
Abstract
The Cheoeum vent field (CVF) is the first example of an inactive ultramafic-hosted seafloor massive sulphide (SMS) deposit identified in the middle part of the Central Indian Ridge. Here, we report on the detailed mineralogy and geochemistry of ultramafic-hosted sulphide sample atop a chimney, together with a few small fragments. Hydrothermal chimneys are characterised by high concentrations of Au (up to 17.8 ppm) and Sn (up to 1720 ppm). The sulphide mineralisation in the CVF shows (1) early precipitation of anhedral sphalerite and pyrite–marcasite aggregates under relatively low-temperature (< 250 °C) fluid conditions; (2) intensive deposition of subhedral pyrrhotite, isocubanite, chalcopyrite, Fe-rich sphalerite (Sp-III), and electrum from high-temperature (250–365 °C) and reduced fluids in the main mineralisation stage; and (3) a seawater alteration stage distinguished by the mineral assemblage of marcasite pseudomorphs, altered isocubanite phase, covellite, amorphous silica, and Fe-oxyhydroxides. Electrum (< 2 μm in size) is the principal form of Au mineralisation and is mainly associated with the main mineralisation stage. The consistently high fineness of electrum (801 to 909‰) is indicative of the selective saturation of Au over Ag in the fluid during high-temperature mineralisation, which differs from the Au mineralisation associated with typical basaltic-hosted hydrothermal systems on mid-ocean ridges. Tin is mainly substituted in structures of sphalerite, isocubanite, and chalcopyrite as a solid solution, and not as mineral inclusions. The continuously ascending hydrothermal fluids enable the early formed Sn-bearing sulphide to be dissolved and reprecipitated, producing significantly Sn-enriched replacement boundaries between isocubanite and Sp-III. This study suggests that Au–Sn mineralisation could be facilitated by the low redox potential of ultramafic-hosted hydrothermal systems such as in the CVF, which may be a common occurrence along slow-spreading mid-ocean ridges.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Seafloor massive sulphide (SMS) deposits distributed along mid-ocean ridges (MORs) are generally discriminated by their mineralogical and geochemical features, according to different types of ridges formed by different spreading rates (Mozgova et al. 2008; Hannington et al. 2010; Nayak et al. 2014; Wang et al. 2014). Compared with fast-spreading ridges, the deeply penetrating faults at slow to intermediate spreading ridges result in more heterogeneous substrates (i.e. lower crustal and upper mantle rocks) and allow the circulation of hydrothermal fluids down to much greater depths (McCaig et al. 2007; Escartín et al. 2008). These differences ultimately influence the composition of hydrothermal fluids expelled on the seafloor due to variable fluid–rock interactions (Allen and Seyfried 2003; Hannington et al. 2005). Notably, hydrothermal fluids circulating through ultramafic rocks are responsible for the production of H2 and CH4 that result in highly reducing conditions, thereby providing a more suitable formation environment for reduced sulphide assemblages, such as pyrrhotite–isocubanite–Fe-rich sphalerite (Charlou et al. 2002, 2010; Nakamura et al. 2009).
Unlike the slow-spreading Mid-Atlantic Ridge (MAR), a small number of SMS deposits (MESO, Edmond, DoDo, Solitaire, Kairei, and Yokoniwa) have been reported along the Central Indian Ridge (CIR) (Halbach et al. 1998; Gamo et al. 2001; Van Dover et al. 2001; Nakamura et al. 2012; Fujii et al. 2016). These are all located in the southern part of the CIR (20–25° S) between the Marie Celeste Fracture Zone and Rodriguez Triple Junction. The mineralisation types and hydrothermal processes have been described for a few vent sites along the CIR, but ultramafic-hosted mineralisation is restricted to the Kairei field (Münch et al. 1999; Wang et al. 2014; Wu et al. 2016).
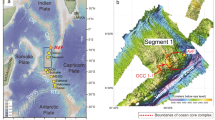
The Korea Institute of Ocean Science & Technology (KIOST) has carried out exploration for deep-sea mineral resources along the middle part of the CIR (MCIR; 8–18° S; ~ 700 km of ridge length) over twelve cruises since 2010. We conducted high-resolution mapping, plume survey, and geological sampling in order to trace the seafloor hydrothermal system (Son et al. 2014; Pak et al. 2017). A total of 11 ultramafic-hosted dome-like structures (oceanic core complexes; OCCs) were recognised throughout the survey areas, and all but one of the OCCs commonly show hydrothermal plume signatures (Pak et al. 2017). In OCC 4–1 (12.4° S) where methane represent the highest content (42.3 nmol/l) in the MCIR, a new hydrothermal vent field, the Cheoeum vent field (CVF), was discovered by deep-towed camera (Fig. 1). Sulfide deposits with an ultramafic affinity typically display elevated concentrations of Cu, Zn, Au, Ni, Co, and Sn (Fouquet et al. 2010; Hannington et al. 2010; Firstova et al. 2016; Knight et al. 2018). In particular, large enrichments of Sn are unique to ultramafic-hosted SMS deposits, when compared with mid-ocean ridge basalt (MORB)-hosted sulphide deposits or those from volcanic arcs (de Ronde et al. 2005; Hannington et al. 2005; Maslennikov et al. 2017; Melekestseva et al. 2017). This might be an enigmatic aspect because Sn typically behaves as a highly incompatible element during igneous processes (Jochum et al. 1993; Badullovich et al. 2017).

a Regional bathymetric map of the 4th segment of the middle part of Central Indian Ridge. The location of the Cheoeum vent field is marked by a yellow star, where hydrothermal samples were collected along with mantle rocks. Dotted black lines represent the fracture zones. The inset (online data from GeoMapAPP: www.geomapapp.org) shows the study area with major tectonic boundaries in the Indian Ocean. b Detailed distribution of hydrothermal vents in the CVF at the 12.4°S oceanic core complex (OCC). Abbreviations: CR = Carlsberg Ridge; MCFZ = Marie Celeste Fracture Zone; NTD = Non-transform discontinuities; RTJ = Rodriguez Triple Junction; SEIR = Southeast Indian Ridge; SWIR = Southwest Indian Ridge
Only a few studies have investigated the geochemistry and origin of Sn-bearing deep-sea hydrothermal deposits along MORs; gold-rich and Sn-bearing hydrothermal deposits hosted in ultramafic rocks along the MAR have been documented (Evrard et al. 2015; Firstova et al. 2016; Dekov et al. 2018), but little is known about the mineralogical and geochemical features of Au and Sn mineralisation along the CIR associated with ultramafic rocks.
A unique aspect of the CVF is that it is one of the few ultramafic-hosted hydrothermal deposits in the CIR that has high grades of both Au and Sn. The aims of our study, therefore, were to document the nature of hydrothermal mineralisation in the CVF and understand the ore-forming processes in the ultramafic-hosted environment. This study thus provides new insights into the first example of Au–Sn mineralisation along the MCIR.
Geological setting and samples
The CIR is located between the Carlsberg Ridge and Rodriguez Triple Junction (25° 30′ S, 70° 06′ E), extending ~ 2000 km from 3° N to 25° S (Fig. 1a). It has a slow to intermediate spreading rate (34–55 mm/year) and commonly has a rough and deep axial valley morphology similar to the MAR (DeMets et al. 1990, 1994; Escartín et al. 2008). The individual segments of the CIR are cut by transform faults and nontransform discontinuities (NTD), with lengths varying between a few and several tens of kilometres (Münch et al. 1999). Pak et al. (2017) categorised six first-order segments and seven second-order segments along the MCIR (8–18° S). They showed that the MCIR is a slow-spreading ridge with average full spreading rates from 33.7 to 45.1 mm/year, with most areas (~ 96%) undergoing asymmetric accretion. A total of 11 OCCs are observed in these asymmetrical spreading centres. A systematic hydrothermal plume survey showed that most OCCs (~ 91%) in this area are commonly associated with hydrothermal activity, which is evident from high methane concentrations, oxidation–reduction potential (ORP) anomalies, and variations in nephelometric turbidity units (△NTUs) (Son et al. 2014; Pak et al. 2017).
Our study area is in segment 4 (12.0–13.3° S) of the MCIR, which is located north of the Argo fracture zone (Pak et al. 2017). Three OCCs (OCC 4–1, 4–2, and 4–3) occur in this segment that is subdivided into segments 4–1 and 4–2 by NTD 4–1 (Fig. 1a). OCC 4–1 and 4–2 are topographically linked to NTD 4–1, and OCC 4–3 are located at the corner of segment 4–2 and the Argo fracture zone. The CVF hydrothermal vent site, with dimensions of 650 × 350 m at a depth of ~ 3000 m, is hosted in OCC 4–1, from which we recovered Au- and Sn-rich hydrothermal sulphide with mantle-derived rocks by dredging (Fig. 1b).
The hydrothermal vents (chimneys) in the CVF are mostly inactive, although weakly diffusing fluids emanating from some mound structure are intermittently observed (Fig. 2). Most chimneys are up to ~ 1 m high and are covered with Fe-oxyhydroxides (Fig. 2a). Fewer chimneys are > 2 m high and merge with each other (Fig. 2b). Short chimneys (< 50 cm high) with a single spire are often coalesced into a cluster without an underlying mound structure (Fig. 2c). By contrast, large chimneys sit on mounds covered by fallen spires (Fig. 2d). Most areas are covered with metalliferous sediments of various colours (Fig. 2e, f). The evidence of life, such as vent fauna and bacteria mats, is not identified in the CVF, but some debris of seashell is intermittently observed on the surface of some mound structures (Fig. 2). This would suggest that the CVF is in the waning stages of hydrothermal activity.

Photographs of the Cheoeum vent field from the deep-sea camera survey. a Example of a dominant inactive chimney, which are commonly up to ~ 1 m in height. b Chimneys that are > 2 m in height and merged with each other. c Small chimney cluster with no mound structure. d Fallen spires around a large chimney. e Black-coloured metalliferous sediments near a collapsed chimney. f Mound structure that is < 5 m in diameter covered with Fe–oxyhydroxides. All scale bars are 50 cm in length
Analytical methods
Mineral identification and textural interpretation of polymetallic sulphide were undertaken with an optical microscope and X-ray diffractometer (XRD) at KIOST, Busan, South Korea. XRD analysis was undertaken with a Panalytical X’Pert-PRO diffractometer with CuKα radiation generated at 40 kV and 30 mA. The XRD patterns were recorded over a 2θ range from 5 to 65°, with a 0.02° step size at a scan rate of 3°/min (ESM 1 Fig. S1). Bulk chemical analyses were performed on selected mineralised samples at Actlabs (Ancaster, Ontario, Canada) using the “Au–Ag Fire Assay”, “4-Acid Digestion (Code 8 ICP-OES)”, and “Peroxide Fusion Package (Ultratrace 7)”.
