
Abstract
Aims/hypothesis
It was shown that maternal endothelial nitric oxide synthase (eNOS) deficiency causes fatty liver disease and numerically lower fasting glucose in female wild-type offspring, suggesting that parental genetic variants may influence the offspring’s phenotype via epigenetic modifications in the offspring despite the absence of a primary genetic defect. The aim of the current study was to analyse whether paternal eNOS deficiency may cause the same phenotype as seen with maternal eNOS deficiency.
Methods
Heterozygous (+/−) male eNOS (Nos3) knockout mice or wild-type male mice were bred with female wild-type mice. The phenotype of wild-type offspring of heterozygous male eNOS knockout mice was compared with offspring from wild-type parents.
Results
Global sperm DNA methylation decreased and sperm microRNA pattern altered substantially. Fasting glucose and liver glycogen storage were increased when analysing wild-type male and female offspring of +/− eNOS fathers. Wild-type male but not female offspring of +/− eNOS fathers had increased fasting insulin and increased insulin after glucose load. Analysing candidate genes for liver fat and carbohydrate metabolism revealed that the expression of genes encoding glucocorticoid receptor (Gr; also known as Nr3c1) and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (Pgc1a; also known as Ppargc1a) was increased while DNA methylation of Gr exon 1A and Pgc1a promoter was decreased in the liver of male wild-type offspring of +/− eNOS fathers. The endocrine pancreas in wild-type offspring was not affected.
Conclusions/interpretation
Our study suggests that paternal genetic defects such as eNOS deficiency may alter the epigenome of the sperm without transmission of the paternal genetic defect itself. In later life wild-type male offspring of +/− eNOS fathers developed increased fasting insulin and increased insulin after glucose load. These effects are associated with increased Gr and Pgc1a gene expression due to altered methylation of these genes.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.

Introduction
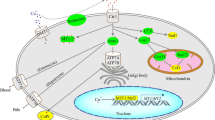
The ‘fetal origin of diseases’ hypothesis proposes that adulthood diseases originate through adaptation of the fetus to environmental conditions in early life [1]. Another mechanism responsible for programming events might be related to maternal genes affecting the fetal phenotype independently of the offspring’s genome [2,3,4,5,6,7,8,9,10,11,12,13,14]. These clinical association studies stimulated the initiation of animal studies to identify the underlying molecular mechanisms. Heterozygous (+/−) female mice in which the Nos3 gene, encoding endothelial nitric oxide synthase (eNOS), was knocked out and wild-type (WT) female mice were bred with male WT mice. Female offspring with normal Nos3 gene status but born to heterozygous female eNOS knockout mice develop hepatic steatosis [15], causally demonstrating that maternal genes can epigenetically alter the offspring’s phenotype without inheritance of the gene itself [15]. Paternal environmental factors prior to mating likewise affect the offspring’s phenotype [1, 16]. It was shown that a pre-conceptional paternal high-fat diet results in impaired glucose tolerance in female offspring [17,18,19,20,21]. There are already studies suggesting that paternal genes without transmission to the offspring might likewise affect the offspring’s phenotype [22,23,24] (Fig. 1).

Paternal programming hypothesis. The paternal genetic defects might impact on the offspring phenotype via genomic–epigenomic interactions without inheritance of the defective paternal genes. The paternal genetic changes might affect the endocrine system and vascular function in testes leading to alterations related to sperm quality and seminal fluid composition, which might in turn trigger early epigenetic modifications in sperm, e.g. miRNAs, tRNA-derived small RNAs (tsRNAs) and DNA methylation [18, 20, 36, 37, 64, 65]. These early epigenetic alterations might impact the offspring leading to late epigenetic changes in target offspring organs with modified gene expression and phenotype without transmittance of the paternal genetic change
We have chosen male heterozygous eNOS knockout mice to test this hypothesis, because eNOS plays an important role in the control of testicular vascular function, and hence heterozygous eNOS deficiency in male mice might create an unfavourable testicular microenvironment. We hypothesised that this could influence the offspring’s phenotype. We analysed the impact of paternal nitric oxide (NO) deficiency on epigenetic alterations in sperm. Next, we analysed the phenotype of WT offspring of male heterozygous eNOS knockout mice, followed by analysis of candidate genes (both gene expression and related epigenetic alterations of differentially expressed genes) potentially responsible for the observed phenotype.
Methods
For detailed methods, please refer to the electronic supplementary material (ESM).
Breeding protocol and study protocol of eNOS-deficient mice
Male heterozygous mice (C57BL/6 J background) in which the Nos3 gene encoding eNOS was knocked out [25] were bred with C57BL/6 J female mice and their WT offspring were compared with offspring from healthy male and female C57BL/6 J mice. The breeding procedure is described in more detail in ESM Fig. 1. Study design and experimental protocols were conducted according to the local institutional guidelines for the care and use of laboratory animals and were approved by the animal welfare ethical committee of the state of Berlin.
Male and female offspring were kept for 24 weeks and analysed separately. Body weight, length, abdominal diameter, blood pressure and plasma creatinine were measured and IPGTT was performed. Experimenters were blind to group assignment and outcome assessment for the entire study.
Effects of NO deficiency on sperm development and epigenetic alterations in the sperm
A total of 30 C57BL/6 J male mice were randomised into three groups and treated with different doses of N(γ)-nitro-l-arginine methyl ester (l-NAME) for 12 consecutive weeks.
Sperm total DNA methylation
Mature sperm were isolated from cauda epididymis. Sperm total DNA methylation was performed as described before [26].
Sperm count and small RNA library construction
Mature sperm were isolated from cauda epididymis of male C57BL/6 J mice and processed for RNA extraction as previously described [27, 28]. Small RNA libraries were constructed. After validation of library quality, sequencing was performed by Illumina HiSeq (Illumina, UK).
