
Abstract
This article summarises the state of the science on the role of the gut microbiota (GM) in diabetes from a recent international expert forum organised by Diabetes, Diabetes Care, and Diabetologia, which was held at the European Association for the Study of Diabetes 2023 Annual Meeting in Hamburg, Germany. Forum participants included clinicians and basic scientists who are leading investigators in the field of the intestinal microbiome and metabolism. Their conclusions were as follows: (1) the GM may be involved in the pathophysiology of type 2 diabetes, as microbially produced metabolites associate both positively and negatively with the disease, and mechanistic links of GM functions (e.g. genes for butyrate production) with glucose metabolism have recently emerged through the use of Mendelian randomisation in humans; (2) the highly individualised nature of the GM poses a major research obstacle, and large cohorts and a deep-sequencing metagenomic approach are required for robust assessments of associations and causation; (3) because single time point sampling misses intraindividual GM dynamics, future studies with repeated measures within individuals are needed; and (4) much future research will be required to determine the applicability of this expanding knowledge to diabetes diagnosis and treatment, and novel technologies and improved computational tools will be important to achieve this goal.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In October 2023, a closed-door, day-long forum organised under the auspices of the journals Diabetes, Diabetes Care, and Diabetologia took place during the European Association for the Study of Diabetes 2023 Annual Meeting in Hamburg, Germany. The express goal of the forum was to create consensus and perform gap analyses to advance research into the role of the gut microbiota (GM) in diabetes. Discussions fell under four main headings: epidemiology; physiology and pathophysiology; technology and methodology; and clinical applications.
The group acknowledged that many of the gaps in understanding of the GM’s role in metabolic diseases are not unique to the diabetes field, but rather reflect broader needs to [1] conduct more well-controlled prospective and retrospective human studies that are followed up mechanistically with model systems studies and [2] refine computational tools and welcome a return of microbiology and molecular biology to our experimental toolkit. Nonetheless, there was agreement that the current reproduced microbiome data represent compelling target areas for future diabetes research. This article presents a distillation of the evidence and recommendations on important microbiome focus areas that would benefit from the attention of young and established diabetes researchers alike. Key knowledge gaps and challenges discussed in this article are summarised in the textbox (see ‘The GM and diabetes: key knowledge gaps and challenges’).

Epidemiological perspectives
Epidemiological associations between the GM and diabetes
The GM is the largest and most complex microbial community of the human body, connecting our external and internal milieu (Fig. 1). The motivation for epidemiological studies of the GM in obesity and cardiometabolic diseases, including type 2 diabetes, emerged from rodent studies demonstrating links among the GM, adiposity, and glucose tolerance [1, 2]. In humans, epidemiological studies have observed decreased microbial diversity in obesity, but no generalisable obesity-associated gut microbial signature has emerged from meta-analyses of small cohorts profiled by 16S rRNA gene sequencing [3, 4] or whole-genome metagenomics [5]. However, a large GM study using deep-sequencing whole-genome metagenomics in 34,057 individuals from Israel and the United States demonstrated consistent GM–phenotype associations and the predictive accuracy of machine-learning models trained on microbiome data for body mass index (BMI) and HbA1c that could be replicated across the cohorts [6]. By subsampling the training cohort, these authors showed increased predictive accuracy with increased cohort size, with ~7500–10,000 individuals optimal for replicable results. This finding highlights the necessity of using large cohorts with hundreds of individuals and deep-sequencing whole-genome metagenomics that adequately represent the embedded interindividual heterogeneity and regional and demographic variation in human GM cross-sectional studies.

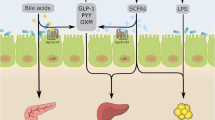
Microecological and physiological differences along the GI tract [103, 234]. Environmental conditions vary along the GI tract depending on physical, nutritional, and biological host factors, which translate into adaptations and differences in the intestinal bacteria inhabiting the different regions and their physiological functions through multidirectional interactions that may affect glucose metabolism and diabetes risk. The main factors affecting the microbial load and composition in the different regions are as follows. (1) pH values increase drastically from the stomach (pH 1.0–4.4) to the small intestine (pH 5.5–7.0) and then more progressively to the colon, where the pH can drop again (pH 5.5) as a consequence of the microbial fermentation of complex carbohydrates (fibre). The pH increases again in faeces (up to pH 7.8). (2) Intestinal transit is shorter and peristaltic movements are more intense in the small intestine than in the large intestine. (3) Small intestinal host epithelial cells (Paneth cells) secrete AMPs, acting as an innate defensive barrier reducing bacterial colonisation, and M cells of Peyer’s patches also pick up bacteria from the intestinal lumen. (4) Oxygen levels are also progressively reduced from the small intestine to the large intestine. (5) Dietary nutrients (proteins, lipids, and simple carbohydrates) are primarily digested by host enzymes and rapidly absorbed in the small intestine, limiting the accessibility of nutrients to intestinal bacteria; in contrast, partially undigested dietary residues (complex carbohydrates and partially hydrolysed proteins/amino acids) accumulate in the large intestine, where they serve as nutrients for bacteria. (6) Host glycans forming part of the mucous layer (produced by goblet cells), which is remarkably thicker in the large intestine than in the small intestine, also represent a nutrient source for intestinal bacteria, supporting their growth. (7) Bile acids are secreted to the small intestine, inhibiting and favouring the growth of specific bacteria that participate in their metabolism and recirculation. Altogether, those abiotic and biotic factors affect the ecological conditions that facilitate the survival of denser populations of bacteria moving to the most distal parts of the intestine (from 102–104 bacterial cells/g in the duodenum to 107–109 in the ileum and 1011–1012 in the colon) and account for differences in bacterial composition, with facultative anaerobes preferentially colonising the small intestine and strict anaerobes dominating the microbiota of the large intestine, including butyrate producers. In the large intestine, EECs, mainly L-cells, are stimulated by SCFAs (butyrate and propionate) to induce the hormones GLP-1 and PYY, which contribute to insulin secretion and glucose homeostasis and regulate appetite. In the small intestine, other EECs, such as I-cells, predominate and produce the hormone CCK, which induces digestive enzymes and bile and suppresses appetite. This is also the main region where nutrient signals are sensed by the enteric neurons and vagal afferents and thus signal to the brain to control energy homeostasis, although knowledge of the role of the gut microbiota in their regulation is limited. SCFAs, especially butyrate, can also induce immunoregulatory T cells (T-regs) that protect against obesity-induced pro-inflammatory macrophages and prevent LPS translocation. AA, amino acid; AMP, antimicrobial peptide; CCK, cholecystokinin; PYY, peptide YY. This figure is available as part of a downloadable slideset
Several observational studies have reported associations between the GM and type 2 diabetes. Consistent features of altered GM composition in type 2 diabetes and impaired glucose tolerance/fasting glucose, found in epidemiological studies worldwide and also occurring in the metabolic syndrome, are reduced diversity and decreased abundance of bacteria that produce the short-chain fatty acid (SCFA) butyrate (Figs 1 and 2) [7,8,9,10,11,12,13,14]. Some studies have also observed an increase of opportunistic pathogens [7, 8, 11], some of which have been linked to subclinical coronary atherosclerosis [15]. The mucus-degrading bacterium Ruminococcus gnavus, which has been linked to inflammatory bowel diseases, recently has also been identified as a predictor of several features of the metabolic syndrome, including low-grade inflammation, large waist circumference, elevated serum triglycerides, elevated HbA1c, and decreased HDL-cholesterol [16]. However, as indicated by meta-analyses of GM alterations across different diseases, including gastrointestinal (GI) and metabolic diseases, several of these features are not disease-specific and might characterise a general GM dysbiosis [4, 5]. Therefore, to disentangle disease-specific microbial signatures beyond differences in race/ethnicity, lifestyle, and other demographic characteristics, it will be important to perform studies in large populations and to include healthy individuals/control participants from different studies as references; these approaches have been shown to increase disease prediction accuracy [5, 6].

GM metabolites and signalling molecules linked to glucose metabolism and type 2 diabetes. Structural and secreted GM proteins are involved in the modulation of immune responses and inflammation, as shown for a protein secreted by F. prausnitzii (microbial anti-inflammatory molecule [MAM]), which is able to inhibit the nuclear factor-κB (Nf-κB) pathway. Another example is Amuc_1100, an outer membrane protein of A. muciniphila, which improves the gut barrier and decreases inflammation. The GM also produces SCFAs, which stimulate the release of incretin hormones and improve peripheral tissue metabolism. In addition, SCFAs modulate immune cell function, improve the gut barrier, and stimulate enteric neuron signalling. The SCFA butyrate also provides energy to colonocytes and increases colonocyte β-oxidation (β-ox) by activating peroxisome proliferator-activated receptor-γ (PPARγ). Bile acid signalling through the bile acid receptors FXR and TGR5 modulates metabolic responses in several different tissues. GM tryptophan metabolites, such as indolepropionic acid (IPA) and indoleacrylic acid (IA), modulate immune and metabolic responses by improving the gut barrier through the pregnane X receptor (PXR) and by signalling through the aryl hydrocarbon receptor (AHR) on intestinal immune cells and increasing the production of interleukin-22 (IL-22). In the bloodstream, IPA and IA also provide antioxidant and anti-inflammatory functions. Imidazole propionate and BCAAs impair insulin signalling through activation of the mechanistic target of rapamycin complex 1 (mTORC1). The GM also produces ethanol, which is linked to fatty liver disease and insulin resistance. IL-10, interleukin-10; IRS1, insulin receptor substrate 1; PYY, peptide YY. © 2019 Canadian Diabetes Association. Adapted from Caesar [235] with permission from Elsevier. This figure is available as part of a downloadable slideset
In addition to a decreased capacity to produce butyrate, GM functional capabilities that are altered in type 2 diabetes are involved in the production of branched-chain amino acids (BCAAs) and the metabolism of B vitamins and simple sugars [7, 12, 17, 18]. Increased levels of circulating BCAAs have been described in insulin-resistant individuals and linked to a higher risk of type 2 diabetes [19]. In line with this, an increased potential to synthesise BCAAs and decreased microbial BCAA uptake and catabolism have been described in the GM of insulin-resistant individuals with normoglycaemia [18]. However, the analyses of GM functions only show an altered potential. Quantification of metabolites has recently been performed to validate these findings. Figure 2 depicts the links between GM metabolites and signalling molecules that have been observed for glucose metabolism and type 2 diabetes.
Associations between the GM or its metabolites and glucose-lowering drug treatments
Evidence supporting the role of the GM in type 2 diabetes has been strengthened by observational and interventional studies demonstrating changes in the relative abundance of multiple bacterial species in the GM of metformin users [10, 20,21,22]. A higher relative abundance of Escherichia coli and a decreased abundance of Intestinibacter bartlettii [10, 20, 21] have been described in multiple cohorts involving individuals being treated with metformin. Additionally, an increase in Escherichia marmotae and a decrease in Romboutsia timonensis have been found in metformin-treated individuals in a recent large metagenomic study [23].
Support for the causal effects of these GM differences in type 2 diabetes has been provided by randomised trials and studies in drug-naive individuals demonstrating that the GM compositional changes translate to enhanced production of propionate and butyrate [20, 21] and modulation of bile acid pools [21], which may mediate some of the glucose-lowering effects of metformin (Fig. 3) [20,21,22, 24]. However, the GM might also be responsible for the transient or persistent intestinal discomfort experienced by ~30% of individuals who take metformin (e.g. through increased gas production by some Escherichia species) [22, 25].

GM interactions with glucose-lowering medications. GM metabolites are involved in the mechanism of action of metformin, including bile acid (BA) signalling through the bile acid receptors FXR and TGR5 and production of SCFAs, which modulate the release of incretin hormones such GLP-1, gastric inhibitory polypeptide (GIP), and peptide YY (PYY) from enteroendocrine cell (K- and L-cells). Other GM-dependent mechanisms involved in the action of metformin include improved glucose sensing through sodium–glucose cotransporter 1 (SGLT1) and an improved gut barrier (e.g. restoration of tight junctions and increase in mucin-producing goblet cells [236, 237]). However, through the expression of DPP-4 isozymes, the GM might decrease GLP-1 activity and affect the efficacy of glucose-lowering drugs. © 2019 Canadian Diabetes Association. Adapted from Caesar [235] with permission from Elsevier. This figure is available as part of a downloadable slideset
With regard to other oral glucose-lowering drugs, studies have shown effects of dipeptidyl peptidase 4 (DPP-4) inhibitors and α-glucosidase inhibitors on the GM and microbial metabolites, but less clear effects of sodium–glucose cotransporter 2 (SGLT2) inhibitors, thiazolidinediones, and glucagon-like peptide 1 (GLP-1) receptor agonists [26,27,28].The majority of studies to date involving SGLT2 inhibitors have been conducted in mouse models, and the few existing human studies have provided contradictory results and were unable to clearly discriminate the effects of the SGLT2 inhibitor from those of previous or concomitant metformin treatment or concurrent lifestyle modifications [29]. GLP-1 receptor agonists may exert anti-inflammatory effects (e.g. through activation of the intraepithelial lymphocyte GLP-1 receptors), which in turn could contribute to modulating the gut microbiota [30, 31]. Although much more research is needed, existing evidence suggests that the GM may mediate some of the benefits of glucose-lowering treatments [26], and certain probiotics or prebiotics might further improve the glucose-lowering effects of these drugs through their effects on the GM or its functions [32]. Further interventional and translational studies are needed to determine whether drug-induced GM changes are causally involved in mediating health effects and to uncover the underlying mechanisms.
Importantly, the GM might also influence the efficacy of glucose-lowering drugs, for example by expressing homologues of human DPP-4, which can decrease the activity of GLP-1 and affect glucose metabolism (Fig. 3) [33, 34]. Because bacterial DPP-4 homologues seem resistant to some drugs targeting human DPP-4 [33], inhibition of bacterial isozymes might be required to improve metabolic responses to current medications.