Quantitative analyses of individual minerals were carried out with a JEOL JXA-8530F electron probe microanalyzer (EPMA) by wavelength dispersive spectrometry (WDS) at Gyeongsang National University, Jinju, South Korea. The analyses were performed under the following conditions accelerating voltage of 15 Kv, beam current of 10 nA, and electron beam diameter of 5 μm, except for Au (1 μm). Natural mineral and synthetic standards and Aztec software using ZAF corrections were used for data calibration. Each spot analysed by EPMA was marked on a back-scattered electron (BSE) image for subsequent trace element analysis of the same mineral grain by laser ablation inductively coupled plasma mass spectrometry (LA–ICP–MS).
LA–ICP–MS analysis was carried out using a 193-nm excimer laser ablation system (ESI NWR 193, USA) coupled to an Agilent 7700 quadrupole ICP-MS, at KIOST. Laser ablation was conducted with a beam diameter of 30 to 50 μm, according to the mineral grain size, 10-Hz laser pulse rate, and 5 J/cm2 laser energy. The following isotopes were measured: 55Mn, 57Fe, 59Co, 60Ni, 65Cu, 66Zn, 69Ga, 74Ge, 75As, 77Se, 95Mo, 109Ag, 111Cd, 118Sn, 121Sb, 182W, 197Au, 205Tl, and 208Pb. The total analysis time for each spot was 80 s, comprising 30 s of background measurement followed by 50 s of sample ablation. The external calibration was performed using STDGL3 (Belousov et al. 2014). The MASS-1 sulphide reference material (also known as PS-1; Wilson et al. 2002) was analysed as an unknown sample to assess data quality (ESM 2 Table S1). Iron, Cu, and Zn determined by EPMA were used as internal standards for quantification of marcasite–pyrrhotite, isocubanite, and sphalerite, respectively. The Ga concentration was calculated using MASS-1 as a primary standard because STDGL3 does not contain Ga. Data reduction was carried out using Iolite software (Paton et al. 2011). The entire dataset is reported in ESM 2 Table S2.
LA–ICP–MS elemental mapping was performed by ablating sets of parallel line rasters in a grid across the sample. The lines were ablated with a beam size of 8 or 15 μm (depending on the target area). The spacing between the lines and scan speed were kept constant to match the laser spot size. A laser frequency of 10 Hz was used at a constant laser energy of 5 J/cm2. The acquisition time for most elements was set to 0.02 s, but the major elements (Fe, Cu, and Zn) were measured for 0.01 s. Images were compiled and processed using Iolite software developed by WaveMetrics (Paton et al. 2011).
A FEI Quanta 3D FEG (Field Emission Gun) was used to prepare cross-sectional samples for field emission–transmission electron microscopy (FE–TEM) at the Korea Basic Science Institute (KBSI), Daejeon, South Korea. Before the specimen was sectioned, the surface morphology was examined by scanning electron microscopy (SEM). TEM foils with a size of ca. 7 × 5 × 0.1 μm were mounted on a Mo grid. A FE–TEM equipped with energy dispersive spectrometers (EDS) was used to identify the mode of occurrence of trace elements (especially Sn) at high-resolution (i.e. nanoinclusions versus solid solution) at KBSI. The high-resolution TEM (HR–TEM) images, scanning TEM (STEM) images, fast Fourier transform (FFT) patterns, and EDS spectra were acquired with a JEOL JEM-2100F operated at 200 kV.
Results
Hydrothermal specimens
Mafic and ultramafic rocks collected with the chimney samples include harzburgite, microgabbro, pegmatitic gabbro, gabbro, and basalt, showing variable degrees of serpentinisation and alteration. These rocks represent the exhumed lower oceanic crust and mantle at OCC 4–1 (Fig. 3a, b). The top part of a typical chimney (e.g. IR 110208-12) is characterised by a bimodal distribution with respect to Zn- and Cu-bearing minerals (Fig. 3c). Wherein the exterior zone is composed mostly of Zn-sulphides, and the proportion of Cu-sulphides increases towards the interior zone (Table 1). Fe-oxyhydroxides cover the outermost surface where a few vent conduits of variable scales (< 1 cm) are locally filled with amorphous silica (Fig. 3d). Small chimney fragments (e.g. IR 110208-13 and IR 110208-1A) also have Zn-dominant assemblages, but no mineral zonation (Fig. 3e, f). Their matrix commonly exhibits a highly porous and/or fragile texture, in which amorphous silica has been precipitated along fracture zones or cavities. All of the sulphide samples consistently represent Zn-rich compositions and do not contain any of the sulphate minerals that are commonly found in MORB-hosted SMS deposits. We selected IR 110208-12 as a typical chimney sample to conduct detailed mineralogical and geochemical analyses because it has well-preserved mineralogical zonation from the exterior to the interior of the chimney.

Photographs of the collected hydrothermal chimneys and ultramafic rocks. a–b Serpentinised ultramafic rocks recovered with the chimney samples. c Top of a hydrothermal chimney (IR 110208-12) characterized by a bimodal distribution of Zn-Cu-minerals from the exterior to inner zone. d A number of vent conduits (white arrows) that are < 1 cm in diameter observed on the outermost surface of c. e Small Zn-rich chimney fragment (IR 110208-13) with a porous texture, where amorphous silica is infilling along fractures. f Small Zn-rich chimney fragments (IR 110208-1A) similar to those in e
Sulphide mineralogy
Hydrothermal chimneys of the CVF are composed mainly of sphalerite, pyrrhotite, isocubanite, chalcopyrite, and pyrite and/or marcasite, and lack sulphates such as anhydrite/gypsum and barite (Table 1). This mineral assemblage is consistent with those of other ultramafic-hosted submarine sulphide from MORs (Mozgova et al. 2008; Fouquet et al. 2010; Melekestseva et al. 2014; Wang et al. 2014). Regarding the mode and texture of the sulphide minerals, the exterior zone of typical chimney IR 110208–12 (Fig. 4a) is characterised by dominant sphalerite that exhibits two distinct textures, colloform and massive. The colloform sphalerite (Sp-I) is generally intergrown with pyrite at the outermost surface of the chimney (Fig. 4b). This mineral assemblage is commonly considered to form during the incipient stages of chimney growth (Melekestseva et al. 2014; Wohlgemuth-Ueberwasser et al. 2015). Dissolution and reprecipitation and/or continuous overgrowth of sphalerite are frequently observed towards the inner zone of the chimney, resulting in sphalerite being more massive (Sp-II) (Fig. 4c). BSE images show submicron-sized galena inclusions irregularly distributed in both Sp-I and Sp-II (Fig. 4b, c). Subhedral marcasite (Mrc-I) and/or pyrite occur along the grain boundaries of Sp-II (Fig. 4c, d). Minor amounts of pyrrhotite+isocubanite+chalcopyrite have locally developed concentric intergrowths around the contact between the exterior and interior parts of the chimney, indicating simultaneous deposition with Sp-II under metastable fluid conditions (Fig. 4e). These minerals are anhedral and/or form small grains mostly < 10 μm in size. In the interior part of the chimney, isocubanite and chalcopyrite are more abundant than in the exterior part, likely as a result of high-temperature mineralisation (Fig. 4f). These mineralogical differences correspond to bulk chemical compositions, with higher Cu and Fe contents in the interior than in the exterior (Table 2). Prismatic marcasite (Mrc-II) mantling relic pyrrhotite is commonly replaced by isocubanite with chalcopyrite exsolution, indicating that the Cu-sulphides postdate pyrrhotite formation (Fig. 4f, g). In some sections, isocubanite has 120° grain boundaries indicative of recrystallisation (Fig. 4h). Chalcopyrite was principally observed as exsolution lamellae within isocubanite, but with little discrete grain (Fig. 4g, h). Main-stage sphalerite (Sp-III) replaced the previously formed minerals after deposition of the Cu-sulphides (Fig. 4f–i).

Photomicrographs of mineral assemblages in a typical chimney (IR 110208-12). a Cross-section displaying two mineralisation zones, which are the exterior (b to e) and interior zones (f to k). Yellow boxes mark the subsampling areas for whole-rock analysis. b Colloform-textured sphalerite (Sp-I) suggestive of rapid crystallization. c Sphalerite with a massive texture (Sp-II) that formed as mineralisation progressed. Nanometre-sized galena inclusions are observed in BSE images (inset). d Association between sphalerite and pyrite aggregates. e Acicular pyrrhotite and isocubanite–chalcopyrite aggregates in contact between the exterior and interior zones. f Typical mineral assemblage in the interior zone. g Sharp contacts between marcasite and isocubanite–chalcopyrite aggregates, which are surrounded by main-stage sphalerite (Sp-III). h Altered isocubanite phase (Aip) formed along fractures zones (120° grain boundaries are marked by red dotted lines), showing a grainy surface in SEM images (inset). i Covellite intermittently present along the grain boundaries of pyrrhotite. j Marcasite converted from a prismatic to dendritic structure by continuous dissolution and reprecipitation. k Pyrrhotite that has undergone considerable volume loss, where Fe-oxyhydroxides have been extensively precipitated along the grain boundaries between pyrrhotite and marcasite. Abbreviations: Aip = altered isocubanite phase, Ams = amorphous silica; Ccp = chalcopyrite; Cv = covellite; Fe-ox = Fe–oxyhydroxides; Gn = galena; Icb = isocubanite; Mrc = marcasite; Po = pyrrhotite; Py = pyrite; Sp = sphalerite. Roman numerals indicate the generation stage, which is related to the texture and mineral assemblages present
Covellite, Fe-oxyhydroxides and amorphous silica replacing the early-formed sulphide are commonly observed regardless of the exterior and interior part of the chimney (Fig. 4b–k). This reflects that seawater alteration extensively occurred throughout the chimney following high-temperature mineralisation (Fig. 5). Isocubanite often shows the altered phase having a grainy texture and darker colour along fracture zones in the isocubanite, or at the grain boundaries of Mrc-II (Fig. 4f, h). This altered phase (Aip) has a stoichiometric composition close to Cu2Fe3S5, similar to Y-phase suggested by Mozgova et al. (2005). Micron-sized voids are often filled with covellite and Fe-oxyhydroxides (commonly goethite) along the grain boundaries between pyrrhotite and Mrc-II (Fig. 4f–k). Next, Mrc-II appears to transform into a needle-shaped form by coupled dissolution and reprecipitation (Fig. 4j). The amorphous silica found at the boundaries of all the early formed sulphide might be expected for hydrothermal fluid experiencing drastic changes in temperature and redox conditions (Fig. 4f–k).

