
Abstract
Aims/hypothesis
Higher maternal BMI during pregnancy is associated with higher offspring birthweight, but it is not known whether this is solely the result of adverse metabolic consequences of higher maternal adiposity, such as maternal insulin resistance and fetal exposure to higher glucose levels, or whether there is any effect of raised adiposity through non-metabolic (e.g. mechanical) factors. We aimed to use genetic variants known to predispose to higher adiposity, coupled with a favourable metabolic profile, in a Mendelian randomisation (MR) study comparing the effect of maternal ‘metabolically favourable adiposity’ on offspring birthweight with the effect of maternal general adiposity (as indexed by BMI).
Methods
To test the causal effects of maternal metabolically favourable adiposity or general adiposity on offspring birthweight, we performed two-sample MR. We used variants identified in large, published genetic-association studies as being associated with either higher adiposity and a favourable metabolic profile, or higher BMI (n = 442,278 and n = 322,154 for metabolically favourable adiposity and BMI, respectively). We then extracted data on the metabolically favourable adiposity and BMI variants from a large, published genetic-association study of maternal genotype and offspring birthweight controlling for fetal genetic effects (n = 406,063 with maternal and/or fetal genotype effect estimates). We used several sensitivity analyses to test the reliability of the results. As secondary analyses, we used data from four cohorts (total n = 9323 mother–child pairs) to test the effects of maternal metabolically favourable adiposity or BMI on maternal gestational glucose, anthropometric components of birthweight and cord-blood biomarkers.
Results
Higher maternal adiposity with a favourable metabolic profile was associated with lower offspring birthweight (−94 [95% CI −150, −38] g per 1 SD [6.5%] higher maternal metabolically favourable adiposity, p = 0.001). By contrast, higher maternal BMI was associated with higher offspring birthweight (35 [95% CI 16, 53] g per 1 SD [4 kg/m2] higher maternal BMI, p = 0.0002). Sensitivity analyses were broadly consistent with the main results. There was evidence of outlier SNPs for both exposures; their removal slightly strengthened the metabolically favourable adiposity estimate and made no difference to the BMI estimate. Our secondary analyses found evidence to suggest that a higher maternal metabolically favourable adiposity decreases pregnancy fasting glucose levels while a higher maternal BMI increases them. The effects on neonatal anthropometric traits were consistent with the overall effect on birthweight but the smaller sample sizes for these analyses meant that the effects were imprecisely estimated. We also found evidence to suggest that higher maternal metabolically favourable adiposity decreases cord-blood leptin while higher maternal BMI increases it.
Conclusions/interpretation
Our results show that higher adiposity in mothers does not necessarily lead to higher offspring birthweight. Higher maternal adiposity can lead to lower offspring birthweight if accompanied by a favourable metabolic profile.
Data availability
The data for the genome-wide association studies (GWAS) of BMI are available at https://portals.broadinstitute.org/collaboration/giant/index.php/GIANT_consortium_data_files. The data for the GWAS of body fat percentage are available at https://walker05.u.hpc.mssm.edu.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.

Introduction
Higher maternal BMI, an index of general adiposity, is strongly associated with higher offspring birthweight [1]. Mendelian randomisation (MR) studies support these associations as causal [2, 3]. High birthweight is associated with adverse perinatal and neonatal outcomes [4].
A likely mechanism for the association of higher maternal BMI with higher offspring birthweight is via its adverse metabolic consequences. For example, higher maternal BMI results in greater maternal insulin resistance and consequently higher maternal circulating glucose levels. As glucose crosses the placenta via facilitated diffusion, this results in increased insulin secretion by the fetus (maternal insulin cannot cross the placenta [5]). Insulin acts as a growth factor (i.e. higher fetal insulin secretion leads to increased fetal skeletal growth and fat deposition), resulting in higher birthweight [6].
Common genetic variants provide a useful tool to investigate the relationship between maternal adiposity and offspring birthweight. Genetic variants have been identified where one allele is associated with higher insulin resistance and with distributions of metabolic and adiposity traits that are consistent with a ‘metabolically obese’ phenotype (i.e. higher triacylglycerols and higher visceral/subcutaneous adiposity ratio) [7]. These associations are consistent with the ‘tissue expandability’ hypothesis, which states that population-level differences in the association between adiposity and type 2 diabetes are due to differences in the ability of adipocytes to absorb additional fatty acids from circulation, and people with either fewer adipocytes or adipocytes that are less able to increase in size (i.e. less ‘expandable’ adipose tissue) store greater fat in the visceral organs, leading to insulin resistance. The opposite allele at each of the identified genetic variants is associated with a ‘metabolically favourable adiposity’ phenotype linked to higher body fat percentage and a lower risk of type 2 diabetes [8]. In an expanded set of such variants, the metabolically favourable adiposity alleles were also associated with higher subcutaneous adipose tissue and lower ectopic liver fat [9].