Associations between GM metabolites and diabetes-related traits
SCFAs
The GM ferments plant-based dietary carbohydrates and fibre, as well as peptides that reach the large intestine, to produce SCFAs—mainly acetate, propionate, and butyrate. After hepatic metabolism, ~70% of colonic acetate, but only small amounts of propionate and butyrate (<2% for butyrate), reach the circulation [35]. As described in more detail in ‘Physiological and pathophysiological perspectives’, SCFAs regulate several processes, including intestinal motility and pH, gut barrier immune responses and systemic metabolism through pathways affecting gluconeogenesis, insulin sensitivity, and insulin secretion (Fig. 2) [36]. However, human studies show extensive variation in the levels of different SCFAs in the stools and/or blood of individuals with type 2 diabetes, which is likely because of methodological limitations [36]. The strongest support for the role of SCFAs in the regulation of glucose metabolism is provided by animal studies and one recent human study using the Mendelian randomisation (MR) statistical method [36, 37] (see ‘Role of MR in elucidating causal effects’ below).
Bile acids
Bile acids are amphipathic molecules that mediate the absorption of dietary fats and lipid-soluble vitamins. These molecules are also recognised as major players in regulating lipid, glucose, and energy metabolism. Consequently, alterations in bile acid pools have been found in type 2 diabetes and other obesity-related diseases and identified as possible contributors to the pathophysiology of type 2 diabetes (Fig. 2) [24, 38,39,40,41].
Increased levels of 12α-hydroxylated bile acids [41] and decreased levels of 6α-hydroxylated bile acids [42, 43] are linked to insulin resistance and occur in people with type 2 diabetes. Increased levels of 6α-hydroxylated bile acids are observed after gastric bypass surgery and can predict type 2 diabetes remission [42]. The GM can deconjugate and transform bile acids, thus contributing to a highly variable but important portion of human bile acid pools (Fig. 1) [44]. For example, circulating levels of 6α-hydroxylated bile acids are found to co-vary with levels of specific Clostridia species in the gut [43].
Intervention studies have also investigated the potential importance of bile acids in human metabolism. Elevated systemic bile acid levels and intestinal signalling to stimulate the release of GLP-1 have been demonstrated after bariatric surgery, with postprandial increases found to be particularly important [45]. However, exaggerated bile acid responses have been found in some individuals with cholecystectomy and are associated with further increased GLP-1 and insulin responses [46]. In people with type 2 diabetes, metformin has been shown to improve glucose metabolism via a decreased abundance of Bacteroides fragilis, which has been linked to increased levels of glycoursodeoxycholic acid in the gut and inhibition of the farnesoid X receptor (FXR) [24]. However, our understanding of direct interactions between the GM and bile acids and their associations with the development and treatment of type 2 diabetes and related diseases is still limited, and more human studies are warranted.
Other metabolites
GM-produced amino acid metabolites have also been linked to type 2 diabetes (Figs 1 and 2). Increased circulating levels of 3-indolepropionic acid, a tryptophan catabolite, have been associated with improved insulin secretion and sensitivity and a decreased risk of type 2 diabetes [47]. Furthermore, increased plasma levels of imidazole propionate, a bacterial product of histidine metabolism, have been reported in individuals with insulin resistance and type 2 diabetes [48]. These metabolites and several others derived from GM catabolism of aromatic amino acids have also been associated with incident cardiovascular risk and mortality in independent cohorts from Europe and the United States [49, 50]. Finally, GM ethanol production has been associated with fatty liver disease [51,52,53] and might be linked to insulin resistance (Fig. 2).
Role of MR in elucidating causal effects
The GM can affect and interact with host health in numerous ways, and the arrow of causality is often bidirectional or even multidirectional. GM features at different levels (e.g. community, species, pathway, gene, and metabolite) can affect a host phenotype (e.g. altering the risk of obesity), while the development of a phenotype (e.g. obesity) can, in turn, change the GM.
MR is a statistical method that uses human genetic variants related to exposures to discriminate causal effects on disease outcomes from associations that result from confounding, reverse causation, or something else. To apply MR to investigate the connection between the GM and type 2 diabetes, the GM feature in question needs to be affected by a human genetic variant or multiple variants strongly enough to allow their use as instruments in instrumental variant analysis.
Although several genome-wide association studies have been performed on different GM features such as gut bacterial taxa relative abundances and human faecal microbial metabolites, large MR studies investigating their causal role in type 2 diabetes have been limited and are not yet confirmed in replication studies. Sanna et al [37] identified human genetic variants that associate with faecal SCFA levels and reported evidence for a potential causal connection between the GM’s butyrate production potential (i.e. genes responsible for GM butyrate production) and improved response to insulin during an oral glucose tolerance test. These authors also found a causal link between abnormal faecal propionate levels and increased type 2 diabetes risk [37]. Another MR study reported that type 2 diabetes and kidney disease increased plasma levels of the GM-dependent metabolite trimethylamine oxide (TMAO) and proposed that the earlier observational evidence of elevated risk of cardiovascular diseases with higher TMAO levels might have been the result of confounding or reverse causality rather than a causal effect [54]. Another recent study suggested that certain bacterial genera could have a causal link to type 2 diabetes [55]. Considering the limitations of both MR (e.g. pleiotropy and problems related to weak instrumental variables) and GM research (e.g. methodological differences, interindividual heterogeneity, and intraindividual variability), large, high-quality studies are needed to assess the ability of host genetic variants using MR to mimic specific GM features—whether specific bacterial species, genera, or metabolite products—to understand causal connections with type 2 diabetes pathogenesis.
Associations among diet, GM, and diabetes
Decreased dietary fibre intake has been associated repeatedly with increased risk of type 2 diabetes; accordingly, new dietary recommendations for diabetes management encourage high consumption of minimally processed plant foods such as whole grains, vegetables, whole fruit, legumes, nuts, and seeds [56]. Diet is a driver of the GM ecosystem, and microbially accessible carbohydrates promote GM diversity and SCFA generation, which decreases inflammation and supports the maintenance of the gut barrier [57].
In relation to the GM and glucose metabolism, increased fibre intake has been associated with increased levels of distinct species, for example Prevotella copri [58] (now renamed Segatella copri). Studies have also shown that the beneficial effects of fibres on HbA1c may be mediated by the specific baseline GM composition and diversity of fibre-promoted SCFA-producing bacteria [59]. However, variable effects are observed even in well-controlled dietary interventions [60], and given the high interindividual variability of the GM, dietary responses of the GM are highly individualised [61]. Precision, or ‘personalised’, nutrition is an evolving field based on identifying individual-specific response-predictive features that can be used to design dietary interventions [62]. Using personal data on GM composition and other information such as blood biomarkers and dietary habits, machine-learning approaches have been applied to predict postprandial glycaemic responses to standardised meals with greater accuracy than other predictive methods [63, 64]. These studies have revealed that the specific composition of the GM contributes to the specific response of its host (i.e. the response to the diet differs in the presence of different bacteria). Hence, the GM determines, at least in part, metabolic heterogeneity among humans. Being modifiable and highly metabolically active, the GM offers possibilities for more precise lifestyle interventions and novel treatments.
Knowledge gaps, challenges, and possibilities
Several large, high-quality reference genome catalogues now exist [65,66,67] and greatly facilitate taxonomic assignment and functional characterisation of the GM in human studies. However, these databases are not without limitations (see textbox). For epidemiological analyses, GM data are fraught with challenges, including great inter- and intraindividual variability, high dimensionality (i.e. the number of observed GM features may be larger than the number of samples and subjects), and sparsity (i.e. GM features such as species are only detected in part of the samples) [68]. At the population level, the GM is composed of thousands of interacting species, each harbouring genetic diversity across hosts and within a host over time; yet, commonly performed analyses often ignore such non-independence, the complex additive and interaction effects among the microbes, and the modifiability and fluctuations of the GM. However, some recent analyses have revealed different patterns of intraindividual variation and adaptation to host physiology for different bacterial species [14, 69, 70].
Other challenges relate to the remarkable number of phenotypic and environmental factors that the GM may influence and to which it may respond. The requirement of large cohorts has unquestionably been demonstrated in human genetics; most polygenic traits are known to be affected by many genetic variants with small effect sizes, which nonetheless can be summed to powerful polygenic risk scores of clinical importance [71, 72]. Similarly, as evidenced by the findings of the large metagenomic study from Israel and the United States [6], single bacterial species might have associations of low effect size with human phenotypes or be present in low abundance. Thus, large sample sizes for adequate statistical power and coverage of interindividual variability are necessary to obtain replicable results and high prediction accuracy.
To better understand the long-term influence of GM variation and dynamics on type 2 diabetes, prospective studies are crucial. In the few prospective studies published to date, GM features have been associated with incident type 2 diabetes in a geographically diverse Chinese population [73] and a subset of a Spanish clinical trial [74], both studies using 16S rRNA gene sequencing. GM features were also linked to type 2 diabetes in a large population-based Finnish cohort with 18 years of follow-up using shallow metagenomic sequencing [75]. However, in these studies, the number of incident cases was restricted, and the analyses had limited resolution (i.e. were restricted to the most dominant GM taxa), as none of the studies used deep-sequencing whole-genome metagenomics.
The importance of subspecies- and strain-level resolution in metagenomic studies may have been undervalued and is an important limitation to harnessing the GM for human health. For example, Faecalibacterium prausnitzii is among the most promising candidates for next-generation probiotics, but there are also other promising candidates, such as Akkermansia muciniphila and P. copri [76]. With regard to F. prausnitzii, several potential subspecies have been found in the human gut, harbouring different functional potential for the use of complex polysaccharides [77]. In line with this observation, several F. prausnitzii subspecies were also identified in the large metagenomic study from Israel and the United States, and negative associations with BMI were observed only for a subset of them [6]. In the case of P. copri, both positive and negative associations have been found with the host metabolic phenotype (e.g. visceral fat and glucose responses).
These inconsistent findings could be partially explained by intra- and interspecies diversity [78, 79]. The current code of nomenclature defines bacterial species based on genome similarity, with conspecific genomes having ≥70% similarity by DNA–DNA hybridisation and an average nucleotide identity ≥94% in the core genome and ≥96% in universal marker genes [80]. However, these genomic variations can translate into important phenotypic differences. For example, these differences may define a strain within the same species as commensal or pathogenic, as in the cases of B. fragilis and Clostridium difficile, depending on whether the strain encodes virulence factors [80]. Overall, the studies mentioned above demonstrate that differences at the strain or even substrain level are highly meaningful, and low-resolution analyses (such as 16S rRNA gene sequencing) miss key information.
Another knowledge gap concerns the viral component of the GM, predominantly comprising viruses that infect bacteria, known as bacteriophages (or, more simply, ‘phages’). Although these phages have not been well studied in the epidemiological setting, they may be important for understanding the bacterial dynamics of the GM that may affect its interactions with the host. To date, only a few epidemiological studies have reported associations between the gut phageome and type 2 diabetes [81, 82] or the metabolic syndrome [83]. Although initially promising, the conclusiveness of the results is limited because of the restricted sample sizes. Future studies of the role of phages as regulators of the GM and cardiometabolic health are warranted but will face challenges related to, among other issues, virome isolation and the limitations of current databases [84, 85].
Integrative multi-omics studies might be needed to investigate the intricate connections among environmental factors, the GM, the virome/phageome, and cardiometabolic phenotypes. Some pioneering examples of reasonably large studies have recently demonstrated the power of such approaches [14, 86,87,88]. The interactions are multifactorial and multidirectional and demand untargeted, large-sized, multi-omics and longitudinal approaches of high depth and resolution.
Physiological and pathophysiological perspectives
Current understanding of the role of the GM in the pathophysiology of diabetes
During their evolution, mammals had to adapt to a world rich in microbes, viruses, and fungi [89]. During and immediately after birth from a sterile intrauterine environment, mammals are exposed to potentially harmful microbes [90,91,92]. Evolution has created substantial barriers, including the GI transit process [93], immunoglobulin A (IgA) [94,95,96], mucus [97], the epithelial layer [98], the endothelial barrier [99], lymph nodes [100], and the liver [101], all of which prevent microbial translocation into the body but create an optimal reservoir for the microbial ecosystem [102]. Low microbe numbers are present in the upper GI tract. At the same time, high microbial density and richness are observed in the large intestine, along with physiological changes in the pH and aerobic/anaerobic conditions from the small to the large intestine, with anaerobic conditions in the large intestine (Fig. 1) [103].
Essential functions of microbes
Besides being a potential deleterious threat for mammals, gut microbes also provide essential functions for mammals, including the education of the immune system, protection from pathogens (i.e. colonisation resistance) [104], metabolic functions, and the supply of nutrients (e.g. vitamins [105]), gut motility, and detoxification of xenobiotics [106]. At the same time, there is competition between microbes and the host for nutrients in the small intestine, and microbially produced macronutrient byproducts are provided to the host. Nutrients (i.e. fibres) and mammalian metabolites such as glucuronides, mucous polysaccharides, and bile acids are fermented or transformed by microbial metabolism (Fig. 1) [107]. Microbial metabolism and microbial cell death and turnover contribute to pools of microbial metabolites in the peripheral blood, where ~30% of all peripheral blood metabolites show associations with the GM and its genes [108, 109]. These microbial metabolites are recognised by receptors such as G-protein-coupled receptors (GPCRs) [110] or the aryl hydrocarbon receptor (AHR) [111] or are further processed by mammalian enzymes such as TMAO to regulate mammalian gene expression by epigenetic modifications, with implications for cardiometabolic health [112,113,114,115].
Roles of non-digestible fibres and their metabolites
Non-digestible carbohydrates are an energy source for specific bacteria in the large intestine that contain enzymes, lacking in the host, that metabolise these fibres and promote the production of SCFAs. Numerous studies have demonstrated that exogenous administration of SCFAs, particularly propionate and butyrate, is beneficial in rodent models of diabetes-like phenotypes [116,117,118]. However, the evidence from clinical trials in both type 1 and type 2 diabetes is less clear [119,120,121,122,123].
In the colon, SCFAs activate enteroendocrine cells (EECs) via binding to GPCRs and free fatty acid receptors 2 and 3 to induce the release of gut peptides, mainly GLP-1 and peptide YY (Fig. 1) [124]. In support of this finding, supplementation with prebiotics in rodents and humans, which can improve glucose tolerance and insulin resistance, has been associated with increased levels of gut peptides [125]. In one study, a high-fibre diet improved glucose tolerance in individuals with type 2 diabetes, an effect that was associated with increased faecal butyrate levels and circulating GLP-1 [59]. GLP-1 regulates glucose homeostasis by increasing insulin secretion, promoting insulin sensitivity, and reducing hepatic glucose production (Fig. 1).