Paragenetic sequence of mineralisation in a hydrothermal chimney from the Cheoeum vent field. Formation temperatures of minerals were calculated based on experimental studies (1. Kissin and Scott 1982; 2. Luks and Bray 2002; 3. Schoonen and Barnes 1991; Qian et al. 2011). Abbreviations are as in Fig. 4
More than 100 grains of electrum (El) were identified in 11 polished thin sections, which typically have a rounded shape and are < 2 μm in diameter (Fig. 6). Although some electrum is associated with Sp-II at the contact between the exterior and interior zones of the chimney (Fig. 6a), most electrum has a close relationship with the high-temperature pyrrhotite–isocubanite–chalcopyrite–Sp-III mineral assemblage and thus is confined to the interior of the chimney sample (Fig. 6b–d). Most electrum occurs as inclusions in Sp-III, followed by isocubanite and chalcopyrite (Fig. 6e, f). This is consistent with the bulk chemistry, which shows Au enrichment towards the interior of the sample (Table 2). Furthermore, electrum was commonly seen precipitated along Sn-rich zones, as determined by EPMA (Fig. 6b–e). Some electrums occur close to grain boundaries between Aip and Sp-III, but Aip does not contain any electrum (Fig. 6g). This suggests that Au mineralisation occurred prior to the transformation of isocubanite to Aip. In rare cases, isolated electrum grains are enclosed by amorphous silica surrounding Sp-III (Fig. 6h).

Reflected light photomicrographs and back-scattered electron images of electrum (El) grains. a Sparse electrum grains intermittently observed in Sp-II. b–d The dominant occurrence of electrum with isocubanite + chalcopyrite + Fe-rich sphalerite (Sp-III) assemblages. Electrum is occasionally associated with Sn-rich replacement boundaries. Some electrum grains are enclosed by e isocubanite and f chalcopyrite, or g precipitated along grain boundaries between altered isocubanite phase (Aip) and Sp-III. h Electrum is rarely trapped within amorphous silica. Abbreviations are as in Fig. 4
Tin-rich bands are closely related to sphalerite in both the exterior and interior parts of the chimney, but their distribution is somewhat different in each part when BSE images are viewed (Fig. 7). For example, in the exterior of the chimney, Sn-rich bands are mainly associated with growth boundaries of Sp-II, which may be indicative of Sn enrichment by zone refining (Fig. 7a). Conversely, relatively thick Sn-rich bands are confined to the replacement boundaries between isocubanite with chalcopyrite exsolution and Sp-III in the interior part of the chimney (Fig. 7b–d); Sn-rich bands are more common in the chimney interior. Tin-bearing minerals are notably absent in all the studied samples, despite the significant enrichments in this element (Table 2). Focused ion beam slices (FIB) for FE–TEM investigation were then sampled from the Sn-rich bands (Fig. 7).

Back-scattered electron images of Sn enrichment bands. a Sn concentrated along the growth boundaries of Sp-II in the exterior zone. b Sn enrichment bands along the replacement boundaries between isocubanite with chalcopyrite exsolution and Sp-III in the interior zone. c Widening of replacement texture along the grain boundaries. d Relics of isocubanite with chalcopyrite within Sn enrichment band. Red arrows in all figures indicate the focused ion beam sectioned areas for TEM observations. Abbreviations are as in Fig. 4
Bulk chemistry
All samples have Zn concentrations that are much higher than those of Cu and Pb, and plot in the sediment-free MOR field (Fig. 8a). To obtain a better understanding of the distribution of metals, we systematically subsampled a typical chimney (IR110208-12) that is characterised by mineralogical zonation from the exterior (A1 and A2) to interior part (A5, A6, and A7) (Fig. 4a). The highest Zn (40.5 wt.%) and Pb contents (0.22 wt.%) are observed in the exterior part, while Cu (up to 3.47 wt.%) and Fe (up to 22.7 wt.%) are more concentrated in the interior part (Table 2). Enrichments of Au (up to 17.8 ppm) and Sn (up to 1720 ppm) are prominent features (Table 2). Gold concentrations generally tend to increase towards the interior part and are positively correlated with Cu (R2Cu–Au = 0.60), indicating that Au deposition may have occurred at high-temperature conditions (Fig. 8b). Unlike Au, Sn has uniformly high concentrations (mostly > 1000 ppm) in the entire sample and positively correlates with Zn (R2Zn–Sn = 0.51) (Fig. 8c). Although the contact (A3 and A4) between the exterior and interior part of the chimney reflects a mixed composition of both areas due to the thin boundaries, the highest Sn content is observed in A4 (Table 2).

Bulk chemical compositions of hydrothermal chimney samples from the Cheoeum vent field. a Cu–Zn–Pb ternary diagram modified from Fouquet et al. (1993). Log–log plots of b Au versus Cu and c Sn versus Zn. Inset figures show a positive correlation for data from subsamples of IR 110208–12. Detailed comparison with other SMS deposits from MORs: d Au versus (Cu + Zn)/Fe; e Sn versus (Cu + Zn)/Fe; f Au versus Ag. Au–Sn enrichment and high Au/Ag ratios are confined to ultramafic-hosted sulphides. Average compositions of sulphides are from Münch et al. (1999), Hannington et al. (2005), Fouquet et al. (2010), de Ronde et al. (2011), and Wang et al. (2014). Abbreviations: EPR = East Pacific Rise; CIR = Central Indian Ridge; MAR = Mid-Atlantic Ridge; MORB = Mid-Ocean Ridge Basalt
Ultramafic-hosted sulphide at MORs appear to have a distinct geochemical composition when compared with MORB-hosted sulphide. High (Cu + Zn)/Fe ratios (commonly > 1) and Au–Sn enrichments are characteristics of ultramafic-hosted sulphide (Fig. 8d, e). Although Ag contents do not show any relationship with the host rock lithology, high Au/Ag ratios are mainly confined to ultramafic-hosted sulphide (Fig. 8f). Compared with sulphide from MORs, those at intraoceanic arcs (e.g. Kermadec and Izu-Bonin) are commonly enriched in Au and Ag but show low (Cu + Zn)/Fe ratios (Fig. 8d–f).
Mineral chemistry
Sphalerite
Sphalerite has diverse compositional variations in the different mineralisation stages (ESM 2 Table S3), with FeS content ranging from 6.9 to 42.5 mol% (ESM 1 Fig. S2). First-generation sphalerite (Sp-I) has the lowest average FeS contents (average = 9.2 mol%), second-stage sphalerite intermediate (Sp-II; average = 18.9 mol% FeS), with third-stage sphalerite having the highest average contents overall (Sp-III; average = 28.7 mol%) (ESM 2 Table S3). The Sn-rich replacement boundaries have a lower FeS content (average = 22.4 mol%) when compared with those of Sp-III (ESM 1 Fig. S2).
Chalcopyrite disease was rarely observed in the BSE images, although most of the sphalerite contains Cu contents and substantial values (average = 4.74 wt.% Cu) are found mainly at the replacement boundaries (Fig. 7 and ESM 2 Table S3). This implies that Cu is present in sphalerite as solid solution or nanoinclusions. The high Sn concentrations (up to 5.5 wt.%) are generally accompanied by Cu enrichment (ESM 2 Table S3). This also may explain the distributional behaviour of Cu whether it mainly occurs as solid solution or nanoinclusion. The replacement boundaries show a negative correlation between Cu + Sn and Zn (ESM 1 Fig. S3a). In addition, Cu and Sn are positively correlated (R2 = 0.805) and closely follow the stoichiometric ratio of Cu:Sn = 2:1 (ESM 1 Fig. S3b). Given that Sn-minerals were not identified (Figs. 4, 6, and 7), these observations may suggest that invisible Cu–Sn-bearing inclusions exist in sphalerite.
Lead contents are mostly close to the detection limit, although relatively high values (up to 3.2 wt.%) are found in Sp-I and Sp-II from the exterior part of the sample (ESM 2 Table S3). The contents of Cd and Mn are typically < 0.3 wt.% (ESM 2 Table S3).
Isocubanite
Isocubanite is very sensitive to temperature, pressure, and compositional variations (Caye et al. 1988). All isocubanite analyses in this study are characterised by variable metal contents with 19.4–24.8 wt.% Cu, 37.9–43.9 wt.% Fe, 0.75–2.20 wt.% Zn, and 0.02–0.67 wt.% Sn (ESM 2 Table S3). Cu/Fe atomic ratios of isocubanite vary from 0.39 to 0.57 and deviate from its theoretical value towards Fe-enriched compositions (ESM 1 Fig. S4). The corresponding mineral formula is close to Cu0.89Fe2.06Zn0.05Sn0.001S2.99 when the analyses are normalised to six atoms, indicating that isocubanite formed under mineralising conditions similar to pyrrhotite (Sugaki et al. 1975). The Zn content of isocubanite is temperature-dependent, and thus ZnS contents can be used as an independent estimate of the depositional temperature of the pyrrhotite–isocubanite–sphalerite assemblage (Hutchison and Scott 1981). We obtained an average formation temperature of ~ 365 °C (ESM 2 Table S3). This is consistent with a previous experimental study in which isocubanite began to form, intergrown with chalcopyrite, pyrite, and pyrrhotite, at 335 °C (Lusk and Bray 2002).
Chalcopyrite
Chalcopyrite exhibits variable Cu/Fe atomic ratios ranging from 0.71 to 1.05 (ESM 2 Table S3). It differs in composition from the stoichiometry of theoretical chalcopyrite and has a more Fe-rich composition (ESM 1 Fig. S4), indicating a mineralogical association with isocubanite (Lafitte et al. 1985). In particular, some chalcopyrite has a composition similar to Aip. This suggests there may be Aip inclusion in the chalcopyrite. Most chalcopyrite contains variable amounts of Zn and Sn (ESM 2 Table S3). Zinc contents are mostly > 1 wt.%, with a maximum value of 3.04 wt.%. Tin contents are mostly lower than 0.5 wt.% but can be as high as 1.46 wt.% in some grains that are replaced by Sp-III (Fig. 6f).
Pyrrhotite
The chemical composition of pyrrhotite shows limited variations, with Fe contents ranging from 60.5 to 62.3 wt.% (ESM 2 Table S3). The chemical formula ranges from Fe0.91S to Fe0.95S on the basis of one sulphur atom. Lafitte et al. (1985) suggested that the atomic Fe content in pyrrhotite can be used to classify the structural type of pyrrhotite. This is consistent with the experimental study of Kissin and Scott (1982) that showed (1) hexagonal pyrrhotite contains ~ 48 at.% Fe, and (2) monoclinic pyrrhotite contains ~46 at.% Fe. Pyrrhotite from CVF mostly corresponds to hexagonal type (ESM 2 Table S3) that precipitates in relatively high-temperature condition (> 250 °C; Kissin and Scott 1982). Minor Cu (0.13–0.77 wt.%) and Zn (0.20–0.41 wt.%) are only found in pyrrhotite enclosed by isocubanite (Fig. 6b).
Marcasite
Marcasite is subdivided into two types based on texture: subhedral (Mrc-I) and prismatic pseudomorphs (Mrc-II) (Fig. 4). Their compositions are close to pure marcasite, although Mrc-II, enclosed by isocubanite with chalcopyrite exsolution (Fig. 4g), often has high Cu (up to 1.53 wt.%) and Zn contents (up to 3.48 wt.%) (ESM 2 Table S3).