Testicular morphology
Testes were fixed, processed and stained with haematoxylin and eosin, followed by computer-aided image analysis.
Liver morphology
Livers were fixed, embedded in paraffin and cut into slices. Haematoxylin and eosin staining, Oil Red O staining and immunohistochemistry were performed, followed by computer-aided image analysis.
Pancreas morphology
Pancreases were fixed, embedded in paraffin and cut into slices. Haematoxylin and eosin staining and immunohistochemistry were performed, followed by computer-aided image analysis.
Liver glycogen content
Glycogen content was determined using the amyloglucosidase method [29].
Quantitative real-time PCR
Quantitative real-time RT-PCR was used to determine the relative expression levels of mRNAs as described recently [15]. Sequences of primers used are listed in ESM Table 1.
Quantification of gene-specific DNA methylation
Quantification of gene-specific DNA methylation was achieved with methylated genomic DNA immunoprecipitation (MeDIP), with minor modifications as described by Weber et al [30].
Statistics
For the statistical analysis of IPGTT glucose and insulin, two-way ANOVA test followed by Bonferroni post hoc test was conducted. The unpaired Student’s t test and Pearson correlation analysis were applied for normally distributed data, while the Mann–Whitney U test and Spearman correlation analysis were used for non-normally distributed data. To correct for multiple testing in the gene expression analysis, a false discovery rate (FDR) cut off was set at 0.05 [31, 32]. Statistically significant differences were considered as p≤0.05.
Results
Effects on sperm under conditions of NO deficiency
Sperm total DNA methylation in +/− eNOS fathers was lower than that in WT fathers (Fig. 2a). Mature sperm has a haploid chromosome set. Half of the spermatozoa from heterozygous eNOS knockout mice therefore have an inactivated Nos3 gene; the remaining half have a normal Nos3 gene. We therefore treated male WT mice with the identical genetic background as the eNOS knockout mice with l-NAME and then analysed the sperm. This sperm is a well-suited model to analyse effects of reduced eNOS activity in the testes on the maturation of genetically healthy sperm. We have chosen two dosages of l-NAME. The lower dose does not increase blood pressure, whereas the higher dose does. The mice treated with the lower dose can thus be regarded as a model of heterozygous eNOS knockout mice with sperm having only WT Nos3 genes, because blood pressure is not increased in heterozygous eNOS knockout mice. Total DNA methylation in sperm of mice treated with l-NAME decreased in a dose-dependent manner (Fig. 2b). Sperm count, Johnsen scores and sloughing rate of maturing sperm cells were not altered in mice on low-dose l-NAME (Fig. 2c–e). Twenty-three microRNAs (miRNAs) were downregulated and five miRNAs were upregulated in the low-dose l-NAME group (Fig. 2f). Only six downregulated miRNAs were described previously (miR-615-3p, miR-193a-5p, miR-199b-5p, miR-144-3p, miR-132-3p, miR-8114) (ESM Tables 2–4).

Sperm DNA methylation in WT (n = 10) and +/− eNOS fathers (n = 6) (a), sperm DNA methylation in mice treated with different doses of l-NAME (b), sperm count (c), determination of testicular morphology: Johnsen score (d), sloughing rate of spermatogenic cells (e) and volcano plots of differentially expressed miRNAs: low-dose l-NAME (0.15 mg/ml drinking water [DW], n = 10) group vs control group (n = 10) (f) and high-dose l-NAME (2 mg/ml DW, n = 10) group vs control group (g). *p<0.05 vs WT fathers in (a); *p<0.05 and **p<0.01 vs control group in (b), (c), (d) and (e)
Birth variables and adult body weight
WT offspring born to +/− eNOS fathers and WT mothers showed no differences in birthweight, length and abdominal diameter when compared with controls (ESM Fig. 2). There was no difference among the groups regarding body weight (ESM Fig. 2, ESM Table 5).
Blood pressure and kidney function
Neither blood pressure nor kidney function was different in WT offspring born to +/− eNOS fathers and WT mothers as compared with controls (ESM Table 5).
IPGTT
During IPGTT, no differences in glucose concentrations could be observed (Fig. 3). Higher insulin concentrations after an i.p. glucose load, however, were found in animals born to +/− eNOS fathers and WT mothers. Sex-specific analyses showed higher insulin levels in male animals after 60 min of the IPGTT. The analyses of the insulin AUC for offspring from +/− eNOS fathers and WT mothers showed a higher AUC compared with controls. Considering offspring sex revealed that this effect was significant only in male offspring (Fig. 3).

Plasma glucose (a–c) and insulin concentrations (d–f) during IPGTT in all (a, d), male (b, e) or female (c, f) offspring; blue circles: father WT/mother WT; red squares: father +/− eNOS/mother WT; AUC for IPGTT plasma glucose (g) and IPGTT plasma insulin (h) in all (squares) (35 F:WT; M:WT and 24 F:+/−eNOS; M:WT), male (triangles) (15 F:WT; M:WT and 9 F:+/−eNOS; M:WT) or female (circles) (20 F:WT; M:WT and 15 F:+/−eNOS; M:WT) offspring; *p<0.05 and **p<0.01 vs father WT/mother WT. F:+/−eNOS; M:WT, WT offspring of eNOS heterozygous fathers and WT mothers; F:WT; M:WT, WT offspring of WT fathers and WT mothers; XX, female offspring; XY, male offspring
Fasting plasma glucose and insulin
WT offspring of +/− eNOS fathers showed higher fasting glucose concentrations. Sex-specific analyses revealed a numerically non-significant elevation of fasting glucose in female and male WT offspring of +/− eNOS fathers (Fig. 4a). Moreover, fasting plasma insulin was significantly higher in male WT offspring of +/− eNOS fathers (Fig. 4b).