Additionally, SCFAs are crucial for maintaining overall gut health and the gut barrier, as butyrate is the primary fuel source for colonocytes. In contrast, reduced butyrate drives colonocytes toward anaerobic glycolysis, which increases epithelial oxygenation, disrupting the anaerobic environment of the colon [126]. Although SCFAs can act to increase gut peptide release or improve the gut barrier, additional work has highlighted a glucoregulatory role via their action in intestinal gluconeogenesis and on energy expenditure via brown adipose tissue, as well as direct action at the liver, pancreas, and even brain, all of which requires further exploration [127,128,129,130,131].
The GM produces a plethora of metabolites in addition to SCFAs, which likely play a crucial role in host glucose homeostasis (Fig. 2) [132]. For example, bile acids are known glucoregulatory signalling molecules, and their affinity for both FXR and Takeda GPCR 5 (TGR5) is significantly affected by deconjugation and metabolism into secondary bile acids coordinated by gut microbes [133,134,135,136]. Additionally, the GM converts tryptophan and other nutrients into indoles that act via the AHR to reduce inflammation in the metabolic syndrome, especially at the gut level [137, 138]. Furthermore, other gut-derived molecules such as TMAO and imidazole propionate have been implicated in the development of diabetes [139, 140].
Role of the GM in gut barrier functioning
The GM plays a vital role in gut barrier functioning. Impairment in the gut barrier leads to a leaky gut, contributing to low-grade systemic inflammation, a characteristic of obesity and diabetes [141, 142]. Although the mechanisms have been studied mostly in experimental models, one potential mechanism contributing to systemic inflammation is an increase in circulating lipopolysaccharide (LPS) endotoxins derived from the cell envelope of Gram-negative bacteria, also known as metabolic endotoxaemia (Fig. 1). LPSs can act on a specific pathogen-associated molecular pattern (PAMP)—toll-like receptor 4 (TLR4)—throughout the body to elicit a pro-inflammatory immune response that negatively affects glucose homeostasis. A series of studies have suggested a potential role for A. muciniphila in mediating some of the effects of alterations in the GM on systemic inflammation through actions on TLR4 and the gut barrier; however, less evidence is available on its role in mediating effects on glucose metabolism in metabolic disease [143,144,145,146]. However, much more research is needed to determine whether metabolite sensing by PAMPs other than TLR4 is implicated in regulating host–microbe crosstalk and gut barrier integrity [147] in humans.
In parallel, the accumulation of pro-inflammatory macrophages (Fig. 1), CD8αβ T cell infiltration, and reduced IgA+ immune cells are observed in the intestines of individuals with obesity [148,149,150], contributing to insulin resistance [149, 150]. GM modulation strategies could mitigate the adverse gut immune effects of hypercaloric diets. For example, reducing the proportion of pro-inflammatory macrophages and increasing type 3 innate lymphoid cells and regulatory T cells are associated with improved glucose metabolism. Nonetheless, understanding the precise molecular mechanisms driving microbe–immune interactions in the gut and their translation to humans will also require extensive future research.
Knowledge gaps, challenges, and possibilities
Reductionist approaches are required to progress from correlations of bacterial phylotypes/strains with metabolic phenotype (e.g. diabetes) to mechanistic and causal relationships. However, many of the earlier analyses performed in epidemiological studies do not have sufficient resolution (i.e. 16S rRNA gene short amplicon sequencing only provides accurate identification at the genus level but not at the species, strain, and functional levels) [151], and deep-sequencing whole-genome metagenomics, coupled with strain-level analyses, are needed to identify bacteria and their functions that are linked to diseases and thereby to correctly downsize further causality and mechanistic studies [152].
The complexity of the GM and the multitude of possible phylotypes and microbial networks associated with specific phenotypes in population studies also make it difficult to test all possible hypotheses experimentally. Advanced statistics (e.g. MR and mediation analysis) and machine-learning methods are helping to establish causal inferences in human studies, but further validation stages are still needed to provide direct evidence of causality. For this purpose, the use of rodent models in which confounding variables (e.g. genetic background, microbial communities, and diet) can be controlled is key to clearly identifying the effects caused by the precise microbe investigated (namely, a bacterial strain or metabolite).
The use of whole faecal microbiota transplantation (FMT) or co-housing experiments in which the microbiota of rodents with different phenotypes are inter-exchanged have substantially helped to unravel cause and effect in whole communities of microorganisms [2, 153]. Nonetheless, the results of those trials could be biased by limitations in study design. For example, bias could arise in relation to the limited number of donors selected and the possible variations in rodent response to microbes from a different donor species (e.g. lack of colonisation or persistency of part of the microbial population or failure in replicating host–microbe interactions of the original host in which the microbiota co-evolved) and consequent differences in physiological effects [154].
Moreover, identifying the key microbial actors driving a health or disease phenotype and the non-active players within the community is a rather complex undertaking. Specific components of the GM could play a role per se or in coordination with other community members [155]. Furthermore, correlations and causal relationships between GM components and disease could also depend on the specific host (e.g. disease predisposition) and environmental context (e.g. dietary patterns), which can vary in different geographical locations and population groups [11].
One reductionist approach that could aid in establishing the causal role of microbes in metabolic disease is the use of defined microbial communities in gnotobiotic mouse models of diabetes [156]. This approach consists of assembling a defined community of well-characterised and genetically tractable microbes, which is then used to colonise gnotobiotic mice. This so-called ‘defined microbiota’ approach allows for the manipulation of a specific microbial feature in the background of a complex, yet manageable, microbial community to determine whether specific microbial functions play a causative role in the pathogenesis of diabetes and related complications.
Intervention studies with specific bacteria in the ecosystem, as well as more sophisticated strategies that deplete specific microbial components or functions, are ideal for providing evidence of causality [157, 158]. However, even if a particular bacterial genus and species has been correlated with a specific disease phenotype in a relatively reproducible manner in epidemiological studies [159] and proven to be causally involved in an intervention trial, differences between species, and even between strains, may also lead to different outcomes. As explained above, small genomic and phenotypic differences between strains belonging to the same species can translate into functional differences affecting the host phenotype (e.g. with regard to their immunomodulatory effects) [160]. Therefore, the results of efficacy and mechanistic analyses deduced from studies performed with a specific strain cannot be systematically generalised to all strains of the given species.
Historically, the study of the impact of GM on human disease has been focused on the large intestine microbiota because the human colon is the site in the body with the highest abundance of microbes and the most accessible intestinal section. The contribution of the large intestine microbiota to the pathogenesis of metabolic disease has been demonstrated by FMT studies in mouse models of obesity and related complications [2, 153]. However, it is important to note that the small intestine overshadows the large intestine with regard to metabolic regulation. The small intestine is home to EECs that produce GLP-1 and other incretin hormones, which are key glucose metabolism regulators [161]. The small intestine epithelium also plays an essential role in glucose and fat uptake and metabolism, protecting the host from features of metabolic dysfunction [162, 163], and the small intestine microbiota is a regulator of EEC function and nutrient absorption, metabolism, and secretion [164, 165]. Thus, microbiota–host interactions in the small intestine would be expected to contribute to diabetes pathogenesis. Recent work in mouse models has determined that specific members of the small intestine microbiota can inhibit lipid secretion by enterocytes and limit serum triglyceride concentrations during the consumption of a Western-style obesogenic diet [166]. Additionally, the small intestine microbiota in rodents has been demonstrated to affect nutrient-induced gut–brain signalling, which regulates glucose homeostasis [167, 168].
Despite their importance, host–microbiota interactions in the small intestine and their relevance to diabetes are understudied because of limitations in the process of acquiring small intestine microbiota samples from humans and the reduced abundance of microbes in this portion of the digestive tract. Novel technologies to overcome technical limitations in the study of the small intestinal microbiota are discussed further in ‘Technological and methodological advancements’.
GM fluctuation, especially related to dietary intake, should be considered when establishing what are normal and what are dysfunctional microbiota changes for metabolic health. The GM mirrors individuals’ habitual diet and daily choices. Therefore, longitudinally considering dietary history and GM variations over multiple days could help to fine-tune associations and infer causal relationships regarding the metabolic health of individuals [169]. Moreover, daily oscillations of the GM related to eating patterns also affect its functional roles, such as appetite regulation and postprandial responses to food intake, with potential long-term effects on metabolic health and diabetes risk. For example, mouse studies have shown that daily oscillations in GM composition are required to maintain the circadian release of GLP-1, which in turn is required to achieve appropriate circadian control of metabolic homeostasis [170]. In humans, type 2 diabetes and obesity are correlated with alterations of GM circadian rhythms [171], suggesting that daily oscillations are relevant to understanding the role of the GM in controlling energy homeostasis.
Sex also seems to contribute to GM variations, although its relevance for predicting health associations and their underlying mechanisms are under-investigated. The epidemiology and pathophysiology of obesity and associated cardiometabolic disorders such as type 2 diabetes have a sex dimorphism that may be related to not only the role of sex hormones in fat distribution, metabolism, and immunity, but also differences in the GM [172]. Studies in mouse models suggest reciprocal interactions between sex hormones and the GM. On one hand, the GM may regulate the production and/or metabolism of sex hormones (i.e. testosterone and oestrogens), as proven in a non-obese diabetic mouse model of type 1 diabetes [173]. On the other hand, physiological effects of sex hormones (e.g. on immunity and intestinal transit) may affect the GM [174]. Therefore, sex should be considered a confounding variable in epidemiological studies and in the design of mechanistic studies using mouse models because, to date, most preclinical studies have been carried out exclusively in males.
Technological and methodological advancements
Separating phenomenology from actual biology in the microbiome field requires tools and approaches to identify mechanisms that deconvolute whether the microbiome may be a driver of, or offer therapeutic opportunities for, metabolic diseases. Here, we discuss the most promising technological developments for advancing the field.
Model systems
When comparing model systems for studying the relationship between the GM and metabolic diseases, it is essential to consider both traditional models (e.g. germ-free [GF] and gnotobiotic mice) and emerging technologies (e.g. organs-on-a-chip and nonmurine GF models such as zebrafish and pigs).
GF animals have been used widely to investigate the role of the human GM in obesity and diabetes [1, 173, 175]. These animals, which are born without any microbiota, allow for the interrogation of interventions in the absence of a microbiome. As a result, we can gain insight into whether the microbiome is necessary for a given biological process.
Gnotobiotic disease models are established by colonising GF mice with either an entire GM via donor stool or specific isolated bacterial strains [176]. Studies have demonstrated that GF animals, when inoculated with faecal microbiota from individuals with obesity and type 2 diabetes, successfully replicated disease phenotypes, providing evidence for the involvement of the GM in metabolic diseases [2, 59, 177]. Additionally, an overgrowing endotoxin-producing bacterium, Enterobacter cloacae B29, isolated from the gut of a person with morbid obesity and diabetes, induced obesity, fatty liver, and insulin resistance in GF C57BL/6J mice that were otherwise resistant to high-fat diet-induced metabolic defects. Knocking out the endotoxin-producing gene in the B29 bacterial strain or the Tlr4 gene in C57BL/6J mice prevented the metabolic defects, underscoring the causal relationship between specific gut bacteria and host responses in the initiation and progression of metabolic disease [178,179,180].
However, certain concepts have been perpetuated about GF mice that are the result of studying only one genotype. For example, GF C57BL/6J mice are resistant to diet-induced obesity [175], whereas GF Swiss Webster mice are not [181]; therefore, because the majority of GF mouse studies use C57BL/6J mice, it has been stated as fact that GF mice in general have to eat more than conventional mice to maintain weight. The divergent responses of these models to high-fat diets underscore the importance of genetic background in research outcomes [182].
The availability of additional GF models, such as pigs and zebrafish, complement the use of GF mice. GF pigs and piglets offer more human-relevant insights than do mice when developing human microbiota-associated gnotobiotic models [183], although the space required to house them is prohibitive for many institutions or limits studies to using just a few animals. GF zebrafish, on the other hand, have proven useful for studies of the GM and distinct host cellular developmental stages [184]. The transparency of the fish body and the ability to fluorescently tag and image different cell types in the presence of different bacteria, as well as the ease of housing and propagating zebrafish, is advantageous for investigating specific questions [185]. These models do not fully replicate human physiology, but they allow longitudinal and invasive sampling in tightly controlled conditions, which is important when asking mechanistic questions.
Organs-on-a-chip, such as the gut-on-a-chip, offer more human-relevant systems because they can be derived directly from human tissue or blood-derived induced pluripotent stem cells, which retain the genetic signature of the host; thus, they enable the study of complex human tissues and cellular interactions in a controlled environment [186]. Recent efforts have demonstrated the ability to seed the gut-on-a-chip with microbiota in a semi-anaerobic environment [187], and many groups are now testing the efficiency of seeding increasingly complex communities on these chips. Although the gut-on-a-chip model lacks some key cell types such as immune cells, major advances include the ability to connect different organ chips such as the gut-chip and neuron-chip [188] to model gut–brain interactions. Creative uses of organs-on-a-chip to study the microbiome will continue to emerge and are likely to fill important gaps to complement animal models.
Understanding of bacterial genes and functions
The ability to sequence and assemble whole genomes of bacteria is an enormously powerful approach for identifying lineages and the relatedness of bacterial strains and for identifying putative pathways involved in a given bacterial phenotype that may have relevance in human health or disease. If we think about the mechanisms of human disease that have been elucidated from the study of genetically manipulated mice, it is not hard to imagine the wealth of information to be gained from doing the same in bacteria. The ability to knock out and manipulate bacterial genes is not new. Nearly 80 years of bacterial genetics have clarified how pathogens colonise the gut epithelium and secrete toxins, leading to diseases such as cholera, how they share information with each other to adapt to different environments, and how nutritional selection drives their composition in a host. E. coli can be considered the bacterial version of the C57BL/6 mouse; its genetics are well-defined and easily engineered [189,190,191], and it has become the workhorse for testing the effect of modifications in a given environment. However, the commensal GM consists of far more diversity than just E. coli; thus, researchers are actively seeking a deeper understanding of GM genetics, using, for example, Bacteroides and Clostridium as representative organisms [192, 193], as numerous human and mouse studies have demonstrated the important roles of these organisms in health and disease.