Electrum
The EPMA results for electrum are affected to some extent by the surrounding host sulphide domain due to the small grain size of electrum (mostly < 2 μm). However, it is reasonable to assume that the Ag content is solely from the electrum because the host minerals do not contain Ag above the detection limit (ESM 2 Table S3). We conducted an offline correction to semiquantitatively evaluate the Au and Ag contents of electrum. All of the electrum grains have Au contents significantly higher than those of Ag, with a high electrum fineness (1000 × weight ratio Au/[Au + Ag]) of 801 to 909‰, regardless of its mineralogical relationship (ESM 1 Fig. S5). This suggests that all electrum was simultaneously deposited from hydrothermal fluids with a limited range of Au/Ag values.
Trace elements in sulphide
The LA–ICP–MS trace element data (103 analyses) for the sulphide minerals are shown in ESM 2 Table S2 and plotted in Fig. 9. Compared with other minerals, sphalerite tends to have higher contents of most elements with some distinct variations, according to the genetic sequence. For example, colloform Sp-I has distinctly higher contents of Ge (average = 349 ppm), As (average = 857 ppm), Ag (average = 944 ppm), Au (average = 1.28 ppm), Tl (average = 2.35 ppm), and Pb (average = 6316 ppm), but is relatively poor in Mn, Cu, Ga, Cd, Sn, and Sb when compared with Sp-II and Sp-III. The highest average contents of Cu (3743 ppm) and Sn (1237 ppm) are associated with Sp-II, while those of Mn (103 ppm), Ga (120 ppm), Cd (1558 ppm), and Sb (204 ppm) are associated with Sp-III. Most sphalerite has smooth LA–ICP–MS time-resolved elemental profiles indicative of the elements being in solid solution (Cook et al. 2009; ESM 1 Fig. S6a, b, c). However, some profiles show spikes of Cu, Pb, Sn, Ag, Ge, Sb, As, and Ga, suggesting the presence of inclusions (ESM 1 Fig. S6d). These spikes were avoided when the trace element composition of the sulphide was calculated.

Box-and-whisker plots of the trace element chemistry of the dominant sulphide minerals. Red stars represent the mean values for Sn-rich replacement boundaries. Horizontal dashed lines indicate the limits of detection (LOD) for pyrrhotite. Abbreviations are as in Fig. 4
Trace element abundances in sphalerite are shown in Fig. 10. Copper has a positive correlation with Sb, Ga, and Sn (Fig. 10a–c). In particular, the molar ratios of Cu to Sn are close to ~ 2, consistent with the EPMA results for replacement boundaries (Fig. 10c and ESM 1 S3b). Tin also shows strong positive correlations with Ga and Cd (Fig. 10d, e). Most individual analyses lie on, or close to, the 1:1 M ratio line between Cd and Sn. Silver is positively correlated with Ge, As, and Pb, with molar ratios of Ag:Ge = 2:1, Ag:As = 1:1, and Ag:Pb = 1:2 (Fig. 10f–h), but it is negatively correlated with Cu, Sb, Cd, Ga, and Sn, suggesting the possible influence of mineral inclusion as well as solid solution.

Plots of a Sb versus Cu, b Ga versus Cu, c Cu versus Sn, d Ga versus Sn, e Cd versus Sn, f Ag versus Ge, g Ag versus As, h Ag versus Pb, and i Ga + Ge + Sn + Sb (tri- and tetravalent cations) versus Cu + Ag (monovalent cations) in sphalerite. Black lines show correlation trends. Abbreviations are as in Fig. 4
Isocubanite contains Ga (up to 31.3 ppm), Ag (up to 300 ppm), and Sn (up to 1177 ppm), but is commonly depleted in Mn, Ge, As, Cd, Sb, and Pb (Fig. 9 and ESM 2 Table S2). Time-resolved LA–ICP–MS depth profiles are commonly flat, although some show coincident Zn, Pb, Sn, Ga, Ge, Sb, Ag, and As spikes, likely reflecting mineral inclusions (ESM 1 Fig. S6e). Gold spikes in the spectra are consistent with the presence of electrum inclusions in isocubanite (Fig. 6e).
The concentrations of most trace elements in pyrrhotite are close to, or below, the limit of detection, although some analyses show anomalously high Cu concentrations up to 1389 ppm (Fig. 9 and ESM 2 Table S2). Where pyrrhotite is replaced by covellite, Cu peaks with most other elements (e.g. Zn, Pb, Sn, Ga, Ge, Sb, Ag, Au, and As) (Fig. 4i and ESM 1 S6g).
Early formed Mrc-I is enriched in Mn (average = 340 ppm) and Tl (average = 103 ppm), whereas Cu, Ag, Sn, Au, and Cd are more concentrated in late-stage Mrc-II (Fig. 9 and ESM 2 Table S2). Probable electrum inclusions are inferred from time-resolved LA–ICP–MS depth profiles of Mrc-II, which show Au spikes (ESM 1 Fig. S6h).
LA–ICP–MS elemental mapping was carried out to investigate the distribution of trace elements and their partitioning amongst the coexisting sulphide (Figs. 11 and 12). Sp-II considered to have formed under relatively low temperatures from the exterior zone of the chimney shows extensive compositional zoning, even in a single grain (Fig. 11). Most of the trace elements are concentrated in the Fe-depleted parts of the sphalerite. Copper enrichments are often accompanied by enrichments in Ga, Cd, Sn, and Sb, whereas Pb shows a strong affinity with Ge, As, and Ag. Molybdenum, Tl, and Au are most abundant along the grain boundaries of Sp-II.

Reflected light photomicrograph (top left) and LA–ICP–MS element maps for selected elements in Sp-II from the exterior zone of the chimney. Units are in counts per second (× 103). Abbreviations are as in Fig. 4

Reflected light photomicrograph (top left) and LA–ICP–MS element maps for selected elements in typical assemblage (pyrrhotite–Mrc-II–isocubanite–chalcopyrite–Aip–Sp-III) from the interior zone of the chimney. Units are in counts per second (× 103). Abbreviations are as in Fig. 4
Figure 12 shows element maps of the interior zone of the chimney with a typical mineral assemblage of pyrrhotite + isocubanite + chalcopyrite + Sp-III + Mrc-II + Aip. The Sp-III primarily hosts Ga, Ge, Sn, and Sb, while As, Mo, Ag, W, Au, Tl, and Pb are preferentially incorporated into Mrc-II and/or isocubanite with chalcopyrite exsolution. The Ga and Sn contents appear to be zoned, with the highest concentrations observed in the replacement boundaries between isocubanite with chalcopyrite exsolution and Sp-III. It should be noted that invisible Au is mainly hosted in Mrc-II rather than primary sulphide and shows little relationship with Ag content.
FE–TEM–EDS analysis
The FIB–SEM image of TEM foil 2 from Sp-II shows obvious colour zonation where nanometre-sized inclusions were not observed (Fig. 7a and ESM 1 S7a). The high-angle annular dark field (HAADF)-STEM image with EDS analysis shows that this colour zonation is mainly associated with Fe and Zn compositions (ESM 1 Fig. S7b). In particular, the relatively high Cu and Sn contents are concentrated in Fe-poor areas, which is consistent with the LA–ICP–MS elemental maps (Fig. 11). The discrete grains of Sp-II are aggregated, which is suggestive of continuous overgrowth or recrystallisation (ESM 1 Fig. S7c). A number of nanometre-scale inclusions are present along the grain boundaries of Sp-II (ESM 1 Fig. S7d, f). These inclusions are distinguished from Sp-II by their slightly higher Cu contents, but there are no discernible differences between the inclusions and Sp-II with respect to Sn concentrations (ESM 1 Fig. S7e). This suggests that these are a Cu-rich phase rather than a Sn-bearing mineral. Rarely, a Sn-rich phase occurs in pores within Sp-II (ESM 1 Fig. S7f). It is composed mainly of Cu and S and has a composition similar to covellite. This is consistent with the time-resolved LA–ICP–MS depth profile that also identified Sn-rich covellite (ESM 1 Fig. S6g).
TEM foil 4 sampled the replacement boundaries between isocubanite with chalcopyrite exsolution and sph-III (Fig. 7c and ESM 1 S7g). The FIB–SEM image does not show any inclusion, and Sn is heterogeneously distributed in the replacement boundaries (ESM 1 Fig. S7h). In HAADF-STEM images, the replacement boundaries consist of several domains with varying Sn enrichment, which are bright Sn-rich and dark Sn-poor domains (ESM 1 Fig. S7i, j, k). These are clearly identifiable from the Sn counts in the EDS spectra (ESM 1 Fig. S8b). Native Cu often occurs along the fractures in isocubanite with chalcopyrite exsolution (ESM 1 Fig. S7l). A HR–TEM investigation was conducted on both Sn-rich and Sn-poor domains of the replacement boundaries in TEM foil 4 to assess whether Sn enrichment was due to the presence of nanocrystals (ESM 1 Fig. S8c, d). No nanocrystals were observed in HR–TEM images with FFT patterns, which demonstrates that both domains are well-ordered sphalerite and not a Sn-phase (ESM 1 Fig. S8c, d). The lattice spacing for the exposed faces of the Sn-rich domain is ~ 3.1 Å and for the Sn-poor domain ~ 1.9 and 1.6 Å. These values are consistent with the {1 1 1}, {2 2 0}, and {3 1 1} planes of the sphalerite lattice structure, respectively. The strong correlations between Sn and Cu, and the absence of Sn-bearing inclusions, indicate the presence of Sn in solid solution in the sphalerite lattice (ESM 1 Fig. S8e).
Discussion
Mineralisation process
The sulphide paragenesis of the Zn-rich chimneys from the CVF indicates significant changes in mineralising conditions from a low- to a high-temperature stage, overprinted by a seawater alteration stage (Fig. 5). The low-temperature hydrothermal activity (< 250 °C) is represented by the deposition of Fe-poor, colloform sphalerite (Sp-I) followed by pyrite/marcasite (Mrc-I) ± galena (Fig. 4b). Continuous replacement, or recrystallisation, changed Sp-I to more massive sphalerite (Sp-II) with a higher FeS content, which suggests that the oxidation state of the hydrothermal fluid was lowered to more reducing conditions (Fig. 4c and ESM 1 S2).
The high-temperature mineralisation (> 250 °C and < 365 °C) is represented by a mineral assemblage of pyrrhotite + isocubanite + chalcopyrite + Fe-rich sphalerite (Sp-III) (Fig. 4f). The pyrrhotite is mostly hexagonal and indicates formation temperatures of > 250 °C (ESM 2 Table S3; Kissin and Scott 1982). As the fluid temperature increased, pyrrhotite occurred in association with isocubanite + chalcopyrite + Sp-III. This assemblage indicates highly reducing conditions with formation temperatures of > 300 °C (Kojima and Sugaki 1985; Herzig and Hannington 1995; Luks and Bray 2002; Melekestseva et al. 2014; Kawasumi and Chiba 2017). This temperature is consistent with the average temperature of ~ 365 °C calculated from isocubanite composition (ESM 2 Table S3). The Fe-rich Cu–Fe–S solid solution is indicative of low sulphidation conditions (Kojima and Sugaki 1985; Evans et al. 2020; ESM 1 Fig. S4), which may be due to hydrothermal alteration associated with ultramafic rocks. It was during this stage that electrum largely formed, suggesting that Au mineralisation originated from a high-temperature, reducing fluid.