Main phenotypic changes in the offspring: fasting plasma glucose (a), fasting plasma insulin (b) and liver glycogen (c) in all (squares) (35 F:WT; M:WT and 24 F:+/−eNOS; M:WT), male (triangles) (15 F:WT; M:WT and 9 F:+/−eNOS; M:WT) or female (circles) (20 F:WT; M:WT and 15 F:+/−eNOS; M:WT) offspring. Correlation analysis between liver glycogen and AUC of plasma glucose (d), AUC of plasma glucose and AUC of plasma insulin (e), and AUC of plasma insulin and liver glycogen (f) in male offspring. *p<0.05, **p<0.01 and ***p<0.001 vs WT (F:WT; M:WT). F:+/−eNOS; M:WT, WT offspring of eNOS heterozygous fathers and WT mothers; F:WT; M:WT, WT offspring of WT fathers and WT mothers; XX, female offspring; XY, male offspring
Liver phenotype
Liver weights, liver lobule dimensions, lobular inflammation connective tissue content and hepatic lipid content were similar in all groups (ESM Table 6). Liver glycogen content in both sexes, however, was higher in animals born to +/− eNOS fathers and WT mothers (p<0.001) (Fig. 4c). In male offspring, no significant correlation was found between liver glycogen and AUC of plasma glucose, or between AUC of plasma glucose and AUC of plasma insulin (Fig. 4d, e). However, AUC of plasma insulin was positively correlated with liver glycogen (r = 0.452, p=0.03) (Fig. 4f). For more details see ESM Table 7.
Liver eNOS/iNOS expression
Liver eNOS (Nos3) and inducible nitric oxide synthetase (iNOS; encoded by Nos2) expression were comparable in animals born to +/− eNOS fathers (ESM Table 8).
Pancreas morphology
Size and density of pancreatic islets of Langerhans and beta cell content of islets were similar in all groups (Fig. 5).

Determination of pancreas morphology: example of insulin stain in beta cells of pancreatic islets of Langerhans (scale bar, 50 μm) (a), comparison of islet density (b), mean islet area (c) and beta cell content (d) of islets in all (squares) (35 F:WT; M:WT and 24 F:+/−eNOS; M:WT), male (triangles) (15 F:WT; M:WT and 9 F:+/−eNOS; M:WT) or female (circles) (20 F:WT; M:WT and 15 F:+/−eNOS; M:WT) offspring. F:+/−eNOS; M:WT, WT offspring of eNOS heterozygous fathers and WT mothers; F:WT; M:WT, WT offspring of WT fathers and WT mothers; XX, female offspring; XY, male offspring
Quantitative real-time PCR in the liver
WT offspring of both sexes born to +/− eNOS fathers and WT mothers showed an altered expression of genes involved in lipid and carbohydrate metabolism. Seventeen genes were differentially expressed (Table 1). When analysing male offspring born to +/− eNOS fathers and WT mothers (p<0.05 and FDR <0.05), 19 genes were differentially expressed (Table 2), with Gr (which encodes glucocorticoid receptor [GR]; also known as Nr3c1) and Pgc1a (which encodes peroxisome proliferator-activated receptor gamma coactivator 1-alpha [PGC-1α]; also known as Ppargc1a) showing the lowest p values and FDR. Analysing female offspring born to +/− eNOS fathers and WT mothers revealed no differences (Table 3).
MeDIP methylation analysis in the liver
MeDIP analysis revealed lower Gr exon 1A and Pgc1a promoter DNA methylation in WT male offspring of eNOS +/− fathers compared with controls (Figs 6, 7). Correlation analysis between the gene expression of liver Gr and Pgc1a and DNA methylation of Gr exon 1A and Pgc1a promoter in WT male offspring of eNOS +/− fathers revealed an inverse correlation.

MeDIP methylation analysis of Gr (Nr3c1) gene exon 1A and Gr expression in the liver: (a) genomic organisation of Gr exon 1A region with putative transcription start site (TSS) and beyond the position of CpG dinucleotides; amplified sequence is shown in the box (primer binding sites are underlined and analysed CpG dinucleotides are in bold letters); (b) degree of DNA methylation in amplified region; (c) hepatic expression of Gr in all (squares) (35 F:WT; M:WT and 24 F:+/−eNOS; M:WT), male (triangles) (15 F:WT; M:WT and 9 F:+/−eNOS; M:WT) or female (circles) (20 F:WT; M:WT and 15 F:+/−eNOS; M:WT) offspring (**p<0.01 and ***p<0.001 vs F:WT; M:WT); and correlation of DNA methylation and gene expression in all (d), male (e) or female (f) offspring. F:+/−eNOS; M:WT, WT offspring of eNOS heterozygous fathers and WT mothers; F:WT; M:WT, WT offspring of WT fathers and WT mothers; XX, female offspring; XY, male offspring

MeDIP methylation analysis of Pgc1a (Ppargc1a) promoter and Pgc1a expression in the liver: (a) genomic organisation of Pgc1a proximal promoter region with putative transcription start site (TSS/+1) and beyond the position of CpG dinucleotides; amplified sequence is shown in the box (primer binding sites are underlined and analysed CpG dinucleotides are in bold letters); (b) degree of DNA methylation in amplified region; (c) hepatic expression of Pgc1a in all (squares) (35 F:WT; M:WT and 24 F:+/−eNOS; M:WT), male (triangles) (15 F:WT; M:WT and 9 F:+/−eNOS; M:WT) or female (circles) (20 F:WT; M:WT and 15 F:+/−eNOS; M:WT) (**p<0.01 and ***p<0.001 vs F:WT; M:WT); and correlation of DNA methylation and gene expression in all (d), male (e) or female (f) offspring. F:+/−eNOS; M:WT, WT offspring of eNOS heterozygous fathers and WT mothers; F:WT; M:WT, WT offspring of WT fathers and WT mothers; XX, female offspring; XY, male offspring
Discussion
To test the advanced fetal programming hypothesis [2,3,4,5,6,7,8,9,10,11,12,13] for paternal genes, we used a comparable approach as in our previous study [15] by breeding male heterozygous eNOS knockout mice with female WT mice and comparing the phenotype of their WT offspring with the phenotype of offspring with WT parents. NO deficiency in male mice reduces sperm global DNA methylation and leads to complex changes in non-coding miRNAs in sperm. WT male offspring of +/− eNOS fathers had increased fasting insulin, increased insulin after glucose load and increased liver glycogen content. Since there were no alterations in the endocrine pancreas and liver is the main site of insulin clearance [33, 34], we focused on the liver in our analysis. We found 19 genes differentially expressed in adult male offspring born to +/− eNOS fathers and WT mothers, with Gr and Pgc1a showing the lowest p value and FDR, whereas no differences in gene expression were seen in female offspring. DNA methylation of Gr exon 1A and Pgc1a promoter in male WT offspring of eNOS +/− fathers was decreased.