Advanced computational tools, including artificial intelligence, have shed new light on unannotated parts of a bacterial genome by predicting the three-dimensional structures of proteins, a task greatly advanced by technologies such as AlphaFold2 [194, 195]. By analysing these structures, researchers can infer possible functions based on their shapes and binding sites. These potential roles can be confirmed by experimental validation in biochemical and microbiological studies [196]. This knowledge, especially regarding how proteins influence metabolic pathways, is crucial for linking microbial activity to health conditions such as diabetes, offering insights into disease mechanisms and potential therapeutic targets.
Reference-free data analysis
The most critical issue with current database-dependent approaches in microbiome sequencing analysis is their limitation in detecting novel or understudied microbes [197]. When microbial community samples are analysed using databases based on reference genomes from well-characterised bacteria, sequences that do not match are overlooked or misclassified. This process results in a biased view of the microbial ecosystem, potentially missing crucial components that could have significant roles in health and disease, including diabetes. Therefore, advancing microbiome research necessitates the development and use of methods that can uncover and characterise these underrepresented microbial entities.
Assembling genomes de novo from metagenomic sequencing data is a powerful approach in microbiome research that involves constructing genomes directly from sequencing reads without relying on reference databases [197]. This method uses advanced computational algorithms to piece together DNA fragments from a sample, allowing the identification of genetic material from a wide range of organisms, including those not previously sequenced or catalogued. By assembling these genomes, researchers can discover novel species and uncover new gene functions, significantly expanding our understanding of microbial diversity and its potential roles in various environments, including the human body. This approach is particularly useful in revealing the full spectrum of microbial life, including rare or unknown species that might play crucial roles in health and disease.
Access to the small intestine microbiota
The small intestine is the primary site of nutrient uptake, enterohepatic recycling, and intestinal hormone stimulation; thus, it is essential to gain a deep understanding of microbial function in this region of the body. However, most of our knowledge of human microbiomes has been based on stool samples and the colonic microbiota because accessing the small intestine microbiota is challenging, even with modern endoscopy methods.
Recent advances use innovative methods such as ingestible capsules that sample intestinal material throughout the GI tract [198]. Because each capsule is triggered by a different pH along the gut, this method can provide a microbial atlas of intestinal communities. These tools are being further refined and commercially developed for use in both diagnostics and research. One caveat, however, is that there is potential for microbes to continue growing after the sample has been collected within a capsule, thus giving an inaccurate representation of the native microbiome community. Additionally, these and other capsules have been developed for sampling in the fasting state, leaving the study of postprandial responses still limited, although these responses are likely important to reach a complete understanding of microbial contributions to the regulation of glucose metabolism. Addressing these issues is crucial for ensuring the reliability and accuracy of microbiome studies with such devices.
Isozyme and small molecule screens
Isozyme and small molecule screens in microbiome research are crucial for identifying specific bacterial products that can be targeted therapeutically. Microbial isozymes are enzymes that have different molecular structures but catalyse the same reaction as the host enzymes. Screening these products can reveal variations in microbial metabolism that might influence health and potentially interfere with medications, as in the case of bacterial DPP-4 isozymes [33]. Small molecule screens focus on identifying bioactive compounds produced by microbes [199]. These compounds can have significant effects on host pathophysiology [140, 200]. By identifying specific isozymes and small molecules, researchers can target them for degradation or enhancement, offering potential therapeutic strategies for diseases such as diabetes.
Potential GM-based diagnostics and therapies in diabetes
As described above, no diagnostic and generalised faecal microbiota taxonomic signature has been found for type 1 or type 2 diabetes [86, 201]. Future research should therefore move toward strain-level studies in large prospective populations and, when possible, focus on functional profiling of intestinal microbes along the GI tract [198], with special attention to stable isotope precursors to study production and substrate fluxes of important microbially produced metabolites in different GI regions [202].
High-fibre diets and SCFA-based treatments
With regard to GM-based therapies for diabetes, high-fibre diets have been shown to be effective in controlling blood glucose levels and reducing insulin resistance in both type 1 and type 2 diabetes [203, 204]. Although the direct mode of action of dietary fibre via the GM remains to be shown, these trials underscore the potential importance of including GM modulation strategies as part of diabetes intervention trials, especially for the production of beneficial metabolites such as SCFAs [205]. However, as noted above, intervention trials of oral SCFA butyrate supplementation have shown no effect on glycaemic control or other markers of diabetes regulation in either type 1 or type 2 diabetes [116, 122, 123, 206], probably because the site of delivery does not mimic endogenous production. For other SCFAs, including propionate and acetate, data are too scarce to draw any conclusions regarding possible effects on metabolic regulation.
Conventional and next-generation probiotics
Probiotic therapies for diabetes can be divided into conventional probiotics, particularly Lactobacillus and Bifidobacterium strains, which have a history of use for human consumption in fermented foods or supplements to promote health, and next-generation probiotics, which are strains of new bacterial species recently identified as indigenous members of the human GM. These strains are associated with health, and their presence is diminished in disease settings [76, 207]. With regard to the conventional probiotics, prospective randomised controlled trials (RCTs) in new-onset type 1 diabetes are ongoing (NCT03961854, NCT03961347, NCT04769037, and NCT05767450), and a smaller trial has shown only moderate effects in longstanding type 1 diabetes [208]. However, an open-label trial of probiotics (strains of Bifidobacterium, Lactobacillus, and Streptococcus salivarius) found beneficial effects on susceptibility to and progression of type 1 diabetes in siblings of people with type 1 diabetes [209]. In type 2 diabetes, a recent meta-analysis described some efficacy of these probiotic strains in metabolic control and reduced insulin resistance [210].
With regard to next-generation probiotics, fewer data have been generated in humans. For example, despite specific strains (e.g. Akkermansia) having been associated with a healthy metabolic phenotype [211], an RCT intervention with A. muciniphila did not identify strong metabolic effects [143]. This finding could be the result of a lack of a causal role of these tested strains in the metabolic syndrome, reduced viability upon passage through the stomach [212, 213], inadequate dosages, or a lack of engraftment when introduced in the human gut [214]. Because the small intestine is important for the pathophysiology of both type 1 and type 2 diabetes, further analyses of small intestinal microbiota from individuals with type 1 [215] and type 2 [216, 217] diabetes are needed, with defined combinations of next-generation probiotic strains studied as possible interventions for diabetes. However, this effort should consider conditions of bacterial strain engraftment, ecological or functional dependencies on other bacterial members, and potential redundancies in functionality, as shown by a meta-analysis of FMT [218].
Donor FMT
Until such investigations with defined combinations of strains are completed, donor FMT might provide insight into the magnitude of effect of modulating the GM and the effect of such modulation on the pathophysiology and potential reversibility of diabetes. de Groot et al [219] recently published research on the efficacy of fresh FMT in maintaining residual beta cell function and dampening autoimmunity in new-onset type 1 diabetes. Other studies have been performed for type 2 diabetes and insulin resistance, showing a modest effect of FMT on insulin resistance and non-alcoholic fatty liver disease [216, 217, 220,221,222,223], whereas one study showed no effect on these parameters [224]. Additionally, a combined intervention of encapsulated donor FMT and fibre supplementation showed beneficial effects on glucose metabolism, suggesting the possible need to design interventions not only with synthetic bacterial strain consortia, but also with dietary support (e.g. fibre to nourish the bacterial strains) [221]. Finally, studies evaluating whether autologous FMT after lifestyle intervention could help prevent weight regain have suggested that diet-induced changes in low-abundance bacteria might be responsible for weight loss maintenance, which could guide more precise interventions with less ethical burden and lower risks of transmitting diseases [225].
Overall, donor FMT is a more diffuse approach than interventions with targeted strains or metabolites [226]. Additionally, there are differences in mode of faecal material administration (capsules vs fresh FMT), intestinal pH (e.g. due to antacids), and colonic transit time in existing datasets, and the amount of faecal microbiota administered also seems to affect engraftment of donor bacterial strains [218, 227]. These factors preclude generalisation of the results of studies to date. We therefore advocate further standardisation of intestinal microbiota composition measurements [228], with strict dietary monitoring. Also, better standardisation is needed in human studies of FMT-based interventions. In this context, production of lyophilised capsules for FMT must follow Good Manufacturing Practices to maintain viability and ensure adequate shelf life [229].
Nevertheless, based on its wide availability and general safety (provided that donors are adequately screened [230]), FMT could provide clinicians with new treatment modalities for diabetes until interventions with defined combinations of strains are available, especially if next-generation probiotics can be spiked in donor faecal microbiota to boost therapeutic efficacy [231]. However, these interventions should adhere to the international Nagoya Protocol on Access to Genetic Resources and the Fair and Equitable Sharing of Benefits Arising From Their Utilization to the Convention on Biological Diversity [232], which seeks to prevent researchers or their institutions from financially capitalising (at the expense of vulnerable individuals or populations) on identified bacterial strains as next-generation probiotics. With regard to trial outcomes for diabetes and GM-based therapies, using dynamic measurements of glucose metabolism over time (e.g. mixed-meal tests or continuous glucose monitoring) could provide better insights into the interactions between the GM, diet, and glucose homeostasis during both FMT and administration of defined strain combinations.
New insights into the GM are increasingly associating it with diabetes in humans, although the microbiome of the small intestine remains understudied. Intervention studies with FMT in humans have been able to dissect associations from causality and have indeed shown some clinical benefit, although the contrast between, on average, relatively small therapeutic effects and ethical concerns [233] preclude widespread practical use of this treatment option in diabetes clinical care. Additional studies are thus needed of prospective associations between the GM and diabetes in multiethnic cohorts. Alongside this effort, the therapeutic potential of synthetic GM-derived bacterial strains and/or communities and engineered systems for targeting intestinal delivery of identified metabolites in diabetes should be explored.
Conclusion
Over the past two decades, alterations in the GM have been associated with aberrant glucose metabolism and steatosis in individuals with diabetes. Larger sample sizes in epidemiological studies have now started to show the magnitude and possible consistency of correlations between the GM and human metabolic traits of relevance to obesity and/or type 2 diabetes; however, for type 1 diabetes, the picture is much less clear.
Interaction with diabetes medications in relation to ethnicity and dietary intake should be taken into account more rigorously in future studies. Moreover, in recent years, more insights have been gained into the function of the GM beyond just its composition, and this information nicely dovetails with earlier reports of links between specific metabolites, including SCFAs, BCAAs and bile acids, and obesity and diabetes.
With regard to GM composition, only a few studies have addressed the role of phages and fungi and the interactions between these inhabitants and bacterial strains in diabetes. It is clear that future studies also need to focus on small intestine microbiota function, as well as developing adequate bioinformatic pipelines and correctly assembling genomes (see textbox).
We must also take into account that most data to date have been generated in mouse studies, whose relevance to human diabetes needs further confirmation because of the large differences between mice and humans in diet, genetics, and life span. Nevertheless, human intervention studies of single strains and FMT in the setting of human diabetes have shown a range of clinical metabolic effects (compared with the more consistent effects of medications), yet without serious side effects. In conclusion, after almost two decades of study, we must still look to future efforts to illuminate the clinical diagnostic and therapeutic applicability of GM research to human diabetes.