During the seawater alteration stage, recrystallisation of primary pyrrhotite to marcasite pseudomorphs (Mrc-II) occurred extensively as hydrothermal activity waned and temperature decreased (Fig. 4f–k). This secondary Fe sulphide is considered to be a product of the low-temperature reaction between relatively oxidised hydrothermal fluids and pyrrhotite (Koski et al. 1988). Previous experimental studies have reported that marcasite rather than pyrite replaces pyrrhotite at temperatures below ~ 200 °C (Schoonen and Barnes 1991; Qian et al. 2011). Thus, the presence of marcasite pseudomorphs reflects an increase in ƒO2 with a decrease in fluid temperature. Given that alteration of isocubanite to Aip occurred mainly along microfracture zones in the isocubanite, or along grain boundaries of recrystallised marcasite (Mrc-II), the Aip was formed by relatively low-temperature and oxidised fluids as compared with isocubanite. This is supported by the LA–ICP–MS elemental mapping, which showed that Mo is mainly concentrated in the Aip and Mrc-II (Fig. 12). This is consistent with the Aip and Mrc-II having formed at relatively low-temperature conditions. Considering Mo enrichments can be used as a proxy for hydrothermal fluid–seawater mixing (Keith et al. 2016), the formation of Aip is therefore indicative of a seawater alteration stage. The occurrence of native Cu, covellite, and Fe-oxyhydroxides in this stage also supports a low-temperature alteration process (Herzig et al. 1993; de Ronde et al. 2005; Wu et al. 2016; Cao et al. 2018).
The mineralogical variations in the CVF chimney sample studied here represent significant changes in the temperature and oxidation state of the mineralising fluids. A wide variation of FeS contents in sphalerite (6.9–42.5 mol%) shows that temperature of the hydrothermal fluid periodically changed during mineralisation (ESM 1 Fig. S2; Keith et al. 2014; Kawasumi and Chiba 2017). Given that vent fluids are typically enriched in H2 and CH4 in ultramafic-hosted hydrothermal systems (Charlou et al. 2002, 2010; Nakamura et al. 2009), this variable mineralisation is readily achieved by mixing between the strongly reducing hydrothermal fluids and ambient oxidised seawater during chimney growth. Hence, the substantial redox inversion of the vent fluids from pyrite → pyrrhotite → pyrite stability fields could be a primary feature of ultramafic-hosted mineralisation.
Gold mineralisation
Gold-rich electrum is the principal form of Au mineralisation in the CVF, and precipitated during main-stage mineralisation along with pyrrhotite + isocubanite + chalcopyrite + Fe-rich sphalerite (Sp-III), reflecting formation at low ƒS2 and relatively high temperatures (Fig. 5). Apart from pyrrhotite, most sulphide minerals contain some invisible Au (Fig. 9). The amount of invisible Au tends to decrease as mineralisation progressed, apart from in marcasite pseudomorphs (Mrc-II) after pyrrhotite (Fig. 9). Relatively high Au concentrations (0.4–3.4 ppm) were observed in early-stage sphalerite (Sp-I) rather than in main-stage sphalerite (Sp-III) (ESM 1 Fig. S9a). The concentration of invisible Au and absence of electrum in Sp-I suggest that Au was undersaturated in the fluid during the early, relatively low-temperature (< 250 °C) mineralisation. However, the precipitation of electrum at high-temperatures (> 250 °C and < 365 °C) without the formation of Ag-bearing minerals indicates that Au was selectively oversaturated during main-stage mineralisation. This implies that the relatively depleted concentrations of invisible Au in Sp-III would result from the equilibration between Sp-III and a fluid with a low Au content, likely due to the deposition of electrum (ESM 1 Fig. S9a). High concentrations of invisible Ag are also confined to Sp-I (ESM 1 Fig. S9b), with a similar distribution to that of invisible Au. Although the coupled substitution between Ag and Ge can affect Ag concentrations in sphalerite (Fig. 10f), most of the Ag enrichment can be attributed to Ag-bearing, submicroscopic galena inclusions (Figs. 4b, c and 10g, h).
The colloform aggregates of Sp-I and pyrite formed during early, relatively low-temperature mineralisation, indicate that the hydrothermal fluids were extensively mixed with seawater, which led to a rapid decrease in temperature and increased ƒO2 and pH of the fluids (Fig. 4b). In general, these changes result in effective precipitation of Au due to oversaturation of the dominant Au species (e.g. Au(HS)0, Au(HS)2−, and AuCl2−) (Pal’yanova 2008; Fuchs et al. 2019). Given the incorporation of invisible Au into early-stage sulphide without discrete Au-mineral inclusions, these aqueous Au species were still undersaturated in the fluid during early, low-temperature mineralisation. This resulted from increasing Au solubility in response to the higher ƒO2 due to seawater mixing. By contrast, oversaturation of Au occurred during the main-stage mineralisation when electrum precipitated with the high-temperature assemblage of Sp-III, isocubanite, and chalcopyrite (Fig. 6).
In general, seafloor hydrothermal deposits with an ultramafic affinity have low fluid H2S contents, corresponding to the low S activity in highly reduced fluids (Schmidt et al. 2007, 2011). Low S contents of fluids in the CVF are evident from the Fe-rich sphalerite (Sp-III) and pyrrhotite assemblage. It is unlikely that desulfidation of a fluid triggered precipitation of Cu–Fe sulphide during main-stage mineralisation. Given that euhedral to subhedral and medium to large sulphide grains in this stage reflect a relatively closed fluid system, an alternative process could be conductive cooling of the mineralising fluid. In terms of electrum precipitation, an effective mechanism for Au mineralisation at the CVF might have been caused by simple cooling of hydrothermal fluid during the formation of the high-temperature mineral assemblages. This would be consistent with recent thermodynamic modelling that showed Au mineralisation in ultramafic-hosted SMS deposits occurs mainly at high temperatures during conductive cooling of fluid (Fuchs et al. 2019).
During the seawater alteration stage, Au enrichment also occurred along the grain boundaries of marcasite pseudomorphs (Mrc-II) after pyrrhotite (Fig. 12). Pyrrhotite grains formed during main-stage mineralisation have Au contents below the detection limit (Fig. 9). This indicates that invisible Au in Mrc-II did not originate from this pyrrhotite. However, time-resolved LA–ICP–MS depth profiles of Mrc-II show occasional Au spikes (ESM 1 Fig. S6h). This suggests that Au mineralisation may have occurred around Mrc-II during the seawater alteration stage. Given that recrystallisation of pyrrhotite to Mrc-II and abundant Fe-oxyhydroxides occurred extensively during seawater alteration (Fig. 4f–k), the Au mineralisation in this stage could be explained by the following reaction (Eq. 1) (Törmänen and Koski 2005):
The residual Fe2+ resulting from pyrrhotite alteration could have induced the reduction of Au(III) along with Fe-oxyhydroxide deposition during this seawater alteration stage. The strong relationship between Au and Tl in LA–ICP–MS elemental maps suggests that Au mineralisation at this stage occurred in low-temperature conditions (Fig. 12), given that Tl is generally considered to be a good proxy for low-temperature mineralisation (Wang et al. 2017). Hence, hydrothermal mixing may have also caused low-temperature Au mineralisation in this stage, which is clearly different from the high-temperature Au mineralisation of the main stage.
Finally, in the CVF, the selective saturation and precipitation of Au over Ag during main-stage mineralisation resulted in high fineness electrum (ESM 1 Fig. S5). This is similar to other ultramafic-hosted SMS deposits (Murphy and Meyer 1998; Melekestseva et al. 2010). In particular, electrum associated with amorphous silica in the vicinity of Sp-III has a similar range of fineness as those in Sp-III. This implies a constant fluid composition during electrum deposition. We suggest that the predominance of high fineness electrum may have largely contributed to the Au enrichment in the CVF. This may explain the distinct Au–Ag grades between ultramafic- and MORB-hosted SMS deposits (Fig. 8f).
Tin mineralisation
Sulfide mineralisation from the CVF is characterised by substantial enrichments of Sn (up to 1720 ppm), comparable to other seafloor hydrothermal deposits hosted in ultramafic rocks (Fig. 8e). In the CVF, Sn in sulfide is present in solid solution as given by absence of Sn-minerals (ESM 1 Fig. S6-8). Tin is mainly present in sphalerite, followed by chalcopyrite, isocubanite, and marcasite, and its distribution systematically changed during mineralisation (ESM 2 Tables S2 and S3). Initially, low concentrations of Sn (average = 270 ppm) precipitated as a solid solution in early-stage colloform sphalerite (Sp-I). As the sphalerite became more massive, due to coupled dissolution and reprecipitation, Sn was redistributed from Sp-I to early-stage massive sphalerite (Sp-II; average = 1237 ppm Sn). The preferential incorporation of Sn into Sp-II would have resulted in marcasite (Mrc-I) being depleted in Sn (average = 1.5 ppm). During the main-stage mineralisation (> 250 °C and < 365 °C), Sn was primarily incorporated into the main-stage sphalerite (Sp-III; average = 1079 ppm Sn) and isocubanite (average = 517 ppm Sn); discrete chalcopyrite grains enclosed by Sp-III rarely have high Sn concentrations (up to 1.5 wt.%). In particular, significant enrichments of Sn (up to 5.5 wt.%) are found along the replacement boundaries of isocubanite with chalcopyrite exsolution and Sp-III (Fig. 12). This suggests that Sn could no longer be incorporated into the Cu-sulphide lattice via coupled dissolution and reprecipitation, resulting in Sn-rich bands formed along the replacement boundaries (Fig. 7). These were readily mobilised by later hydrothermal fluids, resulting in higher Sn concentrations in the newly formed Sp-III rather than isocubanite (Fig. 12). Thus, hydrothermal remobilisation of trace elements might have been a major mechanism controlling Sn enrichment in sulphide minerals in the CVF. LA–ICP–MS time-resolved depth profiling of covellite shows enrichments in various elements (e.g. Zn, Pb, As, Sb, Sn, Ga, Ge, Au, and Ag; ESM 1 Fig. S6g), whereas most elements in Aip have higher contents as compared with those in isocubanite (Fig. 12). These results are also consistent with remobilisation of the ore-forming elements being common in the CVF.
Most Sn incorporation into the sphalerite lattice occurred by the coupled substitution of 3Zn2+ ↔ 2Cu+ + Sn4+, along with minor Sn2+ contributions (Fig. 10c–e and ESM 1 S3). This indicates that Sn4+ was important for Sn enrichment in the CVF. However, it is generally thought that the Sn(II) aqueous complex (i.e. SnCl2) is the main carrier of Sn in reducing hydrothermal fluids (Sherman et al. 2000; Müller and Seward 2001; Uchida et al. 2002; Migdisov and Williams-Jones 2005). Considering the colloform and small grain size of early-stage sulphide, and the absence of Cu sulphides (Fig. 4) and low FeS contents of Sp-I (ESM 1 Fig. S2), we suggest that the oxidative transition from Sn2+ to Sn4+ was due to mixing with oxidised seawater, which enabled Sn to be deposited as Sn4+ in sphalerite.