It is unknown whether the metabolically favourable adiposity alleles in pregnant women affect offspring birthweight. Lack of a positive association between maternal metabolically favourable adiposity alleles and offspring birthweight would be compatible with the hypothesis that the effect of maternal BMI on birthweight is driven by the metabolic consequences of general adiposity and not by adiposity per se.
Our aim was to determine the effect of metabolically favourable adiposity on birthweight and to compare this with the effect of maternal general adiposity on birthweight. In our primary analysis, we used alleles associated with higher maternal metabolically favourable adiposity as genetic instruments to measure the effect of maternal adiposity on birthweight when coupled with ‘favourable’ metabolic effects, using data from large genome-wide association studies (GWAS) [2, 9]. We hypothesised that higher maternal metabolically favourable adiposity would either not associate with birthweight or would associate with lower birthweight if it resulted in lower maternal circulating glucose levels. In a secondary (exploratory) study, we used available individual-level data on mothers and babies to explore potential effects of maternal favourable vs general adiposity on birthweight-related metabolic (e.g. maternal glucose, cord insulin) and anthropometric (e.g. head circumference, skinfold thickness) traits.
Methods
To estimate the causal effect of maternal metabolically favourable adiposity or BMI (exposures) on offspring birthweight (outcome), we used two-sample MR with GWAS summary statistics. In this method, estimates of SNP–exposure associations are first obtained for a set of SNPs that are robustly associated with the exposure (metabolically favourable adiposity or BMI). Second, associations between the same SNPs and outcome (birthweight) are extracted from existing GWAS datasets. For each SNP, the SNP–outcome association is then divided by the SNP–exposure association. These ratios are then pooled to give an estimate of the causative effect of the exposure on the outcome.
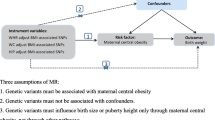
The study design and different data sources are summarised in Fig. 1 and electronic supplemental material (ESM) Fig. 1. Ethical approval and informed consent from participants were obtained in all of the studies included in this research.

Diagram summarising the key data sources and analysis steps for the primary analyses. aThe GWAS data used came from Ji et al. [9] (favourable adiposity), Locke et al. [11] (BMI), Lu et al. [10] (body fat percentage) and Warrington et al. [2] (birthweight). bThe SNP associations were harmonised to the exposure-increasing allele [34]
Data sources
Genetic predictors of metabolically favourable adiposity
Metabolically favourable adiposity SNPs were identified from the most recent GWAS (N = 442,278) [9]. The GWAS of metabolically favourable adiposity uses a composite phenotype characterised by increased body fat percentage and a metabolic profile related to a lower risk of type 2 diabetes, hypertension and heart disease (see ESM Methods: Deriving metabolically favourable adiposity phenotype and genetic variants, and Ji et al. [9] for more details); 14 SNPs associated with higher body fat percentage and a ‘favourable’ metabolic profile were identified at p < 5 × 10−8 and replicated [9]. To facilitate the interpretation of our results we weighted these SNPs by the effect estimates of their association with body fat percentage using the latest GWAS of body fat percentage [10].
Genetic predictors of BMI
We used 76 BMI SNPs as instruments for general adiposity from the most recent GWAS of BMI that did not include the UK Biobank (UKB) sample (N = 322,154), with their weights being extracted from the same GWAS [11]. We did not use the more recent GWAS that did include UKB [12], as that would have resulted in an overlap between sample 1 (genetic instruments-BMI) and sample 2 (genetic instruments-birthweight), which could result in overfitting of the data, and bias towards confounded associations, in the presence of weak instruments [13]. Additional details of the metabolically favourable adiposity and BMI GWAS are provided in ESM Table 1 and the characteristics of the SNPs used in our MR analyses are shown in ESM Table 2.
Genetic associations with offspring birthweight
We used the latest GWAS of offspring birthweight from the Early Growth Genetics (EGG) consortium. A total of 406,063 participants contributed to the weighted linear model (WLM) analyses (see below), of which 101,541 were UKB participants who reported their own birthweight and birthweight of their first child, 195,815 were UKB and EGG participants with their own birthweight data, and 108,707 were UKB and EGG participants with offspring birthweight data. Further details are given in Table 1, and ESM Methods: Study Descriptions and ESM Methods: Defining offspring birthweight for GWAS (which also includes details of participant consent and ethics approval) [2].
Birthweight was standardised within UKB and each of the EGG cohorts so that SNP effect sizes are in SD units (1 SD of birthweight ≈484 g, the median SD for birthweight in 18 studies in an early birthweight GWAS [14]).