Abbreviations
- AHR:
-
Aryl hydrocarbon receptor
- BCAA:
-
Branched-chain amino acid
- BMI:
-
Body mass index
- DPP-4:
-
Dipeptidyl peptidase 4
- EEC:
-
Enteroendocrine cell
- FMT:
-
Faecal microbiota transplantation
- FXR:
-
Farnesoid X receptor
- GF:
-
Germ-free
- GI:
-
Gastrointestinal
- GLP-1:
-
Glucagon-like peptide 1
- GM:
-
Gut microbiota
- GPCR:
-
G-protein-coupled receptor
- LPS:
-
Lipopolysaccharide
- MR:
-
Mendelian randomisation
- PAMP:
-
Pathogen-associated molecular pattern
- RCT:
-
Randomised controlled trial
- SCFA:
-
Short-chain fatty acid
- SGLT2:
-
Sodium–glucose cotransporter 2
- TGR5:
-
Takeda GPCR 5
- TLR4:
-
Toll-like receptor 4
- TMAO:
-
Trimethylamine oxide
References
Bäckhed F, Ding H, Wang T et al (2004) The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A 101:15718–15723. https://doi.org/10.1073/pnas.0407076101
Ridaura VK, Faith JJ, Rey FE et al (2013) Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341:1241214. https://doi.org/10.1126/science.1241214
Sze MA, Schloss PD (2016) Looking for a signal in the noise: revisiting obesity and the microbiome. mBio 7:e01018-16. https://doi.org/10.1128/mBio.01018-16
Duvallet C, Gibbons SM, Gurry T, Irizarry RA, Alm EJ (2017) Meta-analysis of gut microbiome studies identifies disease-specific and shared responses. Nat Commun 8:1784. https://doi.org/10.1038/s41467-017-01973-8
Pasolli E, Truong DT, Malik F, Waldron L, Segata N (2016) Machine learning meta-analysis of large metagenomic datasets: tools and biological insights. PLoS Comput Biol 12:e1004977. https://doi.org/10.1371/journal.pcbi.1004977
Rothschild D, Leviatan S, Hanemann A, Cohen Y, Weissbrod O (2022) An atlas of robust microbiome associations with phenotypic traits based on large-scale cohorts from two continents. PLoS One 17:e0265756. https://doi.org/10.1371/journal.pone.0265756
Qin J, Li Y, Cai Z et al (2012) A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490:55–60. https://doi.org/10.1038/nature11450
Le Chatelier E, Nielsen T, Qin J et al (2013) Richness of human gut microbiome correlates with metabolic markers. Nature 500:541–546. https://doi.org/10.1038/nature12506
Allin KH, Tremaroli V, Caesar R et al (2018) Aberrant intestinal microbiota in individuals with prediabetes. Diabetologia 61:810–820. https://doi.org/10.1007/s00125-018-4550-1
Forslund K, Hildebrand F, Nielsen T et al (2015) Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 528:262–266. https://doi.org/10.1038/nature15766
Karlsson FH, Tremaroli V, Nookaew I et al (2013) Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 498:99–103. https://doi.org/10.1038/nature12198
Wu H, Tremaroli V, Schmidt C et al (2020) The gut microbiota in prediabetes and diabetes: a population-based cross-sectional study. Cell Metab 32:379-390.e3. https://doi.org/10.1016/j.cmet.2020.06.011
Alvarez-Silva C, Kashani A, Hansen TH et al (2021) Trans-ethnic gut microbiota signatures of type 2 diabetes in Denmark and India. Genome Med 13:37. https://doi.org/10.1186/s13073-021-00856-4
Zhou W, Sailani MR, Contrepois K et al (2019) Longitudinal multi-omics of host-microbe dynamics in prediabetes. Nature 569:663–671. https://doi.org/10.1038/s41586-019-1236-x
Sayols-Baixeras S, Dekkers KF, Baldanzi G et al (2023) Streptococcus species abundance in the gut is linked to subclinical coronary atherosclerosis in 8973 participants from the SCAPIS cohort. Circulation 148:459–472. https://doi.org/10.1161/CIRCULATIONAHA.123.063914
Grahnemo L, Nethander M, Coward E et al (2022) Cross-sectional associations between the gut microbe Ruminococcus gnavus and features of the metabolic syndrome. Lancet Diabetes Endocrinol 10:481–483. https://doi.org/10.1016/S2213-8587(22)00113-9
Belda E, Voland L, Tremaroli V et al (2022) Impairment of gut microbial biotin metabolism and host biotin status in severe obesity: effect of biotin and prebiotic supplementation on improved metabolism. Gut 71:2463–2480. https://doi.org/10.1136/gutjnl-2021-325753
Pedersen HK, Gudmundsdottir V, Nielsen HB et al (2016) Human gut microbes impact host serum metabolome and insulin sensitivity. Nature 535:376–381. https://doi.org/10.1038/nature18646
Wang TJ, Larson MG, Vasan RS et al (2011) Metabolite profiles and the risk of developing diabetes. Nat Med 17:448–453. https://doi.org/10.1038/nm.2307
Mueller NT, Differding MK, Zhang M et al (2021) Metformin affects gut microbiome composition and function and circulating short-chain fatty acids: a randomized trial. Diabetes Care 44:1462–1471. https://doi.org/10.2337/dc20-2257
Wu H, Esteve E, Tremaroli V et al (2017) Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat Med 23:850–858. https://doi.org/10.1038/nm.4345
Pryor R, Norvaisas P, Marinos G et al (2019) Host-microbe-drug-nutrient screen identifies bacterial effectors of metformin therapy. Cell 178:1299-1312.e29. https://doi.org/10.1016/j.cell.2019.08.003
Dekkers KF, Sayois-Baixeras S, Baldanzi G et al (2022) An online atlas of human plasma metabolite signatures of gut microbiome composition. Nat Commun 13:5370. https://doi.org/10.1038/s41467-022-33050-0
Sun L, Xie C, Wang G et al (2018) Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat Med 24:1919–1929. https://doi.org/10.1038/s41591-018-0222-4
Bryrup T, Thomsen CW, Kern T et al (2019) Metformin-induced changes of the gut microbiota in healthy young men: results of a non-blinded, one-armed intervention study. Diabetologia 62:1024–1035. https://doi.org/10.1007/s00125-019-4848-7
Gu Y, Wang X, Li J et al (2017) Analyses of gut microbiota and plasma bile acids enable stratification of patients for antidiabetic treatment. Nat Commun 8:1785. https://doi.org/10.1038/s41467-017-01682-2
van Bommel EJM, Herrema H, Davids M, Kramer MHH, Nieuwdorp M, van Raaite DH (2020) Effects of 12-week treatment with dapagliflozin and gliclazide on faecal microbiome: results of a double-blind randomized trial in patients with type 2 diabetes. Diabetes Metab 46:164–168. https://doi.org/10.1016/j.diabet.2019.11.005
Smits MM, Fluitman KS, Herrema H et al (2021) Liraglutide and sitagliptin have no effect on intestinal microbiota composition: a 12-week randomized placebo-controlled trial in adults with type 2 diabetes. Diabetes Metab 47:101223. https://doi.org/10.1016/j.diabet.2021.101223
Bica I-C, Pietroșel VA, Salmen T et al (2023) The effects of cardioprotective antidiabetic therapy on microbiota in patients with type 2 diabetes mellitus: a systematic review. Int J Mol Sci 24:7184. https://doi.org/10.3390/ijms24087184
Moon JS, Hong JH, Jung YJ, Ferrannini E, Nauck MA, Lim S (2022) SGLT-2 inhibitors and GLP-1 receptor agonists in metabolic dysfunction-associated fatty liver disease. Trends Endocrinol Metab 33:424–442. https://doi.org/10.1016/j.tem.2022.03.005
Wong CK, Yusta B, Koehler JA et al (2022) Divergent roles for the gut intraepithelial lymphocyte GLP-1R in control of metabolism, microbiota, and T cell-induced inflammation. Cell Metab 34:1514-1531.e7. https://doi.org/10.1016/j.cmet.2022.08.003
Palacios T, Vitetta L, Coulson S et al (2020) Targeting the intestinal microbiota to prevent type 2 diabetes and enhance the effect of metformin on glycaemia: a randomized controlled pilot study. Nutrients 12:2041. https://doi.org/10.3390/nu12072041
Wang K, Zhang Z, Hang J et al (2023) Microbial-host-isozyme analyses reveal microbial DPP4 as a potential antidiabetic target. Science 381:eadd5787. https://doi.org/10.1126/science.add5787
Olivare M, Schüppel V, Hassan AM et al (2018) The potentialrole of the dipeptidyl peptidase-4-like activity from the gut microbiota on the host health. Front Microbiol 9:1900. https://doi.org/10.3389/fmicb.2018.01900
Boets E, Gomand SV, Deroover L et al (2017) Systemic availability and metabolism of colonic-derived short-chain fatty acids in healthy subjects: a stable isotope study. J Physiol 595:541–555. https://doi.org/10.1113/JP272613
Morrison DJ, Preston T (2016) Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 7:189–200. https://doi.org/10.1080/19490976.2015.1134082
Sanna S, van Zuydam NR, Mahajan A et al (2019) Causal relationships among the gut microbiome, short-chain fatty acids and metabolic diseases. Nat Genet 51:600–605. https://doi.org/10.1038/s41588-019-0350-x
Chávez-Talavera O, Haas J, Grzych G, Tailleux A, Staels B (2019) Bile acid alterations in nonalcoholic fatty liver disease, obesity, insulin resistance and type 2 diabetes: what do the human studies tell? Curr Opin Lipidol 30:244–254. https://doi.org/10.1097/MOL.0000000000000597
Vincent RP, Omar S, Ghozlan S et al (2013) Higher circulating bile acid concentrations in obese patients with type 2 diabetes. Ann Clin Biochem 50:360–364. https://doi.org/10.1177/0004563212473450
Prawitt J, Caron S, Staels B (2011) Bile acid metabolism and the pathogenesis of type 2 diabetes. Curr Diab Rep 11:160–166. https://doi.org/10.1007/s11892-011-0187-x
Haeusler RA, Astiarraga B, Camastra S, Accili D, Ferrannini E (2013) Human insulin resistance is associated with increased plasma levels of 12α-hydroxylated bile acids. Diabetes 62:4184–4191. https://doi.org/10.2337/db13-0639
Zheng X, Chen T, Zhao A et al (2021) Hyocholic acid species as novel biomarkers for metabolic disorders. Nat Commun 12:1487. https://doi.org/10.1038/s41467-021-21744-w
Petersen AØ, Julienne H, Hyötyläinen T et al (2021) Conjugated C-6 hydroxylated bile acids in serum relate to human metabolic health and gut Clostridia species. Sci Rep 11:13252. https://doi.org/10.1038/s41598-021-91482-y
Chen L, van den Munckhof CL, Schraa K et al (2020) Genetic and microbial associations to plasma and fecal bile acids in obesity relate to plasma lipids and liver fat content. Cell Rep 33:108212. https://doi.org/10.1016/j.celrep.2020.108212
Browning MG, Pessoa BM, Khoraki J, Campos GM (2019) Changes in bile acid metabolism, transport, and signaling as central drivers for metabolic improvements after bariatric surgery. Curr Obes Rep 8:175–184. https://doi.org/10.1007/s13679-019-00334-4
van den Broek M, de Heide LJM, Sips FLP et al (2021) Altered bile acid kinetics contribute to postprandial hypoglycaemia after Roux-en-Y gastric bypass surgery. Int J Obes (Lond) 45:619–630. https://doi.org/10.1038/s41366-020-00726-w
de Mello VD, Paananen J, Lindström J et al (2017) Indolepropionic acid and novel lipid metabolites are associated with a lower risk of type 2 diabetes in the Finnish Diabetes Prevention Study. Sci Rep 7:46337. https://doi.org/10.1038/srep46337
Molinaro A, Lassen PB, Henricsson M et al (2020) Imidazole propionate is increased in diabetes and associated with dietary patterns and altered microbial ecology. Nat Commun 11:5881. https://doi.org/10.1038/s41467-020-19589-w
Nemet I, Li XS, Haghikia A et al (2023) Atlas of gut microbe-derived products from aromatic amino acids and risk of cardiovascular morbidity and mortality. Eur Heart J 44:3085–3096. https://doi.org/10.1093/eurheartj/ehad333
Molinaro A, Nemet I, Lassen PB et al (2023) Microbially produced imidazole propionate is associated with heart failure and mortality. JACC Heart Fail 11:810–821. https://doi.org/10.1016/j.jchf.2023.03.008
Meijnikman A, Davids M, Herrema H et al (2022) Microbiome-derived ethanol in nonalcoholic fatty liver disease. Nat Med 28:2100–2106. https://doi.org/10.1038/s41591-022-02016-6
Yuan J, Chen C, Cui J et al (2019) Fatty liver disease caused by high-alcohol-producing Klebsiella pneumoniae. Cell Metab 30:675-688.e7. https://doi.org/10.1016/j.cmet.2019.08.018
Nair S, Cope K, Risby TH, Diehl AM (2001) Obesity and female gender increase breath ethanol concentration: potential implications for the pathogenesis of nonalcoholic steatohepatitis. Am J Gastroenterol 96:1200–1204. https://doi.org/10.1111/j.1572-0241.2001.03702.x
Jia J, Dou P, Gao M et al (2019) Assessment of causal direction between gut microbiota-dependent metabolites and cardiometabolic health: a bidirectional Mendelian randomization analysis. Diabetes 68:1747–1755. https://doi.org/10.2337/db19-0153
Li H, Li C (2023) Causal relationship between gut microbiota and type 2 diabetes: a two-sample Mendelian randomization study. Front Microbiol 14:1184734. https://doi.org/10.3389/fmicb.2023.1184734
Diabetes and Nutrition Study Group (DNSG) of the European Association for the Study of Diabetes (2023) Evidence-based European recommendations for the dietary management of diabetes. Diabetologia 66:965–985. https://doi.org/10.1007/s00125-023-05894-8
Makki K, Deehan EC, Walter J, Backhed F (2018) The impact of dietary fiber on gut microbiota in host health and disease. Cell Host Microbe 23:705–715. https://doi.org/10.1016/j.chom.2018.05.012
Kovatcheva-Datchary P, Nilsson A, Akrami R et al (2015) Dietary fiber-induced improvement in glucose metabolism is associated with increased abundance of Prevotella. Cell Metab 22:971–982. https://doi.org/10.1016/j.cmet.2015.10.001
Zhao L, Zhang F, Ding X et al (2018) Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science 359:1151–1156. https://doi.org/10.1126/science.aao5774
Gardner CD, Trepanowski JF, Del Gobbo LC et al (2018) Effect of low-fat vs low-carbohydrate diet on 12-month weight loss in overweight adults and the association with genotype pattern or insulin secretion: the DIETFITS randomized clinical trial. JAMA 319:667–679. https://doi.org/10.1001/jama.2018.0245