There is growing evidence that ultramafic-hosted SMS deposits such as the CVF, located on slow-spreading ridges, are particularly enriched in Sn (Fouquet et al. 2010; Evrard et al. 2015; Dekov et al. 2018). However, it is still unclear why Sn is strongly enriched in ultramafic-hosted sulphide. The origin of Sn in hydrothermal ore deposits is usually attributed to the leaching of Sn-rich sources (Koski et al. 1994; Barrie et al. 1999) or a progressive Sn input from magmatic fluids (Relvas et al. 2006; Huston et al. 2011). Host rock compositions cannot explain Sn enrichments in ultramafic-hosted sulphide because ultramafic rocks have relatively low concentrations of Sn (0.5 ppm) when compared with MORB (1.4 ppm; Anderson 1989). Also, the low H2S concentrations in ultramafic-hosted vent fluids are not consistent with a magmatic input for the Sn enrichment (Von Damm et al. 1995; Schmidt et al. 2011). Given that serpentinisation is mainly responsible for the high contents of H2 and CH4 in ultramafic-hosted hydrothermal fluids (Charlou et al. 2010; Konn et al. 2015), the highly reducing conditions in an ultramafic-hosted system would enhance the transport capacity of Sn2+ during fluid circulation. This is supported by a recent study that showed the Sn concentration of hydrothermal fluid is two orders-of-magnitude higher in an ultramafic-hosted vent field when compared with a MORB-hosted site (Schmidt et al. 2011). Hence, it is likely that hydrothermal fluid is the principal source of elevated Sn concentrations in the CVF. We also suggest that the low redox potential associated with an ultramafic-hosted hydrothermal system could be one of the most important reasons for the Sn enrichment. That is, compared with a MORB-hosted system, Sn2+ would be readily transported in highly reducing conditions, such as in an ultramafic-hosted system. Thereafter, mixing with seawater may have contributed to a change from Sn2+ to Sn4+, leading to Sn enrichment in the Sn4+ form.
Distribution of trace elements
We investigated the distribution of trace elements in sphalerite, which is the main host for most trace elements in the studied subsamples (Fig. 9 and ESM 2 Table S2). The smooth sphalerite LA–ICP–MS time-resolved depth profiles and rarity of metal inclusions suggest that most of the trace elements are present in sphalerite as solid solution, and not as submicron-scale inclusions (ESM 1 Figs. S6a, b, c, 7, and 8). The nearly constant Cu counts in the depth profiles reflect a lack of chalcopyrite disease (ESM 1 Fig. S6a, b, c). This suggests that Cu substitutes for Zn within the sphalerite lattice. Antimony and Ga show positive correlations with Cu, although the trends are less clear at low Cu contents (Fig. 10a, b). This indicates the coupled substitutions of 2Zn2+ ↔ Cu+ + Sb3+ and 2Zn2+ ↔ Cu+ + Ga3+, respectively (Murakami and Ishihara 2013; Belissont et al. 2014; Bonnet et al. 2016). This is supported by an earlier study that showed Cu is present in sphalerite as a monovalent cation (Cook et al. 2012). Copper is strongly correlated with Sn, while Cu + Sn shows a negative correlation with Zn (ESM 1 Fig. S3), suggesting that the coupled presence of Cu and Sn is responsible for Sn enrichment by its coupled substitution for Zn. In particular, Cu/Sn values are ~ 2 in the replacement boundaries, as well as in the sphalerite domains (Fig. 10c and ESM 1 S3b), which can similarly be explained by the coupled substitution of 3Zn2+ ↔ 2Cu+ + Sn4+ (Cook et al. 2009; Ye et al. 2011; Murakami and Ishihara 2013). Hence, Sn was mainly precipitated in its tetravalent state in sphalerite.
Although Ga is less concentrated in sphalerite relative to Sn, the strong correlation between Sn and Ga suggests the possibility that Sn2+ also had an influence on Sn enrichment. The potential substitution mechanisms could be 3Zn2+ ↔ Cu+ + Sn2+ + Ga3+ or 4Zn2+ ↔ Cu+ + Sn4+ + Ga3+ + □ (□ denotes a vacancy) (Fig. 10d). This is supported by the positive correlation between Cu and Ga (Fig. 10b). In addition, the positive correlation between Cd and Sn at a constant molar ratio of almost 1:1 may indicate the possible involvement of Sn2+, such as substitutions of 2Zn2+ ↔ Sn2+ + Cd2+ or 3Zn2+ ↔ Sn4+ + Cd2+ + □ (Fig. 10e). Thus, Sn2+ appears to have a minor impact on Sn enrichment in the CVF.
The correlation between Ag and Ge is consistent with the LA–ICP–MS elemental maps, in which they are commonly enriched in the Fe-poor domains of sphalerite (Figs. 10f and 11). A previous study demonstrated that Ge4+ is generally present in sphalerite, and there is no evidence to support the presence of Ge2+ from the μ-XANES technique (Belissont et al. 2016). The molar ratio of Ag/Ge of ~ 2 obtained in this study suggests a possible substitution of 3Zn2+ ↔ 2Ag+ + Ge4+ (Fig. 10f; Belissont et al. 2014; Cook et al. 2015; Bauer et al. 2019). Arsenic is homogeneously distributed in the LA–ICP–MS element maps (Fig. 11) and LA–ICP–MS depth profiles (ESM 1 Fig. S6a, b, c). This is indicative of ZnAs ↔ ZnS solid solution (Clark 1970). However, the molar ratios of Ag/As of ~ 1 and Ag/Pb of ~ 0.5 imply that the influence of As-bearing nanoparticles, not resolvable in LA–ICP–MS depth profiles, cannot be ruled out (Fig. 10g, h). The possible coupled substitution could be 2Pb2+ ↔ Ag+ + As3+ (Slater 2016), suggesting that nanometre-sized galena inclusions may have an effect on As–Ag–Pb enrichments at the CVF. This is consistent with the presence of submicron-sized galena inclusions within sphalerite (Fig. 4b, c).
Cu and Ag are responsible for the incorporation of a large range of other trace elements in sphalerite. A bivariate plot of tri- and tetravalent cations versus monovalent cations shows that Cu+ (and to some extent Ag+) could provide the charge-balance in various coupled substitutions related to Ga, Ge, Sn, and Sb (Fig. 10i). However, all of the data are scattered below the (Ga + Ge + Sn + Sb)/(Cu + Ag) = 1:1 line. This might be explained by the influence of Ag on galena, or the coupled substitution between divalent (e.g. Fe and Cd) and monovalent cations (e.g. Cu and Ag). Simple plots in this study cannot differentiate between these possibilities.
Conclusions
Gold-rich (up to 17.8 ppm) and Sn-bearing (up to 1720 ppm) hydrothermal chimneys hosted by ultramafic rocks were collected from the newly discovered CVF on the slow-spreading MCIR. We showed that mineralising conditions in the CVF were characterised by abrupt changes in redox state and a steep temperature gradient. We identified three mineralisation stages and their mineral assemblages; (1) a relatively low-temperature (< 250 °C) stage (Sp-I + Sp-II + Mrc-I ± galena), (2) a high-temperature (> 250 °C and < 365 °C) stage (pyrrhotite + isocubanite + chalcopyrite + Sp-III + electrum), and (3) a seawater alteration stage (Mrc-II + Aip + covellite + amorphous silica + Fe-oxyhydroxides ± native Cu ± invisible gold?). The hydrothermal fluid in the early, low-temperature stage was oxidised, and this increased the solubility of Au and Ag, thereby preventing precious metal precipitation. Subsequently, Au mineralisation was triggered by selective Au precipitation over Ag in high fineness electrum at high temperatures and in reduced fluid conditions as the fluid underwent conductive cooling post peak hydrothermal activity. This differs from a MORB-hosted hydrothermal system where gold saturation commonly occurs later in a relatively low-temperature condition when compared with the ultramafic-hosted system, resulting in low Au/Ag values. The distribution of Sn changed systematically during mineralisation. Our results confirm that Sn enrichments are mostly confined to sphalerite, where Sn exists in lattice-bound solid solution and not as submicron inclusions. This can be explained by the coupled substitution of 3Zn2+ ↔ 2Cu+ + Sn4+, together with the possible influence of Sn2+ (i.e. 3Zn2+ ↔ Cu+ + Sn2+ + Ga3+ or 2Zn2+ ↔ Sn2+ + Cd2+). We suggest that the highly reducing conditions may have enhanced the transportation of Sn in its divalent state, with subsequent seawater mixing having an important role in the oxidative transition from Sn2+ to Sn4+ for Sn precipitation in the CVF. The dissolution and reprecipitation of previously deposited Sn-bearing sulphide was also an important mechanism for Sn enrichment. Consequently, the low redox potential in an ultramafic-hosted hydrothermal system has a large effect on Au solubility and the transport capacity of Sn, which leads to Au mineralisation and substitution of Sn in the sulphide minerals. Potentially, this process may occur at all slow-spreading MORs.