Offspring sex dependency of paternal eNOS deficiency
The phenotype of female WT offspring of +/− eNOS fathers was less pronounced. Only liver glycogen storage was increased. Sex dependency of the offspring’s phenotype is a common phenomenon in fetal programming [1]. One mechanism might be due to offspring sex-dependent transcriptional differences [1, 16]. During preimplantation development, male and female embryos display phenotypic differences that can only be attributed to the transcriptional differences resulting from their different sex chromosomes [35].
Opposite effects of maternal and paternal eNOS deficiency on glucose homeostasis
The same parental stimulus (heterozygous eNOS deficiency that was not transmitted to the next generation) causes different phenotypes in the offspring. Male WT offspring of +/− eNOS fathers developed a phenotype consisting of increased fasting insulin, increased liver glycogen storage and increased insulin secretion after glucose load. It is remarkable that the effect on fasting glucose seems to go in opposite directions in WT offspring of heterozygous eNOS-deficient mothers (see supplementary Table 2 of our previous publication [15] and Fig. 4). Fasting glucose was numerically lower in WT offspring of +/− eNOS mothers. In contrast, fasting glucose was significantly higher in WT offspring of +/− eNOS fathers compared with controls (Figs 3, 4). Epigenetic alterations were likewise different depending on the parental status of eNOS deficiency. In female WT offspring of +/− eNOS mothers liver fat content correlated significantly with fat storage-inducing transmembrane protein 1 (Fitm1) gene expression and Fitm1 methylation was significantly decreased, whereas DNA methylation of Gr exon 1A and Pgc1a promoter in male WT offspring of eNOS +/− fathers in comparison with controls was lower while corresponding gene expression was increased.
Taken together, in parental eNOS deficiency the offspring phenotype strongly depends on whether the genetic defect was present in the mother or in the father. The different consequences of the same parental genetic defect (eNOS deficiency) can probably be explained by different impacts of eNOS deficiency for the maturation of the egg or the intrauterine development of the embryo (Figs 1, 2) [1, 16]. Paternal eNOS deficiency affects maturation and development of the sperm and finally alters the epigenome of the sperm, potentially causing long-lasting secondary epigenetic alterations resulting in an adult phenotype characterised by increased fasting insulin, increased insulin after glucose load and increased liver glycogen content. Studies showing that a pre-conceptional paternal high-fat diet results in an impaired glucose tolerance in female offspring due to epigenetic sperm and target organ alterations [17,18,19, 21] fit well with our findings.
Polymorphisms in the human NOS3 gene (encoding eNOS) are associated with alterations in the composition of seminal plasma. eNOS deficiency-mediated changes in seminal plasma might thus also be a contributing factor [36, 37]. Maternal eNOS deficiency may affect egg maturation and intrauterine development [1, 15]. In this context, it is of note that parental diabetes has opposite effects on offspring birthweight [38], most likely due to the different effects of paternal and maternal diabetes on spermatogenesis and oocyte/intrauterine development, respectively.
Can the phenotype in male offspring be explained by an upregulation of GRs?
The hepatic phenotype in WT male offspring of heterozygous eNOS-deficient fathers is in agreement with studies showing that exposure to hepatic GR inhibition lowers glucose in ob/ob mice [39] and that hepatic GR blockade decreases glucose production and improves insulin resistance [40, 41]. Excess glucocorticoid exposure causes hyperglycaemia and insulin resistance. Our finding of no differences in the glucose response to glucose load in WT male offspring of heterozygous eNOS-deficient fathers vs WT offspring of WT parents but marked differences with respect to insulin levels between WT male offspring of heterozygous eNOS-deficient fathers and controls (Figs 3, 4) suggests that paternal eNOS deficiency causes insulin resistance in the WT male offspring of male heterozygous eNOS knockout mice. In the liver, glucocorticoids increase glycogen storage [42]. We assume that an increased expression of the hepatic GR may have similar consequences. It was reported that GR interacts with insulin degrading enzyme [43]. Since the liver is the primary site for insulin clearance [33, 34], upregulated hepatic GR expression might likewise lead to increased insulin levels. This pathway is androgen-dependent [43]. Hepatic androgen-dependent GR effects on insulin might explain the observed phenotype of increased plasma insulin levels in male WT offspring of male heterozygous eNOS knockout mice.