Genetic associations with other outcomes
Four birth cohorts were used to perform secondary analyses on birth anthropometric and cord-blood outcomes, including the Avon Longitudinal Study of Parents and Children (ALSPAC) [15, 16], Born in Bradford (BiB) [17], the Exeter Family Study of Childhood Health (EFSOCH) [18] and Hyperglycemia and Adverse Pregnancy Outcome (HAPO) study [19] (maximum N = 9323 mother–child pairs).
We used published GWAS summary statistics from the Meta-Analyses of Glucose and Insulin-related traits Consortium (MAGIC) to investigate the effects of the metabolically favourable adiposity and BMI SNPs on fasting glucose. The MAGIC consortium GWAS reported data from 46,186 White European adults (including men and non-pregnant women) on fasting glucose from 17 population cohorts and four case–control studies in the discovery dataset (there were 122,743 adults in the total dataset) [20]. We checked the consistency of these associations with those for pregnancy fasting glucose in BiB, EFSOCH and HAPO (pregnancy fasting glucose data was not available for ALSPAC).
Further information on these cohorts and their contribution to the study can be found in Table 2, ESM Table 3 and ESM Fig. 1. For the cohort descriptions and additional information on participant consent, ethics approval and data extraction, see ESM Methods: Study descriptions. For additional information on how ethnicity was defined for each study, see ESM Methods: Selecting participants of European ancestry. For additional information on genotyping, see ESM Methods: Genotyping. For additional information on phenotype assessment, see ESM Methods: Measuring cord-blood outcomes in selected birth cohorts for secondary analyses; and ESM Methods: Measuring pregnancy glucose outcomes in selected cohorts for secondary analyses.
Data analyses
Our primary analysis was to study the effect of maternal metabolically favourable adiposity and BMI on offspring birthweight in the UKB and EGG meta-analysis. In addition, we undertook exploratory analyses on the effects of metabolically favourable adiposity and BMI on other outcomes (see Table 2 and ESM Fig. 1).
Adjusting for the fetal genotype
To avoid violating the third assumption of MR due to fetal genetic effects, we needed to adjust for the effects of the fetal genotype on the outcomes (see ESM Methods: Potential violation of MR assumptions by the fetal genotype).
For the primary analyses, to ensure our analyses considered only the effect of the maternal genotype and not the correlated fetal genotype, we used the maternal genetic effect on offspring birthweight that had been estimated using a WLM [2]. The WLM is a linear approximation of a structural equation modelling (SEM) technique that was developed to combine data from disparate study designs to estimate independent maternal and fetal genetic effects, equivalent to conditional analysis in mother–child pairs. The WLM/SEM method combines studies with own genotype data available in addition to own birthweight and offspring birthweight data, with data from studies with only their own birthweight or offspring birthweight.
Further details about WLM/SEM, and methods used to confirm that we obtained similar causal effect estimates with both the WLM- and SEM-adjusted summary statistics for birthweight, are given in ESM Methods: Structural equation modelling (SEM) and weighted linear modelling theory and ESM Methods: Extracting own birthweight data in UK Biobank (see also [2, 21]).
For the secondary analyses, the maternal genotype effects on offspring outcomes were directly adjusted for the fetal genotype, as both mother and offspring genotypes were available for the four cohorts used.
MR analysis
For the primary analyses, we performed two-sample MR using Wald ratios [22], which were calculated by dividing each SNP’s effect on offspring birthweight (maternal genetic effect estimated using WLM) by the same SNP’s effect on the exposure (maternal metabolically favourable adiposity or BMI). Standard errors were calculated by dividing the WLM/SEM-defined standard error of the SNP’s effect on offspring birthweight by each SNP’s effect on the exposure.
The resulting effect estimates from our MR analyses are reported as the mean difference in offspring birthweight per 1 SD higher maternal body fat percentage (1 SD of body fat percentage = 6.5%; see ESM Methods: Defining a 1 SD increase in body fat percentage) for metabolically favourable adiposity, and the mean difference in offspring birthweight per 1 SD increase in maternal BMI (1 SD of BMI = 4 kg/m2 [3]) for BMI.
For the secondary analyses, we used fixed effect pooled Wald ratio analysis to determine the effect of maternal metabolically favourable adiposity and BMI on offspring outcomes in each of these studies and compared the pooled result of that with the same result in our main analyses of effects on birthweight.
