aust J Earth Sci 41:429–443
van den Heuvel BH (1969) Sedimentation, stratigraphy and post-depositional changes in the sediments of the upper formations of the Mount Isa group, North-West Queensland. Unpubl. PhD Thesis, University of Queensland 195
Vaughan JR, Hickey KA, Barker SLL (2016) Isotopic, chemical, and textural evidence for pervasive calcite dissolution and precipitation accompanying hydrothermal fluid flow in low-temperature, carbonate-hosted, gold systems. Econ Geol 111:1127–1157
Voigt M, Mavromatis V, Oelkers EH (2017) The experimental determination of REE partition coefficients in the water-calcite system. Chem Geol 462:30–43
Waring CL (1990) Genesis of the Mount Isa copper ore system. Unpub. PhD Thesis, Monash University 295
Waring CL, Andrew AS, Ewers GR (1998a) Use of O, C, and S stable isotopes in regional mineral exploration. AGSO J Aust Geol Geophys 17:301–313
Waring CL, Heinrich CA, Wall VJ (1998b) Proterozoic metamorphic copper deposits. AGSO J Aust Geol Geophys 17:239–246
Webb GE, Nothdurft LD, Kamber BS, Kloprogge JT, Zhao J-X (2009) Rare earth element geochemistry of scleractinian coral skeleton during meteoric diagenesis: a sequence through neomorphism of aragonite to calcite. Sedimentology 56:1433–1463
Williams-Jones AE, Migdisov AA, Samson IM (2012) Hydrothermal mobilisation of the rare earth elements–a tale of “ceria” and “yttria”. Elements 8:355–360
Wittkop C, Swanner ED, Grengs A, Lambrecht N, Fakhraee M, Myrbo A, Bray AW, Poulton SW, Katsev S (2020) Evaluating a primary carbonate pathway for manganese enrichments in reducing environments. Earth Planet Sci Lett 538:116201
Woodhead JD, Hellstrom J, Hergt JM, Greig A, Maas R (2007) Isotopic and elemental imaging of geological materials by laser ablation inductively coupled plasma-mass spectrometry. Geostand Geoanal Res 31:331–343
Zhang J, Amakawa H, Nozaki Y (1994) The comparative behaviors of yttrium and lanthanides in the seawater of the North Pacific. Geophys Res Lett 21:2677–2680
Zhang X, Yang S, Zhao H, Jiang S, Zhang R, Xie J (2019) Effect of beam current and diameter on electron probe microanalysis of carbonate minerals. J Earth Sci 30:834–842
Acknowledgements
Funding for this project was provided by a Helmholtz recruitment initiative grant to S. Gleeson. The geology teams at Mount Isa Mines George Fisher operation and Mount Isa Mines Resource Development are thanked for the support during field work and for access to drill cores. The technical assistance by U. Dittmann and E. Lewerenz (sample preparation) is warmly acknowledged. We would also like to thank P. Rea from Mount Isa Mines Resource Development for an early review of this manuscript. Furthermore, we thank S. Barker and B. Lehmann for their thoughtful and constructive reviews, and B. Lehmann for editorial handling of the manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL. Funding for this project was provided by a Helmholtz Recruitment Initiative grant to S. Gleeson.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Editorial handling: B. Lehmann
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rieger, P., Magnall, J.M., Gleeson, S.A. et al. Differentiating between hydrothermal and diagenetic carbonate using rare earth element and yttrium (REE+Y) geochemistry: a case study from the Paleoproterozoic George Fisher massive sulfide Zn deposit, Mount Isa, Australia. Miner Deposita 57, 187–206 (2022). https://doi.org/10.1007/s00126-021-01056-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00126-021-01056-1