The human GR gene (also known as NR3C1) comprises nine exons in which exons 2 to 9 are the protein-encoding region. This gene has a long complex promoter region (the 5′ untranslated region) which is similar to the mouse and rat Gr gene [44]. The mouse Gr gene has five distinct promoter regions which are 1A, 1B, 1C, 1D and 1E. Exon 1A is found 32 kb upstream from exon 2, and its expression was only detected in tissues with high GR content [45, 46]. Thus, Gr exon 1A was selected for DNA methylation analysis and indeed we saw decreased methylation of Gr exon 1A and increased Gr gene expression in the liver of male WT offspring of eNOS-deficient fathers. The GR gene in particular has been shown to be sensitive to early-life environmental conditions, and this effect has been attributed to epigenetic mechanisms [47].
PGC1a methylation and gene expression
DNA methylation of the PGC1a gene promoter modulates insulin resistance and is strongly associated with plasma fasting insulin [48, 49]. A study in patients with non-alcoholic fatty liver disease showed that PGC1a promoter methylation was inversely correlated with liver PGC1a mRNA expression. In addition, PGC1a promoter methylation was inversely correlated with HOMA-IR, fasting glucose and insulin. PGC1a promoter methylation was also inversely correlated with PGC1a promoter methylation [50]. A study done in a rat fetal programming model likewise found an alteration in DNA methylation and transcription of Pgc1a. The genetic and epigenetic modifications of PGC1a provide a potential mechanism linking early-life nutrition insult to long-term metabolic disease susceptibility [51].
Pathophysiological and clinical implications
Many studies have indicated that insulin resistance can be caused by fetal programming. Also, paternal factors prior to mating may influence the epigenome of the sperm and hence the adult offspring’s phenotype [1, 16, 52, 53], as was observed in our study. Male heterozygous eNOS knockout mice might be a model of the human endothelial dysfunction sometimes observed in elderly fathers or fathers with hypertension [54,55,56,57]. If our findings can be translated to humans, paternal endothelial dysfunction in men might be a risk factor for developing insulin resistance in offspring. Our current study also supports the advanced fetal programming hypothesis as set out in our previous study [15], where we could demonstrate a maternal Nos3 gene-driven epigenetic alteration of the offspring’s phenotype. Our current study now proposes a non-environmental mechanism of fetal programming driven by altered paternal Nos3/NOS3 gene function [58, 59] primarily affecting the sperm epigenome and later in life the methylation of offspring target organ genes, resulting in our case in a liver phenotype.
Our current study and the previous study [15] have some general implications:
-
(1)
They break with the classical laws of inheritance. The phenotype of WT offspring born to either male or female heterozygous eNOS knockout mice should be identical to offspring from WT parents. However, this was not the case for offspring of either heterozygous eNOS knockout fathers as shown in this study or heterozygous eNOS knockout mothers as shown previously [15].
-
(2)
They challenge a key research tool developed to understand gene function: murine transgenic or knockout animal models. Genetically altered animal models may not only reflect causality between a certain genetic alteration and a resulting phenotype. Altered parental genes may additionally induce epigenetic changes affecting the offspring’s phenotype. This notion is supported by human genome-wide association studies [60]. The clinical implications of our study should be further investigated in monogenic inherited diseases such as thalassemia.
Study limitations
We used inbred mice for our experiments (see also ESM Fig. 1). However, founder fathers for the control group (WT offspring of WT fathers and WT mothers) and fathers for the investigated group (WT offspring of heterozygous eNOS fathers and WT mothers) are different. Thus, additional unknown genetic differences in the fathers used to generate the control group and the investigated group cannot be fully excluded. It is a study limitation that epigenetic changes caused by paternal NO deficiency were analysed at only two time points, in sperm and in the adult animal at the time of characterisation of the adult phenotype. Dynamic epigenetic changes, particularly during fetal development, and their impact on the adult phenotype are important topics of follow-up projects.
We performed IPGTT by measuring glucose and insulin at 0, 15 and 60 min and not for a longer duration on account of animal welfare. However, IPGTT for only 60 min showed differences among the groups and this duration for IPGTT was reported as a suitable approach previously [61,62,63]. Although group means for insulin AUC comparing WT male offspring born to heterozygous fathers and WT mothers with controls were clearly different (Fig. 3h), it needs to be mentioned that the variation of individual data in the groups was quite high. This might be due to the fact that this variable is calculated based on several measurements. There might be variations in the amount of glucose injected, the time of blood taking after glucose injection, the body weight of the individual animals as well as variation of the insulin ELISA used. All these variables potentiated the variability of insulin AUC. This is for example completely different from reports of organ weight. Here just the variability of the weight measurement method accounts for the variability of the variable. Moreover, we did not analyse fat tissue and muscles. Given the phenotype, this would have been of interest. Furthermore, we screened for differentially expressed genes by using a large but limited list of candidate genes known to be involved in liver fat and carbohydrate metabolism. We did this because the approach was successfully used in our initial study [15].
Conclusions
This study shows that paternal genes without passing on to the offspring can influence the offspring’s phenotype by altering the epigenome in the sperm and subsequently later in certain organs in adulthood. The same genetic defect in either the father or the mother without transmission to the next generation results in different offspring phenotypes. Our data specifically suggest that heterozygous eNOS deficiency in male mice might cause an unfavourable testicular environment influencing the sperm epigenome. These primary sperm epigenetic alterations may trigger long-lasting epigenetic and subsequent phenotypic alterations in offspring target organs (Fig. 1).