Guthrie L, Spencer SP, Perelman D et al (2022) Impact of a 7-day homogeneous diet on interpersonal variation in human gut microbiomes and metabolomes. Cell Host Microbe 30:863-874.e4. https://doi.org/10.1016/j.chom.2022.05.003
Ordovas JM, Ferguson LR, Tai ES, Mathers JC (2018) Personalised nutrition and health. BMJ 361:bmj.k2173. https://doi.org/10.1136/bmj.k2173
Zeevi D, Korem T, Zmora N et al (2015) Personalized nutrition by prediction of glycemic responses. Cell 163:1079–1094. https://doi.org/10.1016/j.cell.2015.11.001
Berry SE, Valdes AM, Drew DA et al (2020) Human postprandial responses to food and potential for precision nutrition. Nat Med 26:964–973. https://doi.org/10.1038/s41591-020-0934-0
Almeida A, Nayfach S, Boland M et al (2021) A unified catelog of 204,938 reference genomes from the human gut microbiome. Nat Biotechnol 39:105–114. https://doi.org/10.1038/s41587-020-0603-3
Zeng S, Patangia D, Almeida A et al (2022) A compendium of 32,277 metagenome-assembled genomes and over 80 mission genes from the early-life human gut microbiome. Nat Commun 13:5139. https://doi.org/10.1038/s41467-022-32805-z
Leviatan S, Shoer S, Rothschild D, Gorodetski M, Segal E (2022) An expanded reference map of the human gut microbiome reveals hundreds of previously unknown species. Nat Commun 13:3863. https://doi.org/10.1038/s41467-022-31502-1
Tsilimigras MC, Fodor AA (2016) Compositional data analysis of the microbiome: fundamentals, tools, and challenges. Ann Epidemiol 26:330–335. https://doi.org/10.1016/j.annepidem.2016.03.002
Olsson LM, Boulund F, Nilsson S et al (2022) Dynamics of the normal gut microbiota: a longitudinal one-year population study in Sweden. Cell Host Microbe 30:726-739.e3. https://doi.org/10.1016/j.chom.2022.03.002
Chen L, Wang D, Garmaeva S et al (2021) The long-term genetic stability and individual specificiaty of the human gut microbiome. Cell 184:2302-2315.e12. https://doi.org/10.1016/j.cell.2021.03.024
Abdellaoui A, Yengo L, Verweij KJH, Visscher PM (2023) 15 years of GWAS discovery: realizing the promise. Am J Hum Genet 110:179–194. https://doi.org/10.1016/j.ajhg.2022.12.011
Kullo IJ, Lewis CM, Inouye M, Martin AR, Ripatti S, Chatterjee N (2022) Polygenic scores in biomedical research. Nat Rev Genet 23:524–532. https://doi.org/10.1038/s41576-022-00470-z
Wang H, Gou W, Su C et al (2022) Association of gut microbiota with glycaemic traits and incident type 2 diabetes, and modulation by habitual diet: a population-based longitudinal cohort study in Chinese adults. Diabetologia 65:1145–1156. https://doi.org/10.1007/s00125-022-05687-5
Vals-Delgado C, Alcala-Diaz JF, Molina-Abril H et al (2021) An altered microbiota pattern preceded type 2 diabetes mellitus development: from the CORDIOPREV study. J Adv Res 35:99–108. https://doi.org/10.1016/j.jare.2021.05.001
Ruuskanen MO, Erawijantari PP, Havulinna AS et al (2022) Gut microbiome composition is predictive of incident type 2 diabetes in a population cohort of 5,572 Finnish adults. Diabetes Care 45:811–818. https://doi.org/10.2337/dc21-2358
De Filippis F, Esposito A, Ercolini D (2022) Outlook on next-generation probiotics from the human gut. Cell Mol Life Sci 79:76. https://doi.org/10.1007/s00018-021-04080-6
De Filippis F, Pasolli E, Ercolini D (2020) Newly explored Faecalibacterium diversity is connected to age, lifestyle, geography, and disease. Curr Biol 30:4932-4943.e4. https://doi.org/10.1016/j.cub.2020.09.063
Tett A, Huang KD, Asnicar F et al (2019) The Prevotella copri complex comprises four distinct clades underrepresented in westernized populations. Cell Host Microbe 26:666-679.e7. https://doi.org/10.1016/j.chom.2019.08.018
Blanco-Miguez A, Gálvez EJC, Pasolli E et al (2023) Extension of the Segatella copri complex to 13 species with distinct large extrachromosomal elements and associations with host conditions. Cell Host Microbe 31:1804-1819.e9. https://doi.org/10.1016/j.chom.2023.09.013
Van Rossum T, Ferretti P, Maistrenko OM, Bork P (2020) Diversity within species: interpreting strains in microbiomes. Nat Rev Microbiol 18:491–506. https://doi.org/10.1038/s41579-020-0368-1
Ma Y, You X, Mai G, Tokuyasu T, Liu C (2018) A human gut phage catalog correlates the gut phageome with type 2 diabetes. Microbiome 6:24. https://doi.org/10.1186/s40168-018-0410-y
Yang K, Niu J, Zuo T et al (2021) Alterations in the gut virome in obesity and type 2 diabetes mellitus. Gastroenterology 161:1257-1269.e13. https://doi.org/10.1053/j.gastro.2021.06.056
de Jonge PA, Wortelboer K, Scheithauer TPM et al (2022) Gut virome profiling identifies a widespread bacteriophage family associated with metabolic syndrome. Nat Commun 13:3594. https://doi.org/10.1038/s41467-022-31390-5
Lawrence D, Baldridge MT, Handley SA (2019) Phages and human health: more than idle hitchhikers. Viruses 11:587. https://doi.org/10.3390/v11070587
Garmaeva S, Sinha T, Kurilshikov A, Fu J, Wijmenga C, Zhernakova A (2019) Studying the gut virome in the metagenomic era: challenges and perspectives. BMC Biol 17:84. https://doi.org/10.1186/s12915-019-0704-y
Talmor-Barkan Y, Bar N, Shaul AA et al (2022) Metabolomic and microbiome profiling reveals personalized risk factors for coronary artery disease. Nat Med 28:295–302. https://doi.org/10.1038/s41591-022-01686-6
Fromentin S, Forslund SK, Chechi K et al (2022) Microbiome and metabolome features of the cardiometabolic disease spectrum. Nat Med 28:303–314. https://doi.org/10.1038/s41591-022-01688-4
Asnicar F, Berry SE, Valdes AM et al (2021) Microbiome connections with host metabolism and habitual diet from 1,098 deeply phenotyped individuals. Nat Med 27:321–332. https://doi.org/10.1038/s41591-020-01183-8
Hug LA, Baker BJ, Anantharaman K et al (2016) A new view of the tree of life. Nat Microbiol 1:16048. https://doi.org/10.1038/nmicrobiol.2016.48
Bogaert D, van Beveren GJ, de Koff EM et al (2023) Mother-to-infant microbiota transmission and infant microbiota development across multiple body sites. Cell Host Microbe 31:447-460.e6. https://doi.org/10.1016/j.chom.2023.01.018
Dominguez-Bello MG, De Jesus-Laboy KM, Shen N et al (2016) Partial restoration of the microbiota of cesarean-born infants via vaginal microbial transfer. Nat Med 22:250–253. https://doi.org/10.1038/nm.4039
Cox LM, Yamanishi S, Sohn J et al (2014) Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell 158:705–721. https://doi.org/10.1016/j.cell.2014.05.052
Falony G, Joossens M, Vieira-Silva S et al (2016) Population-level analysis of gut microbiome variation. Science 352:560–564. https://doi.org/10.1126/science.aad3503
Rollenske T, Burkhalter S, Muerner L et al (2021) Parallelism of intestinal secretory IgA shapes functional microbial fitness. Nature 598:657–661. https://doi.org/10.1038/s41586-021-03973-7
Kabbert J, Benckert J, Rollenske T et al (2020) High microbiota reactivity of adult human intestinal IgA requires somatic mutations. J Exp Med 217:e20200275. https://doi.org/10.1084/jem.20200275
Li H, Limenitakis JP, Greiff V et al (2020) Mucosal or systemic microbiota exposures shape the B cell repertoire. Nature 584:274–278. https://doi.org/10.1038/s41586-020-2564-6
Nyström EEL, Martinez-Abad B, Arike L et al (2021) An intercrypt subpopulation of goblet cells is essential for colonic mucus barrier function. Science 372:eabb1590. https://doi.org/10.1126/science.abb1590
Li T, Ding N, Guo H et al (2024) A gut microbiota-bile acid axis promotes intestinal homeostasis upon aspirin-mediated damage. Cell Host Microbe 32:191-208.e9. https://doi.org/10.1016/j.chom.2023.12.015
Spadoni I, Zagato E, Bertocchi A et al (2015) A gut-vascular barrier controls the systemic dissemination of bacteria. Science 350:830–834. https://doi.org/10.1126/science.aad0135
Macpherson AJ, Uhr T (2004) Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science 303:1662–1665. https://doi.org/10.1126/science.1091334
Balmer ML, Slack E, de Gottardi A et al (2014) The liver may act as a firewall mediating mutualism between the host and its gut commensal microbiota. Sci Transl Med 6:237ra66. https://doi.org/10.1126/scitranslmed.3008618
Arifuzzaman M, Collins N, Guo C-J, Artis D (2024) Nutritional regulation of microbiota-derived metabolites: implications for immunity and inflammation. Immunity 57:14–27. https://doi.org/10.1016/j.immuni.2023.12.009
McCallum G, Tropini C (2024) The gut microbiota and its biogeography. Nat Rev Microbiol 22:105–118. https://doi.org/10.1038/s41579-023-00969-0
Pi H, Sun R, McBride JR et al (2023) Clostridioides difficile ferrosome organelles combat nutritional immunity. Nature 623:1009–1016. https://doi.org/10.1038/s41586-023-06719-9
Rodionov DA, Vitreschak AG, Mironov AA, Gelfand MS (2003) Comparative genomics of the vitamin B12 metabolism and regulation in prokaryotes. J Biol Chem 278:41148–41159. https://doi.org/10.1074/jbc.M305837200
Lam KN, Alexander M, Turnbaugh PJ (2019) Precision medicine goes microscopic: engineering the microbiome to improve drug outcomes. Cell Host Microbe 26:22–34. https://doi.org/10.1016/j.chom.2019.06.011
Schirmer M, Stražar M, Avila-Pacheco J et al (2024) Linking microbial genes to plasma and stool metabolites uncovers host-microbial interactions underlying ulcerative colitis disease course. Cell Host Microbe 32:209-226.e7. https://doi.org/10.1016/j.chom.2023.12.013
Holmes E, Li JV, Marchesi JR, Nicholson JK (2012) Gut microbiota composition and activity in relation to host metabolic phenotype and disease risk. Cell Metab 16:559–564. https://doi.org/10.1016/j.cmet.2012.10.007
Visconti A, Le Roy CI, Rosa F et al (2019) Interplay between the human gut microbiome and host metabolism. Nat Commun 10:4505. https://doi.org/10.1038/s41467-019-12476-z
Chen H, New P-K, Yang Y et al (2019) A forward chemical genetic screen reveals gut microbiota metabolites that modulate host physiology. Cell 177:1217-1231.e18. https://doi.org/10.1016/j.cell.2019.03.036
Li Y, Innocentin S, Withers DR et al (2011) Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell 147:629–640. https://doi.org/10.1016/j.cell.2011.09.025
Buffa JA, Romano KA, Copeland MF et al (2022) The microbial gbu gene cluster links cardiovascular disease risk associated with red meat consumption to microbiota L-carnitine catabolism. Nat Microbiol 7:73–86. https://doi.org/10.1038/s41564-021-01010-x
Tang WHW, Wang Z, Levison BS et al (2013) Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med 368:1575–1584. https://doi.org/10.1056/NEJMoa1109400
Wang Z, Klipfell E, Bennett BJ et al (2011) Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472:57–63. https://doi.org/10.1038/nature09922
Koeth RA, Wang Z, Levinson BS et al (2013) Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med 19:576–585. https://doi.org/10.1038/nm.3145
Gao F, Lv Y-W, Long J et al (2019) Butyrate improves the metabolic disorder and get microbiome dysbiosis in mice induced by a high-fat diet. Front Pharmacol 10:1040. https://doi.org/10.3389/fphar.2019.01040
Perry RJ, Peng L, Barry NA et al (2016) Acetate mediates a microbiome-brain-β-cell axis to promote metabolic syndrome. Nature 534:213–217. https://doi.org/10.1038/nature18309
Xu Y-H, Gao C-L, Guo H-L et al (2018) Sodium butyrate supplementation ameliorates diabetic inflammation in db/db mice. J Endocrinol 238:231–244. https://doi.org/10.1530/JOE-18-0137
Bouter K, Bakker GJ, Levin E et al (2018) Differential metabolic effects of oral butyrate treatment in lean versus metabolic syndrome subjects. Clin Transl Gastroenterol 9:155. https://doi.org/10.1038/s41424-018-0025-4
Canfora EE, van der Beek CM, Jocken JWE et al (2017) Colonic infusions of short-chain fatty acid mixtures promote energy metabolism in overweight/obese men: a randomized crossover trial. Sci Rep 7:2360. https://doi.org/10.1038/s41598-017-02546-x
van der Beek CM, Canfora EE, Lenaerts K et al (2016) Distal, not proximal, colonic acetate infusions promote fat oxidation and improve metabolic markers in overweight/obese men. Clin Sci (Lond) 130:2073–2082. https://doi.org/10.1042/CS20160263
de Groot PF, Nikolic T, Imangaliyev S et al (2020) Oral butyrate does not affect innate immunity and islet autoimmunity in individuals with longstanding type 1 diabetes: a randomised controlled trial. Diabetologia 63:597–610. https://doi.org/10.1007/s00125-019-05073-8
Tougaard NH, Frimodt-Møller M, Salmenkari H et al (2022) Effects of butyrate supplementation on inflammation and kidney parameters in type 1 diabetes: a randomized, double-blind, placebo-controlled trial. J Clin Med 11:3573. https://doi.org/10.3390/jcm11133573
Howard EJ, Lam TKT, Duca FA (2022) The gut microbiome: connecting diet, glucose homeostasis, and disease. Annu Rev Med 73:469–481. https://doi.org/10.1146/annurev-med-042220-012821
Martinez TM, Meyer RK, Duca FA (2021) Therapeutic potential of various plant-based fibers to improve energy homeostasis via the gut microbiota. Nutrients 13:3470. https://doi.org/10.3390/nu13103470
Byndloss MX, Olsan EE, Rivera-Chávez F et al (2017) Microbiota-activated PPAR-γ signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 357:570–575. https://doi.org/10.1126/science.aam9949
De Vadder F, Kovatcheva-Datchary P, Goncalves D et al (2014) Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell 156:84–96. https://doi.org/10.1016/j.cell.2013.12.016
Frost G, Sleeth ML, Sahuri-Arisoylu M et al (2014) The short-chain fatty acid acetate reduces appetite via a central homeostasis mechanism. Nat Commun 5:3611. https://doi.org/10.1038/ncomms4611