References
Allen DE, Seyfried WE (2003) Compositional controls on vent fluids from ultramafic-hosted hydrothermal systems at mid-ocean ridges: an experimental study at 400°C, 500 bars. Geochim Cosmochim Acta 67:1531–1542
Anderson DL (1989) Theory of earth. Blackwell Scientific Publications, Boston, p 150
Badullovich N, Moynier F, Creech J, Teng F-Z, Sossi PA (2017) Tin isotopic fractionation during igneous differentiation and Earth’s mantle composition. Geochemical Perspect Lett 5:24–28
Barrie CT, Hannington MD, Bleeker W (1999) The Giant Kidd Creek volcanic-associated massive sulfide deposit, Abitibi subprovince, Canada. Rev Econ Geol 8:247–269
Bauer ME, Burisch M, Ostendorf J, Krause J, Frenzel M, Seifert T, Gutzmer J (2019) Trace element geochemistry of sphalerite in contrasting hydrothermal fluid systems of the Freiberg district, Germany: insights from LA-ICP-MS analysis, near-infrared light microthermometry of sphalerite-hosted fluid inclusions, and sulfur isotope geochemistry. Mineral Deposita 54:237–262
Belissont R, Boiron M-C, Luais B, Cathelineau M (2014) LA-ICP-MS analyses of minor and trace elements and bulk Ge isotopes in zoned Ge-rich sphalerites from the Noailhac – Saint-Salvy deposit (France): insights into incorporation mechanisms and ore deposition processes. Geochim Cosmochim Acta 126:518–540
Belissont R, Muñoz M, Boiron M-C, Luais B, Mathon O (2016) Distribution and oxidation state of Ge, Cu and Fe in sphalerite by μ-XRF and K-edge μ-XANES: insights into Ge incorporation, partitioning and isotopic fractionation. Geochim Cosmochim Acta 177:298–314
Belousov I, Danyushevsky LV, Olin PH, Gilbert S, Thompson JM (2014) New calibration standard for LA-ICPMS analysis of sulfide. In AGU Fall Meeting Abstracts
Bonnet J, Mosser-Ruck R, Caumon M-C, Rouer O, Andre-Mayer A-S, Cauzid J, Peiffert C (2016) Trace element distribution (Cu, Ga, Ge, Cd, and Fe) in sphalerite from the Tennessee MVT deposits, USA, by combined EMPA, LA-ICP-MS, Raman spectroscopy, and crystallography. Can Mineral 54:1261–1284
Cao H, Sun Z, Zhai S, Cao Z, Jiang X, Huang W, Wang L, Zhang X, He Y (2018) Hydrothermal processes in the Edmond deposits, slow- to intermediate-spreading Central Indian Ridge. J Mar Syst 180:197–210
Caye R, Cervelle B, Cesbron F, Oudin E, Picot P, Pillard F (1988) Isocubanite, a new definition of the cubic polymorph of cubanite CuFe2S3. Mineral Mag 52:509–514
Charlou JL, Donval JP, Fouquet Y, Jean-Baptiste P, Holm N (2002) Geochemistry of high H2 and CH4 vent fluids issuing from ultramafic rocks at the rainbow hydrothermal field (36°14′N, MAR). Chem Geol 191:345–359
Charlou JL, Donval JP, Konn C, Ondréas H, Fouquet Y, Jean-Baptiste P, Fourré E (2010) High production and fluxes of H2 and CH4 and evidence of abiotic hydrocarbon synthesis by serpentinization in ultramafic-hosted hydrothermal systems on the Mid-Atlantic Ridge. Rona P, Devey C, Dyment J, Murton B (Eds.), Diversity of Hydrothermal Systems on Slow Spreading Ocean Ridges, Geophysical Monograph Series, pp 265–296
Clark AH (1970) Alpha-arsenic sulfide, from Mina Alacràn, Pampa Larga, Chile. Am Mineral 55:1338–1344
Cook NJ, Ciobanu CL, Pring A, Skinner W, Shimizu M, Danyushevsky L, Saini-Eidukat B, Melcher F (2009) Trace and minor elements in sphalerite: a LA-ICPMS study. Geochim Cosmochim Acta 73:4761–4791
Cook NJ, Ciobanu CL, Brugger J, Etschmann B, Howard DL, de Jonge MD, Ryan C, Paterson D (2012) Determination of the oxidation state of Cu in substituted Cu-in-Fe-bearing sphalerite via -XANES spectroscopy. Am Mineral 97:476–479
Cook N, Etschmann B, Ciobanu C, Geraki K, Howard D, Williams T, Rae N, Pring A, Chen G, Johannessen B, Brugger J (2015) Distribution and substitution mechanism of Ge in a Ge-(Fe)-bearing sphalerite. Minerals 5:117–132
Dekov VM, Garbe-Schönberg D, Kamenov GD, Guéguen B, Bayon G, Bindi L, Asael D, Fouquet Y (2018) Redox changes in a seafloor hydrothermal system recorded in hematite-chalcopyrite chimneys. Chem Geol 483:351–371
DeMets C, Gordon RG, Argus DF, Stein S (1990) Current plate motions. Geophys J Int 101:425–478
DeMets C, Gordon RG, Argus DF, Stein S (1994) Effect of recent revisions to the geomagnetic reversal time scale on estimates of current plate motions. Geophys Res Lett 21:2191–2194
de Ronde CEJ, Hannington MD, Stoffers P, Wright IC, Ditchburn RG, Reyes AG, Baker ET, Massoth GJ, Lupton JE, Walker SL, Greene RR, Soong CWR, Ishibashi J, Lebon GT, Bray CJ, Resing JA (2005) Evolution of a submarine magmatic-hydrothermal system: Brothers volcano, southern Kermadec arc, New Zealand. Econ Geol 100:1097–1133
de Ronde CEJ, Massoth GJ, Butterfield DA, Christenson BW, Ishibashi J, Ditchburn RG, Hannington MD, Brathwaite RL, Lupton JE, Kamenetsky VS, Graham IJ, Zellmer GF, Dziak RP, Embley RW, Dekov VM, Munnik F, Lahr J, Evans LJ, Takai K (2011) Submarine hydrothermal activity and gold-rich mineralization at Brothers Volcano, Kermadec Arc, New Zealand. Mineral Deposita 46:541–584
Escartín J, Smith DK, Cann J, Schouten H, Langmuir CH, Escrig S (2008) Central role of detachment faults in accretion of slow-spreading oceanic lithosphere. Nature 455:790–794
Evans GN, Tivey MK, Monteleone B, Shimizu N, Seewald JS, Rouxel OJ (2020) Trace element proxies of seafloor hydrothermal fluids based on secondary ion mass spectrometry (SIMS) of black smoker chimney linings. Geochim Cosmochim Acta 269:346–375
Evrard C, Fouquet Y, Moëlo Y, Rinnert E, Etoubleau J, Langlade JA (2015) Tin concentration in hydrothermal sulfide related to ultramafic rocks along the Mid-Atlantic Ridge: a mineralogical study. Eur J Mineral 27:627–638
Firstova A, Stepanova T, Cherkashov G, Goncharov A, Babaeva S (2016) Composition and formation of gabbro-peridotite hosted seafloor massive sulfide deposits from the Ashadze-1 hydrothermal field, Mid-Atlantic Ridge. Minerals 6:19
Fouquet Y, von Stackelberg U, Charlou JL, Erzinger J, Herzig PM, Muehe R, Wiedicke M (1993) Metallogenesis in back-arc environments; the Lau Basin example. Econ Geol 88:2154–2181
Fouquet Y, Cambon P, Etoubleau J, Charlou JL, Ondréas H, Barriga FJAS, Cherkashov G, Semkova T, Poroshina T, Bohn M, Donval JP, Henry K, Murphy P, Rouxel O (2010) Geodiversity of hydrothermal processes along the Mid-Atlantic Ridge and ultramafichosted mineralization: a new type of oceanic Cu-Zn-Co-Au volcanogenic massive sulfide deposit. Diversity of hydrothermal systems on slow spreading ocean ridges. Geophys Monogr Ser 188:321–367
Fuchs S, Hannington MD, Petersen S (2019) Divining gold in seafloor polymetallic massive sulfide systems. Mineral Deposita 54:789–820
Fujii M, Okino K, Sato T, Sato H, Nakamura K (2016) Origin of magnetic highs at ultramafic hosted hydrothermal systems: insights from the Yokoniwa site of Central Indian Ridge. Earth Planet Sci Lett 441:26–37
Gamo T, Chiba H, Yamanaka T, Okudaira T, Hashimoto J, Tsuchida S, Ishibashi J, Kataoka S, Tsunogai U, Okamura K, Sano Y, Shinjo R (2001) Chemical characteristics of newly discovered black smoker fluids and associated hydrothermal plumes at the Rodriguez Triple Junction, Central Indian Ridge. Earth Planet Sci Lett 193:371–379
Halbach P, Blum N, Münch U, Plüger W, Garbe-Schönberg D, Zimmer M (1998) Formation and decay of a modern massive sulfide deposit in the Indian Ocean. Mineral Deposita 33:302–309
Hannington MD, de Ronde CEJ, Petersen S (2005) Sea-floor tectonics and submarine hydrothermal systems. Econ Geol 100th Anniversary Volume, pp 111–141
Hannington M, Jamieson J, Monecke T, Petersen S (2010) Modern sea-floor massive sulfides and base metal resources: toward an estimate of global sea-floor massive sulfide potential. Rev Econ Geol 15:317–338
Herzig PM, Hannington MD, Fouquet Y, von Stackelberg U, Petersen S (1993) Gold-rich polymetallic sulfides from the Lau back arc and implications for the geochemistry of gold in sea-floor hydrothermal systems of the Southwest Pacific. Econ Geol 88:2182–2209
Herzig PM, Hannington MD (1995) Polymetallic massive sulfides at the modern seafloor a review. Ore Geol Rev 10:95–115
Hutchison MN, Scott SD (1981) Sphalerite geobarometry in the Cu-Fe-Zn-S system. Econ Geol 76:143–153
Huston DL, Relvas JMRS, Gemmell JB, Drieberg S (2011) The role of granites in volcanic-hosted massive sulfide ore-forming systems: an assessment of magmatic–hydrothermal contributions. Mineral Deposita 46:473–507
Jochum KP, Hofmann AW, Seufert HM (1993) Tin in mantle-derived rocks: constraints on earth evolution. Geochim Cosmochim Acta 57:3585–3595
Kawasumi S, Chiba H (2017) Redox state of seafloor hydrothermal fluids and its effect on sulfide mineralization. Chem Geol 451:25–37
Keith M, Haase KM, Schwarz-Schampera U, Klemd R, Petersen S, Bach W (2014) Effects of temperature, sulfur, and oxygen fugacity on the composition of sphalerite from submarine hydrothermal vents. Geology 42:699–702
Keith M, Häckel F, Haase KM, Schwarz-Schampera U, Klemd R (2016) Trace element systematics of pyrite from submarine hydrothermal vents. Ore Geol Rev 72:728–745
Kissin SA, Scott SD (1982) Phase relations involving pyrrhotite below 350°C. Econ Geol 77:1739–1754
Knight DR, Roberts S, Webber AP (2018) The influence of spreading rate, basement composition, fluid chemistry and chimney morphology on the formation of gold-rich SMS deposits at slow and ultraslow mid-ocean ridges. Mineral Deposita 53:143–152
Kojima S, Sugaki A (1985) Phase relations in the Cu-Fe-Zn-S system between 500° and 300°C under hydrothermal conditions. Econ Geol 80:158–171
Konn C, Charlou JL, Holm NG, Mousis O (2015) The production of methane, hydrogen, and organic compounds in ultramafic-hosted hydrothermal vents of the Mid-Atlantic Ridge. Astrobiology 15:381–399
Koski RA, Shanks WC, Bohrson WA, Oscarson RL (1988) Composition of massive sulfide deposits from the sediment-covered floor of Escanaba Trough, Gorda Ridge: implications for depositional processes. Can Mineral 26:655–673