Abbreviations
- eNOS:
-
Endothelial nitric oxide synthase
- FDR:
-
False discovery rate
- GR:
-
Glucocorticoid receptor
- iNOS:
-
Inducible nitric oxide synthetase
- l-NAME:
-
N(γ)-nitro-l-arginine methyl ester
- MeDIP:
-
Methylated genomic DNA immunoprecipitation
- miRNA:
-
microRNA
- NO:
-
Nitric oxide
- PGC-1α:
-
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- WT:
-
Wild-type
References
Reichetzeder C, Dwi Putra SE, Li J, Hocher B (2016) Developmental origins of disease - crisis precipitates change. Cell Physiol Biochem 39(3):919–938 http://doi.org/000447801
Masuda K, Osada H, Iitsuka Y, Seki K, Sekiya S (2002) Positive association of maternal G protein beta3 subunit 825T allele with reduced head circumference at birth. Pediatr Res 52(5):687–691. https://doi.org/10.1203/00006450-200211000-00013
Hocher B, Slowinski T, Stolze T, Pleschka A, Neumayer HH, Halle H (2000) Association of maternal G protein beta3 subunit 825T allele with low birthweight. Lancet. 355(9211):1241–1242. https://doi.org/10.1016/S0140-6736(00)02094-8
Yadav U, Kumar P, Yadav SK, Mishra OP, Rai V (2015) Polymorphisms in folate metabolism genes as maternal risk factor for neural tube defects: an updated meta-analysis. Metab Brain Dis 30(1):7–24. https://doi.org/10.1007/s11011-014-9575-7
Liu J, Zhang Y, Jin L et al (2015) Variants in maternal COMT and MTHFR genes and risk of neural tube defects in offspring. Metab Brain Dis 30(2):507–513. https://doi.org/10.1007/s11011-014-9582-8
van Beynum IM, Kapusta L, den Heijer M et al (2006) Maternal MTHFR 677C>T is a risk factor for congenital heart defects: effect modification by periconceptional folate supplementation. Eur Heart J 27(8):981–987 http://doi.org/ehi815
Wang X, Zuckerman B, Pearson C et al (2002) Maternal cigarette smoking, metabolic gene polymorphism, and infant birth weight. JAMA 287(2):195–202 http://doi.org/joc10264
Cohen IL, Liu X, Lewis ME et al (2011) Autism severity is associated with child and maternal MAOA genotypes. Clin Genet 79(4):355–362 http://doi.org/CGE1471
Torres-Espinola FJ, Altmae S, Segura MT et al (2015) Maternal PPARG Pro12Ala polymorphism is associated with infant's neurodevelopmental outcomes at 18 months of age. Early Hum Dev 91(8):457–462 http://doi.org/S0378-3782(15)00100-0
Miodovnik A, Diplas AI, Chen J, Zhu C, Engel SM, Wolff MS (2012) Polymorphisms in the maternal sex steroid pathway are associated with behavior problems in male offspring. Psychiatr Genet 22(3):115–122. https://doi.org/10.1097/YPG.0b013e328351850b
Tsai HJ, Liu X, Mestan K et al (2008) Maternal cigarette smoking, metabolic gene polymorphisms, and preterm delivery: new insights on GxE interactions and pathogenic pathways. Hum Genet 123(4):359–369. https://doi.org/10.1007/s00439-008-0485-9
Danileviciute A, Grazuleviciene R, Paulauskas A, Nadisauskiene R, Nieuwenhuijsen MJ (2012) Low level maternal smoking and infant birthweight reduction: genetic contributions of GSTT1 and GSTM1 polymorphisms. BMC Pregnancy Childbirth 12:161 http://doi.org/1471-2393-12-161
Wu T, Hu Y, Chen C et al (2007) Passive smoking, metabolic gene polymorphisms, and infant birth weight in a prospective cohort study of Chinese women. Am J Epidemiol 166(3):313–322 http://doi.org/kwm090
Cassina M, Salviati L, Di Gianantonio E, Clementi M (2012) Genetic susceptibility to teratogens: state of the art. Reprod Toxicol 34(2):186–191 http://doi.org/S0890-6238(12)00119-0
Hocher B, Haumann H, Rahnenfuhrer J et al (2016) Maternal eNOS deficiency determines a fatty liver phenotype of the offspring in a sex dependent manner. Epigenetics 11(7):539–552. https://doi.org/10.1080/15592294.2016.1184800
Li J, Tsuprykov O, Yang X, Hocher B (2016) Paternal programming of offspring cardiometabolic diseases in later life. J Hypertens 34(11):2111–2126. https://doi.org/10.1097/HJH.0000000000001051
Ng SF, Lin RC, Laybutt DR, Barres R, Owens JA, Morris MJ (2010) Chronic high-fat diet in fathers programs beta-cell dysfunction in female rat offspring. Nature 467(7318):963–966 http://doi.org/nature09491
Chen Q, Yan M, Cao Z et al (2016) Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science 351(6271):397–400 https://doi.org/10.1126/science.aad7977
Terashima M, Barbour S, Ren J, Yu W, Han Y, Muegge K (2015) Effect of high fat diet on paternal sperm histone distribution and male offspring liver gene expression. Epigenetics 10(9):861–871. https://doi.org/10.1080/15592294.2015.1075691
Yan M, Zhai Q (2016) Sperm tsRNAs and acquired metabolic disorders. J Endocrinol 230(3):F13–F18 http://doi.org/JOE-16-0185
Nembhard WN, Tang X, Li J et al (2018) A parent-of-origin analysis of paternal genetic variants and increased risk of conotruncal heart defects. Am J Med Genet A 176(3):609–617. https://doi.org/10.1002/ajmg.a.38611
Lesch BJ, Tothova Z, Morgan EA et al (2019) Intergenerational epigenetic inheritance of cancer susceptibility in mammals. eLife 8:e39380. https://doi.org/10.7554/eLife.39380
Liu C, Liu H, Zhang H et al (2021) Paternal USP26 mutations raise Klinefelter syndrome risk in the offspring of mice and humans. EMBO J 40(13):e106864. https://doi.org/10.15252/embj.2020106864