Shimizu H, Masujima Y, Ushiroda C et al (2019) Dietary short-chain fatty acid intake improves the hepatic metabolic condition via FFAR3. Sci Rep 9:16574. https://doi.org/10.1038/s41598-019-53242-x
McNelis JC, Lee YS, Mayoral R et al (2015) GPR43 potentiates β-cell function in obesity. Diabetes 64:3203–3217. https://doi.org/10.2337/db14-1938
Li Z, Yi C-X, Katiraei S et al (2018) Butyrate reduces appetite and activates brown adipose tissue via the gut-brain neural circuit. Gut 67:1269–1279. https://doi.org/10.1136/gutjnl-2017-314050
Fogelson KA, Dorrestein PC, Zarrinpar A, Knight R (2023) The gut microbial bile acid modulation and its relevance to digestive health and diseases. Gastroenterology 164:1069–1085. https://doi.org/10.1053/j.gastro.2023.02.022
Wahlström A, Sayin SI, Marschall H-U, Bäckhed F (2016) Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab 24:41–50. https://doi.org/10.1016/j.cmet.2016.05.005
Makki K, Brolin H, Petersen N et al (2023) 6α-hydroxylated bile acids mediate TGR5 signalling to improve glucose metabolism upon dietary fiber supplementation in mice. Gut 72:314–324. https://doi.org/10.1136/gutjnl-2021-326541
Waise TMZ, Lim Y-M, Danaea Z, Zhang S-Y, Lam TKT (2021) Small intestinal taurochenodeoxycholic acid-FXR axis alters local nutrient-sensing glucoregulatory pathways in rats. Mol Metab 44:101132. https://doi.org/10.1016/j.molmet.2020.101132
Zhang S-Y, Li RJW, Lim Y-M et al (2021) FXR in the dorsal vagal complex is sufficient and necessary for upper small intestinal microbiome-mediated changes of TCDCA to alter insulin action in rats. Gut 70:1675–1683. https://doi.org/10.1136/gutjnl-2020-321757
Scott SA, Fu J, Change PV (2020) Microbial tryptophan metabolites regulate gut barrier function via the aryl hydrocarbon receptor. Proc Natl Acad Sci U S A 117:19376–19387
Liu W-C, Chen P-H, Chen L-W (2020) Supplementation of endogenous AHR ligands reverses insulin resistance and associated inflammation in an insulin-dependent diabetic mouse model. J Nutr Biochem 83:108384. https://doi.org/10.1016/j.jnutbio.2020.108384
Yoo W, Zieba JK, Foegeding NJ et al (2021) High-fat diet-induced colonocyte dysfunction escalates microbiota-derived trimethylamine N-oxide. Science 373:813–818. https://doi.org/10.1126/science.aba3683
Koh A, Molinaro A, Ståhlman M et al (2018) Microbially produced imidazole propionate impairs insulin signaling through mTORC1. Cell 175:947-961.E17. https://doi.org/10.1016/j.cell.2018.09.055
Mishra SP, Wang B, Jain S et al (2023) A mechanism by which gut microbiota elevates permeability and inflammation in obese/diabetic mice and human gut. Gut 72:1848–1865. https://doi.org/10.1136/gutjnl-2022-327365
Camillera M, Vella A (2022) What to do about leaky gut. Gut 71:424–435. https://doi.org/10.1136/gutjnl-2021-325428
Depommier C, Everard A, Druart C et al (2019) Supplementation with Akkermansia muciniphila in overweight and obese human volunteers: a proof-of-concept exploratory study. Nat Med 25:1096–1103. https://doi.org/10.1038/s41591-019-0495-2
Dao MC, Everard A, Aron-Wisnewsky J et al (2016) Akkermansia muciniphila and improved metabolic health during a dietary intervention in obesity: relationship with gut microbiome richness and ecology. Gut 65:426–436. https://doi.org/10.1136/gutjnl-2014-308778
Yoon HS, Cho CH, Yun MS et al (2021) Akkermansia muciniphila secretes a glucagon-like peptide-1-inducing protein that improves glucose homeostasis and ameliorates metabolic disease in mice. Nat Microbiol 6:563–573. https://doi.org/10.1038/s41564-021-00880-5
Plovier H, Everard A, Druart C et al (2017) A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat Med 23:107–113. https://doi.org/10.1038/nm.4236
Clasen SJ, Bell MEW, Borbón A et al (2023) Silent recognition of flagellins from human gut commensal bacteria by Toll-like receptor 5. Sci Immunol 8:eabq7001. https://doi.org/10.1126/sciimmunol.abq7001
Rohm TV, Fuchs R, Müller RL et al (2021) Obesity in humans is characterized by gut inflammation as shown by pro-inflammatory intestinal macrophage accumulation. Front Immunol 12:668654. https://doi.org/10.3389/fimmu.2021.668654
Monteiro-Sepulveda M, Touch S, Mendes-Sá C et al (2015) Jejunal T cell inflammation in human obesity correlated with decreased enterocyte insulin signaling. Cell Metab 22:113–124. https://doi.org/10.1016/j.cmet.2015.05.020
Luck H, Khan S, Kim JH et al (2019) Gut-associated IgA+ immune cells regulate obesity-related insulin resistance. Nat Commun 10:3650. https://doi.org/10.1038/s41467-019-11370-y
Lin Y, Xu Z, Yeoh YK et al (2023) Combing fecal microbial community data to identify consistent obesity-specific microbial signatures and shared metabolic pathways. iScience 26:106476. https://doi.org/10.1016/j.isci.2023.106476
Truong DT, Tett A, Pasolli E, Huttenhower C, Segata N (2017) Microbial strain-level population structure and genetic diversity from metagenomes. Genome Res 27:626–638. https://doi.org/10.1101/gr.216242.116
Li J, Zhao F, Wang Y et al (2017) Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome 5:14. https://doi.org/10.1186/s40168-016-0222-x
Walter J, Armet AM, Finlay BB, Shanahan F (2020) Establishing or exaggerating causality for the gut microbiome: lessons from human microbiota-associated rodents. Cell 180:221–232. https://doi.org/10.1016/j.cell.2019.12.025
Sanz Y (2023) Turning cooperative bacteria into probiotics for human health. Nature 620:283–284. https://doi.org/10.1038/d41586-023-02407-w
Lagkouvardos I, Pukall R, Abt B et al (2016) The Mouse Intestinal Bacterial Collection (miBC) provides host-specific insight into cultured diversity and functional potential of the gut microbiota. Nat Microbiol 1:16131. https://doi.org/10.1038/nmicrobiol.2016.131
Hosomi K, Saito M, Park J et al (2022) Oral administration of Blautia wexlerae ameliorates obesity and type 2 diabetes via metabolic remodeling of the gut microbiota. Nat Commun 13:4477. https://doi.org/10.1038/s41467-022-32015-7
Romaní-Pérez M, López-Almela I, Bullich-Vilarrubias C et al (2021) Holdemanella biformis improves glucose tolerance and regulates GLP-1 signaling in obese mice. FASEB J 35:e21734. https://doi.org/10.1096/fj.202100126R
Chaudhari SN, McCurry MD, Devlin AS (2021) Chains of evidence from correlations to causal molecules in microbiome-linked diseases. Nat Chem Biol 17:1046–1056. https://doi.org/10.1038/s41589-021-00861-z
Hajjo H, Geva-Zatorsky N (2021) Strain-level immunomodulatory variation of gut bacteria. FEBS Lett 595:1322–1327. https://doi.org/10.1002/1873-3468.14057
Blandino G, Inturri R, Lazzara F, Di Rosa M, Malaguarnera L (2016) Impact of gut microbiota on diabetes mellitus. Diabetes Metab 42:303–315. https://doi.org/10.1016/j.diabet.2016.04.004
Zembroski AS, D’Aquila T, Buhman KK (2021) Characterization of cytoplasmic lipid droplets in each region of the small intestine of lean and diet-induced obese mice in response to dietary fat. Am J Physiol Gastrointest Liver Physiol 321:G75–G86. https://doi.org/10.1152/ajpgi.00084.2021
Kondo H, Minegishi Y, Komine Y et al (2006) Differential regulation of intestinal lipid metabolism-related genes in obesity-resistant A/J vs. obesity-prone C57BL/6J mice. Am J Physiol Endocrinol Metab 291:E1092–E1099. https://doi.org/10.1152/ajpendo.00583.2005
Martinez-Guryn K, Hubert N, Frazier K et al (2018) Small intestine microbiota regulate host digestive and absorptive adaptive responses to dietary lipids. Cell Host Microbe 23:458-469.e5. https://doi.org/10.1016/j.chom.2018.03.011
El Aidy S, Merrifield CA, Derrien M et al (2013) The gut microbiota elicits a profound metabolic reorientation in the mouse jejunal mucosa during conventionalisation. Gut 62:1306–1314. https://doi.org/10.1136/gutjnl-2011-301955
Araújo JR, Tazi A, Burlen-Defranoux O et al (2020) Fermentation products of commensal bacteria alter enterocyte lipid metabolism. Cell Host Microbe 27:358-375.e7. https://doi.org/10.1016/j.chom.2020.01.028
Bauer PV, Duca FA, Waise TMZ et al (2018) Lactobacillus gasseri in the upper small intestine impacts an ACSL3-dependent fatty acid-sensing pathway regulating whole-body glucose homeostasis. Cell Metab 27:572-587.e6. https://doi.org/10.1016/j.cmet.2018.01.013
Weninger SN, Herman C, Meyer RK et al (2023) Oligofructose improves small intestinal lipid-sensing mechanisms via alterations to the small intestinal microbiota. Microbiome 11:169. https://doi.org/10.1186/s40168-023-01590-2
Johnson AJ, Vangay P, Al-Ghalith GA et al (2019) Daily sampling reveals personalized diet-microbiome associations in humans. Cell Host Microbe 25:789-802.e5. https://doi.org/10.1016/j.chom.2019.05.005
Martchenko SE, Martchenko A, Cox BJ et al (2020) Circadian GLP-1 secretion in mice is dependent on the intestinal microbiome for maintenance of diurnal metabolic homeostasis. Diabetes 69:2589–2602. https://doi.org/10.2337/db20-0262
Reitmeier S, Kiessling S, Clavel T et al (2020) Arrhythmic gut microbiome signatures predict risk of type 2 diabetes. Cell Host Microbe 28:258-272.e6. https://doi.org/10.1016/j.chom.2020.06.004
Brettle H, Tran V, Drummond GR et al (2022) Sex hormones, intestinal inflammation, and the gut microbiome: major influencers of the sexual dimorphisms in obesity. Front Immunol 13:971048. https://doi.org/10.3389/fimmu.2022.971048
Markle JGM, Frank DN, Mortin-Toth S et al (2013) Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science 339:1084–1088. https://doi.org/10.1126/science.1233521
Wang D, Liu J, Zhou L, Zhang Q, Li M, Xiao X (2022) Effects of oral glucose-lowering agents on gut microbiota and microbial metabolites. Front Endocrinol (Lausanne) 13:905171. https://doi.org/10.3389/fendo.2022.905171
Bäckhed F, Manchester JK, Semenkovich CF, Gordon JI (2007) Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci U S A 104:979–984. https://doi.org/10.1073/pnas.0605374104
Cheng AG, Ho P-Y, Aranda-Díaz A et al (2022) Design, construction, and in vivo augmentation of a complex gut microbiome. Cell 185:3617-3636.e19. https://doi.org/10.1016/j.cell.2022.08.003
Zhang C, Yin A, Li H et al (2015) Dietary modulation of gut microbiota contributes to alleviation of both genetic and simple obesity in children. EBioMedicine 2:968–984. https://doi.org/10.1016/j.ebiom.2015.07.007
Fei N, Zhao L (2013) An opportunistic pathogen isolated from the gut of an obese human causes obesity in germfree mice. ISME J 7:880–884. https://doi.org/10.1038/ismej.2012.153
Fei N, Bruneau A, Zhang X et al (2020) Endotoxin producers overgrowing in human gut microbiota as the causative agents for nonalcoholic fatty liver disease. mBio 11:e03263-19. https://doi.org/10.1128/mBio.03263-19
Cani PD, Amar J, Iglesias MA et al (2007) Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56:1761–1772. https://doi.org/10.2337/db06-1491
Logan IE, Bobe G, Miranda CL et al (2020) Germ-free Swiss Webster mice on a high-fat diet develop obesity, hyperglycemia, and dyslipidemia. Microorganisms 8:520. https://doi.org/10.3390/microorganisms8040520
Almind K, Kahn CR (2004) Genetic determinants of energy expenditure and insulin resistance in diet-induced obesity in mice. Diabetes 53:3274–3285. https://doi.org/10.2337/diabetes.53.12.3274
Pang X, Hua X, Yang Q et al (2007) Inter-species transplantation of gut microbiota from human to pigs. ISME J 1:156–162. https://doi.org/10.1038/ismej.2007.23
Rawls JF, Samuel BS, Gordon JI (2004) Gnotobiotic zebrafish reveal evolutionarily conserved responses to the gut microbiota. Proc Natl Acad Sci U S A 101:4596–4601. https://doi.org/10.1073/pnas.0400706101
Melancon E, Gomez De La Torre Canny S, Sichel S et al (2017) Best practices for germ-free derivation and gnotobiotic zebrafish husbandry. Methods Cell Biol 138:61–100. https://doi.org/10.1016/bs.mcb.2016.11.005
Leung CM, de Haan P, Ronaldson-Bouchard K et al (2022) A guide to the organ-on-a-chip. Nat Rev Methods Primers 2:33. https://doi.org/10.1038/s43586-022-00118-6
Jalili-Firoozinezhad S, Gazzaniga FS, Calamari EL et al (2019) A complex human gut microbiome cultured in an anaerobic intestine-on-a-chip. Nat Biomed Eng 3:520–531. https://doi.org/10.1038/s41551-019-0397-0
Trapecar M, Wogram E, Svoboda D et al (2021) Human physiomimetic model integrating microphysiological systems of the gut, liver, and brain for studies of neurodegenerative diseases. Sci Adv 7:eabd1707. https://doi.org/10.1126/sciadv.abd1707
Yang D, Park SY, Park YS, Eun H, Lee SY (2020) Metabolic engineering of Escherichia coli for natural product biosynthesis. Trends Biotechnol 38:745–765. https://doi.org/10.1016/j.tibtech.2019.11.007
Fang H, Li D, Kang J, Jiang P, Sun J, Zhang D (2018) Metabolic engineering of Escherichia coli for de novo biosynthesis of vitamin B12. Nat Commun 9:4917. https://doi.org/10.1038/s41467-018-07412-6
Kolisnychenko V, Plunkett G 3rd, Herring CD et al (2002) Engineering a reduced Escherichia coli genome. Genome Res 12:640–647. https://doi.org/10.1101/gr.217202
Tan Y, Liang J, Lai M, Wan S, Luo X, Li F (2023) Advances in synthetic biology toolboxes paving the way for mechanistic understanding and strain engineering of gut commensal Bacteroides spp. and Clostridium spp. Biotechnol Adv 69:108272. https://doi.org/10.1016/j.biotechadv.2023.108272