Koski RA, Benninger LM, Zierenberg RA, Jonasson IR (1994) Composition and growth history of hydrothermal deposits in Escanaba Trough, Southern Gorda Ridge. In: Morton JL, Zierenberg RA, Reiss CA (eds) Geologic, hydrothermal and biologic studies at Escanaba Trough, Gorda Ridge, offshore Northern California. US Geol Surv Bull 2022:293–324
Lafitte M, Maury R, Perseil EA, Boulegue J (1985) Morphological and analytical study of hydrothermal sulfides from 21° north East Pacific rise. Earth Planet Sci Lett 73:53–64
Lusk J, Bray DM (2002) Phase relations and the electrochemical determination of sulfur fugacity for selected reactions in the Cu–Fe–S and Fe–S systems at 1 bar and temperatures between 185 and 460 °C. Chem Geol 192:227–248
Maslennikov VV, Maslennikova SP, Large RR, Danyushevsky LV, Herrington RJ, Ayupova NR, Zaykov VV, Lein AY, Tseluyko AS, Melekestseva IY, Tessalina SG (2017) Chimneys in Paleozoic massive sulfide mounds of the Urals VMS deposits: mineral and trace element comparison with modern black, grey, white and clear smokers. Ore Geol Rev 85:64–106
McCaig AM, Cliff RA, Escartin J, Fallick AE, MacLeod CJ (2007) Oceanic detachment faults focus very large volumes of black smoker fluids. Geology 35:935–938
Melekestseva IY, Kotlyarov VA, Khvorov PV, Ivanov VN, Beltenev VE, Dobretsova IG (2010) Noble-metal mineralization in the Semenov-2 hydrothermal field (13°31′N), mid-Atlantic ridge. Geol Ore Depos 52:800–810
Melekestseva IY, Tret’yakov GA, Nimis P, Yuminov AM, Maslennikov VV, Maslennikova SP, Kotlyarov VA, Beltenev VE, Danyushevsky LV, Large R (2014) Barite-rich massive sulfides from the Semenov-1 hydrothermal field (Mid-Atlantic Ridge, 13°30.87′ N): evidence for phase separation and magmatic input. Mar Geol 349:37–54
Melekestseva IY, Maslennikov VV, Tret’yakov GA, Nimis P, Beltenev VE, Rozhdestvenskaya II, Maslennikova SP, Belogub EV, Danyushevsky L, Large R, Yuminov AM, Sadykov SA (2017) Gold- and silver-rich massive sulfides from the Semenov-2 hydrothermal field, 13°31.13′N, Mid-Atlantic Ridge: a case of magmatic contribution? Econ Geol 112:741–773
Migdisov AA, Williams-Jones AE (2005) An experimental study of cassiterite solubility in HCl-bearing water vapour at temperatures up to 350 °C. Implications for tin ore formation. Chem Geol 217:29–40
Mozgova NN, Borodaev YS, Gablina IF, Cherkashev GA, Stepanova TV (2005) Mineral assemblages as indicators of the maturity of oceanic hydrothermal sulfide mounds. Lithol Miner Resour 40:293–319
Mozgova NN, Trubkin NV, Borodaev YS, Cherkashev GA, Stepanova TV, Semkova TA, Uspenskaya TY (2008) Mineralogy of massive sulfides from the ASHADZE hydrothermal field, 13°n, Mid-Atlantic Ridge. Can Mineral 46:545–567
Müller B, Seward TM (2001) Spectrophotometric determination of the stability of tin(II) chloride complexes in aqueous solution up to 300°C. Geochim Cosmochim Acta 65:4187–4199
Münch U, Blum N, Halbach P (1999) Mineralogical and geochemical features of sulfide chimneys from the MESO zone, Central Indian Ridge. Chem Geol 155:29–44
Murakami H, Ishihara S (2013) Trace elements of indium-bearing sphalerite from tin-polymetallic deposits in Bolivia, China and Japan: a femto-second LA-ICPMS study. Ore Geol Rev 53:223–243
Murphy PJ, Meyer G (1998) A gold-copper association in ultramafic-hosted hydrothermal sulfides from the Mid-Atlantic Ridge. Econ Geol 93:1076–1083
Nakamura K, Morishita T, Bach W, Klein F, Hara K, Okino K, Takai K, Kumagai H (2009) Serpentinized troctolites exposed near the Kairei hydrothermal field, Central Indian Ridge: insights into the origin of the Kairei hydrothermal fluid supporting a unique microbial ecosystem. Earth Planet Sci Lett 280:128–136
Nakamura K, Watanabe H, Miyazaki J, Takai K, Kawagucci S, Noguchi T, Nemoto S, Watsuji T, Matsuzaki T, Shibuya T, Okamura K, Mochizuki M, Orihashi Y, Ura T, Asada A, Marie D, Koonjul M, Singh M, Beedessee G, Bhikajee M, Tamaki K (2012) Discovery of new hydrothermal activity and chemosynthetic Fauna on the Central Indian Ridge at 18°–20°S. PLoS One 7:e32965
Nayak B, Halbach P, Pracejus B, Münch U (2014) Massive sulfides of Mount Jourdanne along the super-slow spreading Southwest Indian Ridge and their genesis. Ore Geol Rev 63:115–128
Pak S-J, Moon J-W, Kim J, Chandler MT, Kim H-S, Son J, Son S-K, Choi SK, Baker ET (2017) Widespread tectonic extension at the Central Indian Ridge between 8°S and 18°S. Gondwana Res 45:163–179
Pal’yanova G (2008) Physicochemical modeling of the coupled behavior of gold and silver in hydrothermal processes: gold fineness, Au/Ag ratios and their possible implications. Chem Geol 255:399–413
Paton C, Hellstrom J, Paul B, Woodhead J, Hergt J (2011) Iolite: freeware for the visualisation and processing of mass spectrometric data. J Anal At Spectrom 26:2508–2518
Qian G, Xia F, Brugger J, Skinner WM, Bei J, Chen G, Pring A (2011) Replacement of pyrrhotite by pyrite and marcasite under hydrothermal conditions up to 220 °C: an experimental study of reaction textures and mechanisms. Am Mineral 96:1878–1893
Relvas JMRS, Barriga FJAS, Ferreira A, Noiva PC, Pacheco N, Barriga G (2006) Hydrothermal alteration and mineralization in the Neves-Corvo volcanic-hosted massive sulfide deposit, Portugal. I. Geology, mineralogy, and geochemistry. Econ Geol 101:753–790
Schmidt K, Koschinsky A, Garbe-Schönberg D, de Carvalho LM, Seifert R (2007) Geochemistry of hydrothermal fluids from the ultramafic-hosted Logatchev hydrothermal field, 15°N on the Mid-Atlantic Ridge: temporal and spatial investigation. Chem Geol 242:1–21
Schmidt K, Garbe-Schönberg D, Koschinsky A, Strauss H, Jost CL, Klevenz V, Königer P (2011) Fluid elemental and stable isotope composition of the Nibelungen hydrothermal field (8°18′S, Mid-Atlantic Ridge): constraints on fluid–rock interaction in heterogeneous lithosphere. Chem Geol 280:1–18
Schoonen MAA, Barnes HL (1991) Reactions forming pyrite and marcasite from solution: II. Via FeS precursors below 100°C. Geochim Cosmochim Acta 55:1505–1514
Sherman DM, Ragnarsdottir KV, Oelkers EH, Collins CR (2000) Speciation of tin (Sn2+ and Sn4+) in aqueous Cl solutions from 25°C to 350°C: an in situ EXAFS study. Chem Geol 167:169–176
Slater ET (2016) The Cortaderas Zone, Pirquitas Mine, NW Argentina: an example of Miocene epithermal Ag-Zn-Pb-Sn mineralization in the Andean Tin Belt. Unpublished M.Sc. thesis, Sudbury, Canada, Laurentian University, Sudbury, Ontario, pp.1–113
Son J, Pak S-J, Kim J, Baker ET, You O-R, Son S-K, Moon J-W (2014) Tectonic and magmatic control of hydrothermal activity along the slow-spreading Central Indian Ridge, 8°S-17°S. Geochem Geophys Geosyst 15:2011–2020
Sugaki A, Shima H, Kitakaze A, Harada H (1975) Isothermal phase relations in the system Cu-Fe-S under hydrothermal conditions at 350°C and 300°C. Econ Geol 70:806–823
Törmänen TO, Koski RA (2005) Gold enrichment and the bi-Au Association in Pyrrhotite-Rich Massive Sulfide Deposits, Escanaba Trough, Southern Gorda Ridge. Econ Geol 100:1135–1150
Uchida E, Sakamori T, Matsunaga J (2002) Aqueous speciation of lead and tin chlorides in supercritical hydrothermal solutions. Geochem J 36:61–72
Van Dover CL, Humphris SE, Vrijenhoek RC et al (2001) Biogeography and ecological setting of Indian Ocean hydrothermal vents. Science 294(5543):818–823
Von Damm KL, Oosting SE, Kozlowski R, Buttermore LG, Colodner DC, Edmonds HN, Edmond JM, Grebmeier JM (1995) Evolution of East Pacific rise hydrothermal vent fluids following a volcanic eruption. Nature 375:47–50
Wang Y, Han X, Petersen S, Jin X, Qiu Z, Zhu J (2014) Mineralogy and geochemistry of hydrothermal precipitates from Kairei hydrothermal field, Central Indian Ridge. Mar Geol 354:69–80
Wang Y, Han X, Petersen S, Frische M, Qiu Z, Li H, Li H, Wu Z, Cui R (2017) Mineralogy and trace element geochemistry of sulfide minerals from the Wocan hydrothermal field on the slow-spreading Carlsberg Ridge, Indian Ocean. Ore Geol Rev 84:1–19
Wilson SA, Ridley WI, Koenig AE (2002) Development of sulfide calibration standards for the laser ablation inductively-coupled plasma mass spectrometry technique. J Anal At Spectrom 17:406–409
Wohlgemuth-Ueberwasser CC, Viljoen F, Petersen S, Vorster C (2015) Distribution and solubility limits of trace elements in hydrothermal black smoker sulfides: an in-situ LA-ICP-MS study. Geochim Cosmochim Acta 159:16–41
Wu Z, Sun X, Xu H, Konishi H, Wang Y, Wang C, Dai Y, Deng X, Yu M (2016) Occurrences and distribution of “invisible” precious metals in sulfide deposits from the Edmond hydrothermal field, Central Indian Ridge. Ore Geol Rev 79:105–132
Ye L, Cook NJ, Ciobanu CL, Yuping L, Qian Z, Tiegeng L, Wei G, Yulong Y, Danyushevskiy L (2011) Trace and minor elements in sphalerite from base metal deposits in South China: a LA-ICPMS study. Ore Geol Rev 39:188–217
Acknowledgments
The authors thank scientists and crews of the R/V ONNURI and R/V ISABU for their support during the sampling and collection of data. The careful reviews by J. Jameison and C. de Ronde are greatly appreciated, which improved the quality of this manuscript. The authors also thank chief editor G. Breaudoin and T. Monecke for valuable comments on the manuscript.
Funding
This research was financially supported by the Ministry of Oceans and Fisheries of Korea (19992001 and 20170411) and Korea Institute of Ocean Science and Technology (PE99824).
Author information
Authors and Affiliations
Corresponding author
Additional information
Editorial handling: T. Monecke
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Choi, S.K., Pak, S.J., Kim, J. et al. Gold and tin mineralisation in the ultramafic-hosted Cheoeum vent field, Central Indian Ridge. Miner Deposita 56, 885–906 (2021). https://doi.org/10.1007/s00126-020-01012-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00126-020-01012-5