Nelson VR, Spiezio SH, Nadeau JH (2010) Transgenerational genetic effects of the paternal Y chromosome on daughters' phenotypes. Epigenomics 2(4):513–521. https://doi.org/10.2217/epi.10.26
Godecke A, Decking UK, Ding Z et al (1998) Coronary hemodynamics in endothelial NO synthase knockout mice. Circ Res 82(2):186–194. https://doi.org/10.1161/01.res.82.2.186
Reichetzeder C, Dwi Putra SE, Pfab T et al (2016) Increased global placental DNA methylation levels are associated with gestational diabetes. Clin Epigenetics 8:82. https://doi.org/10.1186/s13148-016-0247-9
Peng H, Shi J, Zhang Y et al (2012) A novel class of tRNA-derived small RNAs extremely enriched in mature mouse sperm. Cell Res 22(11):1609–1612. https://doi.org/10.1038/cr.2012.141
Chen Q, Yan M, Cao Z et al (2016) Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science 351(6271):397–400. https://doi.org/10.1126/science.aad7977
Bezborodkina NN, Chestnova AY, Vorobev ML, Kudryavtsev BN (2016) Glycogen content in hepatocytes is related with their size in normal rat liver but not in cirrhotic one. Cytometry A 89(4):357–364. https://doi.org/10.1002/cyto.a.22811
Weber M, Davies JJ, Wittig D et al (2005) Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet 37(8):853–862. https://doi.org/10.1038/ng1598
Xia J, Wishart DS (2016) Using MetaboAnalyst 3.0 for comprehensive metabolomics data analysis. Curr Protoc Bioinformatics 55:14.10.1-14.10.91. https://doi.org/10.1002/cpbi.11
Chong J, Wishart DS, Xia J (2019) Using MetaboAnalyst 4.0 for comprehensive and integrative metabolomics data analysis. Curr Protoc Bioinformatics 68(1):e86. https://doi.org/10.1002/cpbi.86
Duckworth WC, Bennett RG, Hamel FG (1998) Insulin degradation: progress and potential. Endocr Rev 19(5):608–624. https://doi.org/10.1210/edrv.19.5.0349
Najjar SM, Perdomo G (2019) Hepatic insulin clearance: mechanism and physiology. Physiology (Bethesda) 34(3):198–215. https://doi.org/10.1152/physiol.00048.2018
Bermejo-Alvarez P, Rizos D, Lonergan P, Gutierrez-Adan A (2011) Transcriptional sexual dimorphism during preimplantation embryo development and its consequences for developmental competence and adult health and disease. Reproduction 141(5):563–570 http://doi.org/REP-10-0482
Watkins AJ, Dias I, Tsuro H et al (2018) Paternal diet programs offspring health through sperm- and seminal plasma-specific pathways in mice. Proc Natl Acad Sci U S A 115(40):10064–10069. https://doi.org/10.1073/pnas.1806333115
Mostafa T, Rashed LA, Nabil N, Fouad H, Sabry D, El-Saied DM (2015) Endothelial nitric oxide synthase gene polymorphism relationship with semen parameters and oxidative stress in infertile oligoasthenoteratozoospermic men. Urology 85(5):1058–1061 http://doi.org/S0090-4295(15)00092-8
Hypponen E, Smith GD, Power C (2003) Parental diabetes and birth weight of offspring: intergenerational cohort study. BMJ 326(7379):19–20. https://doi.org/10.1136/bmj.326.7379.19
Jacobson PB, von Geldern TW, Ohman L et al (2005) Hepatic glucocorticoid receptor antagonism is sufficient to reduce elevated hepatic glucose output and improve glucose control in animal models of type 2 diabetes. J Pharmacol Exp Ther 314(1):191–200 http://doi.org/jpet.104.081257
Zinker B, Mika A, Nguyen P et al (2007) Liver-selective glucocorticoid receptor antagonism decreases glucose production and increases glucose disposal, ameliorating insulin resistance. Metabolism 56(3):380–387. https://doi.org/10.1016/j.metabol.2006.10.021
Priyadarshini E, Anuradha CV (2017) Glucocorticoid antagonism reduces insulin resistance and associated lipid abnormalities in high-fructose-fed mice. Can J Diabetes 41(1):41–51 http://doi.org/S1499-2671(15)30054-X
Kuo T, McQueen A, Chen TC, Wang JC (2015) Regulation of glucose homeostasis by glucocorticoids. Adv Exp Med Biol 872:99–126. https://doi.org/10.1007/978-1-4939-2895-8_5
Kupfer SR, Wilson EM, French FS (1994) Androgen and glucocorticoid receptors interact with insulin degrading enzyme. J Biol Chem 269(32):20622–20628
Vandevyver S, Dejager L, Libert C (2014) Comprehensive overview of the structure and regulation of the glucocorticoid receptor. Endocr Rev 35(4):671–693. https://doi.org/10.1210/er.2014-1010
Strahle U, Schmidt A, Kelsey G et al (1992) At least three promoters direct expression of the mouse glucocorticoid receptor gene. Proc Natl Acad Sci U S A 89(15):6731–6735. https://doi.org/10.1073/pnas.89.15.6731
Purton JF, Monk JA, Liddicoat DR et al (2004) Expression of the glucocorticoid receptor from the 1A promoter correlates with T lymphocyte sensitivity to glucocorticoid-induced cell death. J Immunol 173(6):3816–3824 http://doi.org/173/6/3816
Turner JD, Alt SR, Cao L et al (2010) Transcriptional control of the glucocorticoid receptor: CpG islands, epigenetics and more. Biochem Pharmacol 80(12):1860–1868 http://doi.org/S0006-2952(10)00471-5
Sookoian S, Pirola CJ (2012) DNA methylation and hepatic insulin resistance and steatosis. Curr Opin Clin Nutr Metab Care 15(4):350–356. https://doi.org/10.1097/MCO.0b013e3283546f9f