Whitaker WR, Shepherd ES, Sonnenburg JL (2017) Tunable expression tools enable single-cell strain distinction in the gut microbiome. Cell 169:538-546.e12. https://doi.org/10.1016/j.cell.2017.03.041
Jumper J, Evans R, Pritzel A et al (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596:583–589. https://doi.org/10.1038/s41586-021-03819-2
Varadi M, Anyango S, Deshpande M et al (2022) AlphaFold Protein Structure Database: massively expanded the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res 50:D439–D444. https://doi.org/10.1093/nar/gkab1061
Wu G, Zhao N, Zhang C, Lam YY, Zhao L (2021) Guild-based analysis for understanding gut microbiome in human health and diseases. Genome Med 13:22. https://doi.org/10.1186/s13073-021-00840-y
Bowers RM, Kyrpides NC, Stepanauskas R et al (2017) Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat Biotechnol 35:725–731. https://doi.org/10.1038/nbt.3893
Shalon D, Culver RN, Grembi JA et al (2023) Profiling the human intestinal environment under physiological conditions. Nature 617:581–591. https://doi.org/10.1038/s41586-023-05989-7
Donia MS, Fischbach MA (2015) Small molecules from the human microbiota. Science 349:1254766. https://doi.org/10.1126/science.1254766
Zhai L, Xiao H, Lin C et al (2023) Gut microbiota-derived tryptamine and phenethylamine impair insulin sensitivity in metabolic syndrome and irritable bowel syndrome. Nat Commun 14:4986. https://doi.org/10.1038/s41467-023-40552-y
Vatanen T, Franzosa EA, Schwager R et al (2018) The human gut microbiome in early-onset type 1 diabetes from the TEDDY study. Nature 562:589–594. https://doi.org/10.1038/s41586-018-0620-2
Berry D, Loy A (2018) Stable-isotope probing of human and animal microbiome function. Trends Microbiol 26:999–1007. https://doi.org/10.1016/j.tim.2018.06.004
Reynolds AN, Akerman AP, Mann J (2020) Dietary fibre and whole grains in diabetes management: systematic review and meta-analysis. PLoS Med 17:e1003053. https://doi.org/10.1371/journal.pmed.1003053
Chandalia M, Garg A, Lutjohann D, von Bergmann K, Grundy SM, Brinkley LJ (2000) Beneficial effects of high dietary fiber intake in patients with type 2 diabetes mellitus. N Engl J Med 342:1392–1398. https://doi.org/10.1056/NEJM200005113421903
Blaak EE, Canfora EE, Theis S et al (2020) Short chain fatty acids in human gut and metabolic health. Benef Microbes 11:411–455. https://doi.org/10.3920/BM2020.0057
Khosravi Z, Hadi A, Tutunchi H et al (2022) The effects of butyrate supplementation on glycemic control, lipid profile, blood pressure, nitric oxide level and glutathione peroxidase activity in type 2 diabetic patients: a randomized triple-blind, placebo-controlled trial. Clin Nutr ESPEN 49:79–85. https://doi.org/10.1016/j.clnesp.2022.03.008
O’Toole PW, Marchesi JR, Hill C (2017) Next-generation probiotics: the spectrum from probiotics to live biotherapeutics. Nat Microbiol 2:17057. https://doi.org/10.1038/nmicrobiol.2017.57
Wang C-H, Yen H-R, Lu W-L et al (2022) Adjuvant probiotics of Lactobacillus salivarius subsp. salicinius AP-32, L. johnsonii MH-68, and Bifidobacterium animalis subsp. lactis CP-9 attenuate glycemic levels and inflammatory cytokine in patients with type 1 diabetes mellitus. Front Endocrinol (Lausanne) 13:754401. https://doi.org/10.3389/fendo.2022.754401
Cabrera SM, Coren AT, Pant T et al (2022) Probiotic normalization of systemic inflammation in siblings of type 1 diabetes patients: an open-label pilot study. Sci Rep 12:3306. https://doi.org/10.1038/s41598-022-07203-6
Tao Y-W, Gu Y-L, Mao X-Q, Zhang L, Pei Y-F (2020) Effects of probiotics on type II diabetes mellitus: a meta-analysis. J Transl Med 18:30. https://doi.org/10.1186/s12967-020-02213-2
Zhou Q, Zhang Y, Wang X et al (2020) Gut bacteria Akkermansia is associated with reduced risk of obesity: evidence from the American Gut Project. Nutr Metab (Lond) 17:90. https://doi.org/10.1186/s12986-020-00516-1
Gilijamse PW, Hartstra AV, Levin E et al (2020) Treatment with Anaerobutyricum soehngenii: a pilot study of safety and dose-response effects on glucose metabolism in human subjects with metabolic syndrome. NPJ Biofilms Microbiomes 6:16. https://doi.org/10.1038/s41522-020-0127-0
Koopen A, Witjes J, Wortelboer K et al (2022) Duodenal Anaerobutyricum soehngenii infusion stimulates GLP-1 production, ameliorates glycaemic control and beneficially shapes the duodenal transcriptome in metabolic syndrome subjects: a randomized double-blind placebo-controlled cross-over study. Gut 71:1577–1587. https://doi.org/10.1136/gutjnl-2020-323297
Hanssen NMJ, do Vos WM, Nieuwdorp M (2021) Fecal microbiota transplantation in human metabolic diseases: from a murky past to a bright future? Cell Metab 33:1098–1110. https://doi.org/10.1016/j.cmet.2021.05.005
Pellegrini S, Sordi V, Bolla AM et al (2017) Duodenal mucosa of patients with type 1 diabetes shows distinctive inflammatory profile and microbiota. J Clin Endocrinol Metab 102:1468–1477. https://doi.org/10.1210/jc.2016-3222
Vrieze A, Van Nood E, Holleman F et al (2012) Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 143:913-916.e7. https://doi.org/10.1053/j.gastro.2012.06.031
Kootte RS, Levin E, Salojärvi J et al (2017) Improvement of insulin sensitivity after lean donor feces in metabolic syndrome is driven by baseline intestinal microbiota composition. Cell Metab 26:611-619.e6. https://doi.org/10.1016/j.cmet.2017.09.008
Schmidt TSB, Li SS, Maistrenko OM et al (2022) Drivers and determinants of strain dynamics following fecal microbiota transplantation. Nat Med 28:1902–1912. https://doi.org/10.1038/s41591-022-01913-0
de Groot P, Nikolic T, Pellegrinin S et al (2021) Faecal microbiota transplantation halts progression of human new-onset type 1 diabetes in a randomised controlled trial. Gut 70:92–105. https://doi.org/10.1136/gutjnl-2020-322630
Rinott E, Youngster I, Meir AY et al (2021) Effects of diet-modulated autologous fecal microbiota transplantation on weight regain. Gastroenterology 160:158-173.e10. https://doi.org/10.1053/j.gastro.2020.08.041
Mocanu V, Zhang Z, Deehan EC et al (2021) Fecal microbial transplantation and fiber supplementation in patients with serve obesity and metabolic syndrome: a randomized double-blind, placebo-controlled phase 2 trial. Nat Med 27:1272–1279. https://doi.org/10.1038/s41591-021-01399-2
Witjes JJ, Smits LP, Pekmez CT et al (2020) Donor fecal microbiota transplantation alters gut microbiota and metabolites in obese individuals with steatohepatitis. Hepatol Commun 4:1578–1590. https://doi.org/10.1002/hep4.1601
Ng SC, Xu Z, Mak JWY et al (2022) Microbiota engraftment after faecal microbiota transplantation in obese subjects with type 2 diabetes: a 24-week, double-blind, randomized controlled trial. Gut 71:716–723. https://doi.org/10.1136/gutjnl-2020-323617
Yu EW, Gao L, Stastka P et al (2020) Fecal microbiota transplantation for the improvement of metabolism in obesity: the FMT-TRIM double-blind placebo-controlled pilot trial. PLoS Med 17:e1003051. https://doi.org/10.1371/journal.pmed.1003051
Kamer O, Rinott E, Tsaban G et al (2023) Successful weight regain attenuation by autologous fecal microbiota transplantation is associated with non-core gut microbiota changes during weight loss; randomized controlled trial. Gut Microbes 15:2264457. https://doi.org/10.1080/19490976.2023.2264457
Fessler J, Matson V, Gajewski TF (2019) Exploring the emerging role of the microbiome in cancer immunotherapy. J Immunother Cancer 7:108. https://doi.org/10.1186/s40425-019-0574-4
Ianiro G, Punčochář M, Karcher N et al (2022) Variability of strain engraftment and predictability of microbiome composition after fecal microbiota transplantation across different diseases. Nat Med 28:1913–1923. https://doi.org/10.1038/s41591-022-01964-3
Attaye I, Warmbrunn MV, Boot ANAF et al (2022) A systematic review and meta-analysis of dietary interventions modulating gut microbiota and cardiometabolic diseases: striving for new standards in microbiome studies. Gastroenterology 162:1911–1932. https://doi.org/10.1053/j.gastro.2022.02.011
European Medicines Agency. Faecal microbiota transplantation: EU-IN horizon scanning report. Available from https://www.ema.europa.eu/en/documents/report/faecal-microbiota-transplantation-eu-horizon-scanning-report_en.pdf. Accessed 7 February 2024
Bénard MV, de Bruijn CMA, Fenneman AC et al (2022) Challenges and costs of donor screening for fecal microbiota transplantations. PLoS One 17:e0276323. https://doi.org/10.1371/journal.pone.0276323
Groen AK, Nieuwdorp M (2017) An evaluation of the therapeutic potential of fecal microbiota transplantation to treat infectious and metabolic diseases. EMBO Mol Med 9:1–3. https://doi.org/10.15252/emmm.201607035
Pascual J, Tanner K, Vilanova C, Porcar M, Delgado A (2021) The microbial terroir: open questions on the Nagoya protocol applied to microbial resources. Microb Biotechnol 14:1878–1880. https://doi.org/10.1111/1751-7915.13839
Bojanova DP, Bordenstein SR (2016) Fecal transplants: what is being transferred? PLoS Biol 14:e1002503. https://doi.org/10.1371/journal.pbio.1002503
Kovatcheva-Datchary P, Tremaroli V, Bäckhed F (2013) The gut microbiota. In: Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F (eds) The prokaryotes: human microbiology, 4th edn. Springer-Verlag, Berlin, Germany, pp 3–24. https://doi.org/10.1007/978-3-642-30144-5_87
Caesar R (2019) Pharmacologic and nonpharmacologic therapies for the gut microbiota in type 2 diabetes. Can J Diabetes 43:224–231. https://doi.org/10.1016/j.jcjd.2019.01.007
Zhou Z-Y, Ren L-W, Zhan P, Yang H-Y, Chai D-D, Yu Z-W (2016) Metformin exerts glucose-lowering action in high-fat fed mice via attenuating endotoxemia and enhancing insulin signaling. Acta Pharmacol Sin 37:1063–1075. https://doi.org/10.1038/aps.2016.21
Shin N-R, Lee J-C, Lee H-Y et al (2014) An increase in the Akkermansia spp. Population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut 63:727–735. https://doi.org/10.1136/gutjnl-2012-303839
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Acknowledgements
Editorial support services for this article were provided by Debbie Kendall of Kendall Editorial in Richmond, VA. The authors also thank Robert Caesar and Anna Hallen of Wallenberg Laboratory, Department of Molecular and Clinical Medicine, Institute of Medicine, University of Gothenburg, Gothenburg, Sweden, for their help with the figures. Portions of the content of this article were presented at a symposium at the 59th Annual Meeting of the European Association for the Study of Diabetes on 6 October 2023 in Hamburg, Germany.
Funding
The 2023 Diabetes, Diabetes Care, and Diabetologia expert forum ‘The microbiome and diabetes: research, translation and clinical applications’, including a subsequent symposium presented on this topic and the preparation of this article, was supported by a grant from the Novo Nordisk Foundation (NNF23SA0085453). MB is a Howard Hughes Medical Institute Freeman Hrabowski Scholar and is supported by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) grant NIDDK R01DK131104, the Pew Charitable Trusts (2022-A-19568), and the Burroughs Wellcome Fund Investigators in the Pathogenesis of Infectious Disease (022792). SD is supported by NIDDK grants NIDDK R01DK123446 and 1DP1 DK130687. JHN is supported by the Swiss National Science Foundation (SNSF) grant 310030_219210. MN is supported by a personal ZONMW-VICI grant 2020 (09150182010020) and a DFN-DON grant 2020 (2020.10.002). YS is supported by grants from the Spanish Ministry of Science, Innovation and Universities MICIU/AEI (PID2020-119536RB-I00) and Severo Ochoa Center of Excellence (ref. CEX2021- 001189-S/10.13039/501100011033). VT is a co-principal investigator of a research project funded by the European Union’s Innovative Medicines Initiative (IMI grant 875534).
Authors’ relationships and activities
JHN has served as an advisor for Abbvie, Bristol Myers Squibb, Falk, Janssen, Roche, and Takeda. MN is the founder and a scientific advisory board member of Caelus Pharmaceuticals and Advanced Microbiome Interventions in Amsterdam, the Netherlands. YS is co-author of patents based on the use of intestinal bacteria for obesity and metabolic comorbidities. VT holds a patent for the use of microbiota in type 2 diabetes. LZ is a co-founder of Notitia Biotechnologies, a microbiome company providing products related to diabetes. All other authors declare that there are no relationships or activities that might bias, or be perceived to bias, their work.
Contribution statement
All authors contributed to writing, reviewing, and revising the manuscript and approved the final version for submission. MO-M is the guarantor of this work and, as such, takes responsibility for the integrity of the review.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is being simultaneously published in Diabetes (https://doi.org/10.2337/dbi24-0028), Diabetes Care (https://doi.org/10.2337/dci24-0052) and Diabetologia (https://doi.org/10.1007/s00125-024-06198-1) by the ADA and the EASD.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Byndloss, M., Devkota, S., Duca, F. et al. The gut microbiota and diabetes: research, translation, and clinical applications – 2023 Diabetes, Diabetes Care, and Diabetologia Expert Forum. Diabetologia (2024). https://doi.org/10.1007/s00125-024-06198-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00125-024-06198-1