Sookoian S, Pirola CJ (2013) Epigenetics of insulin resistance: an emerging field in translational medicine. Curr Diab Rep 13(2):229–237. https://doi.org/10.1007/s11892-012-0361-9
Sookoian S, Rosselli MS, Gemma C et al (2010) Epigenetic regulation of insulin resistance in nonalcoholic fatty liver disease: impact of liver methylation of the peroxisome proliferator-activated receptor gamma coactivator 1alpha promoter. Hepatology 52(6):1992–2000. https://doi.org/10.1002/hep.23927
Xie X, Lin T, Zhang M et al (2015) IUGR with infantile overnutrition programs an insulin-resistant phenotype through DNA methylation of peroxisome proliferator-activated receptor-gamma coactivator-1alpha in rats. Pediatr Res 77(5):625–632. https://doi.org/10.1038/pr.2015.32
Gluckman PD, Hanson MA, Cooper C, Thornburg KL (2008) Effect of in utero and early-life conditions on adult health and disease. N Engl J Med 359(1):61–73. https://doi.org/10.1056/NEJMra0708473
Ravelli AC, van der Meulen JH, Michels RP et al (1998) Glucose tolerance in adults after prenatal exposure to famine. Lancet 351(9097):173–177 http://doi.org/S0140673697072449
Khandwala YS, Baker VL, Shaw GM, Stevenson DK, Lu Y, Eisenberg ML (2018) Association of paternal age with perinatal outcomes between 2007 and 2016 in the United States: population based cohort study. BMJ 363:k4372. https://doi.org/10.1136/bmj.k4372
Kasman AM, Zhang CA, Li S et al (2021) Association between preconception paternal health and pregnancy loss in the USA: an analysis of US claims data. Hum Reprod 36(3):785–793. https://doi.org/10.1093/humrep/deaa332
Kasman AM, Zhang CA, Li S, Stevenson DK, Shaw GM, Eisenberg ML (2020) Association of preconception paternal health on perinatal outcomes: analysis of U.S. claims data. Fertil Steril 113(5):947–954. https://doi.org/10.1016/j.fertnstert.2019.12.026
Crump C (2020) An overview of adult health outcomes after preterm birth. Early Hum Dev 150:105187. https://doi.org/10.1016/j.earlhumdev.2020.105187
Hocher B, Slowinski T, Bauer C, Halle H (2001) The advanced fetal programming hypothesis. Nephrol Dial Transplant 16(6):1298–1299. https://doi.org/10.1093/ndt/16.6.1298
Hocher B (2014) More than genes: the advanced fetal programming hypothesis. J Reprod Immunol 104-105:8–11. https://doi.org/10.1016/j.jri.2014.03.001
Warrington NM, Beaumont RN, Horikoshi M et al (2019) Maternal and fetal genetic effects on birth weight and their relevance to cardio-metabolic risk factors. Nat Genet 51(5):804–814. https://doi.org/10.1038/s41588-019-0403-1
Wang Q, Chen K, Liu R et al (2010) Novel GLP-1 fusion chimera as potent long acting GLP-1 receptor agonist. PLoS One 5(9):e12734. https://doi.org/10.1371/journal.pone.0012734
Kim BJ, Zhou J, Martin B et al (2010) Transferrin fusion technology: a novel approach to prolonging biological half-life of insulinotropic peptides. J Pharmacol Exp Ther 334(3):682–692. https://doi.org/10.1124/jpet.110.166470
Du X, Kosinski JR, Lao J et al (2012) Differential effects of oxyntomodulin and GLP-1 on glucose metabolism. Am J Physiol Endocrinol Metab 303(2):E265–E271. https://doi.org/10.1152/ajpendo.00142.2012
Chen JC, Johnson BA, Erikson DW et al (2014) Seminal plasma induces global transcriptomic changes associated with cell migration, proliferation and viability in endometrial epithelial cells and stromal fibroblasts. Hum Reprod 29(6):1255–1270 http://doi.org/deu047
Rodgers AB, Morgan CP, Leu NA, Bale TL (2015) Transgenerational epigenetic programming via sperm microRNA recapitulates effects of paternal stress. Proc Natl Acad Sci U S A 112(44):13699–13704 http://doi.org/1508347112
Data availability
All data generated or analysed during this study are included in this published article (and its supplementary information files).
Funding
Open Access funding enabled and organized by Projekt DEAL. This study was supported by the Deutsche Forschungsgemeinschaft to BH and partially by the National Natural Science Foundation of China (grant no. 81873861) as well as the Huxiang Young Talents project (2021RC3094).
Authors’ relationships and activities
The authors declare that there are no relationships or activities that might bias, or be perceived to bias, their work.
Contribution statement
BH designed the study, wrote grant applications and got funding for this study. AAH and BH wrote the manuscript. BH, Y-PL, CR, XZ, OT, JR, LX, JL, LH, BKK and AAH provided feedback to the initial draft. Y-PL, CR, XZ, OT and JR did animal breeding and all animal procedures. Y-PL, CR, XZ, OT, JR, LX, JL and LH performed experiments. Data analysis was done by JR, LH, BKK, AAH and BH. All authors gave final approval of the version to be published. BH is the guarantor of the entire study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM
(PDF 240 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hocher, B., Lu, YP., Reichetzeder, C. et al. Paternal eNOS deficiency in mice affects glucose homeostasis and liver glycogen in male offspring without inheritance of eNOS deficiency itself. Diabetologia 65, 1222–1236 (2022). https://doi.org/10.1007/s00125-022-05700-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-022-05700-x