Sensitivity and additional analysis
For both the primary and secondary analyses, we performed additional sensitivity analyses to assess the validity of the genetic instrumental variables. This mostly included methods to test the assumptions of two-sample MR (such as Cochran’s Q and I2, leave-one-out analyses [23], MR-Egger [24], weighted-median [25] and Radial MR [26]). To further explore the hypothesis that glucose mediates the effect of BMI on birthweight, we performed a multivariable MR analysis [27] for adjusting the effect of BMI on birthweight. This could not be done for metabolically favourable adiposity, due to the fact that it is a composite trait in which insulin resistance is one of the defining features. Further details are provided in the following ESM Methods sections: Overview of tests to explore potential violations of two-sample Mendelian randomisation; Sensitivity analyses to explore horizontal pleiotropy and additional sources of invalid instruments; BMI SNP validation; Collider bias test; Cross exposure analyses; Testing potential confounders and mediators; Multivariable MR analyses for potential confounders and mediators; and Multivariable MR analyses for glucose mediation.
Results
The associations between the SNPs analysed in the primary study and UKB + EGG birthweight are shown in ESM Table 4 and the association between the same SNPs and all the outcomes analysed for the secondary study (fasting glucose, pregnancy fasting glucose, pregnancy 2 h glucose, cord insulin, cord C-peptide, cord leptin, cord adiponectin, birthweight, birth length, ponderal index, head circumference, triceps skinfold thickness, subscapular skinfold thickness and sum of skinfold thickness) are shown in ESM Tables 5–7.
Maternal metabolically favourable adiposity and maternal general adiposity, indexed by BMI, have opposite effects on offspring birthweight
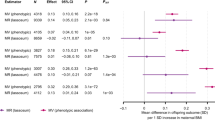
We found evidence that higher maternal metabolically favourable adiposity causes lower offspring birthweight (Fig. 2). The main estimate (−94 g [95% CI −150, −38]) of difference in mean birthweight per 1 SD (6.5%) higher maternal metabolically favourable body fat percentage (p = 0.001) was consistent with both the MR-Egger and weighted-median estimates (Fig. 2). There was evidence of heterogeneity between the Wald ratios across the SNPs (Cochran’s Q = 33.46 [degrees of freedom (d.f.) =13], I2 = 61.1%, p = 0.001), yet results were consistent across leave-one-out analysis (ESM Fig. 2). Using the SEM method to adjust for fetal genotype effects also gave very similar results (ESM Fig. 3).

Causative effect estimates for maternal metabolically favourable adiposity on offspring birthweight. The methods (Wald ratio, MR-Egger and weighted-median analysis) used WLM-adjusted estimates to account for offspring genotype. The x-axis shows the change in birthweight (g) per 1 SD increase in body fat percentage (6.5%)
The two-sample MR estimates for BMI are consistent with higher maternal general adiposity leading to higher offspring birthweight (Fig. 3). The main MR estimate was 35 g (95% CI 16, 53) of difference in mean birthweight per 1 SD (4 kg/m2) higher maternal BMI (p = 0.0002). MR-Egger (20 g [95% CI −52, 92], p = 0.576) and weighted-median (14 g [95% CI −20, 48], p = 0.424) estimates were directionally the same, though smaller than the main estimate (Fig. 3). In this analysis, there was also evidence of heterogeneity between the Wald ratios for each SNP (Cochran’s Q = 178.42 (d.f. = 75), I2 = 58%, p = 2 × 10−10), yet again results were consistent across leave-one-out analysis (ESM Fig. 4). Using the SEM method to adjust for fetal genotype gave very similar results (ESM Fig. 5).

Causative effect estimates for maternal BMI on offspring birthweight. The methods (Wald ratio, MR-Egger and weighted-median analysis) used WLM-adjusted estimates to account for offspring genotype. The x-axis shows the change in birthweight (g) per 1 SD increase in maternal BMI (4 kg/m2)
Using Radial MR, we identified four MR-Egger outlier SNPs for the metabolically favourable adiposity effect (ESM Fig. 6, ESM Fig. 7) and 18 outlier SNPs for BMI (ESM Fig. 8, ESM Fig. 9). In both cases the Radial MR results were consistent with the main results with the outliers removed.
Secondary study results
Higher metabolically favourable adiposity resulted in lower fasting glucose in men and non-pregnant women, while higher maternal BMI resulted in higher fasting glucose (Fig. 4). The relationship between maternal metabolically favourable adiposity or BMI and fasting glucose in pregnancy were consistent with this, as was 2 h postprandial glucose in pregnancy (Fig. 4). The point estimate for the effect of metabolically favourable adiposity on 2 h postprandial glucose was greater than that for fasting glucose but the CIs were wider (Fig. 4).

Causative effect estimates for maternal BMI and metabolically favourable adiposity on maternal pregnancy glucose outcomes. The x-axis shows the change in glucose outcomes per 1 SD increase in body fat percentage (6.5%) and BMI (4 kg/m2)
Higher maternal metabolically favourable adiposity consistently resulted in lower neonatal anthropometric measures, in particular lower infant head circumference (Fig. 5). In contrast, evidence suggested that maternal BMI consistently resulted in higher neonatal anthropometric measures (Fig. 5).

Causative effect estimates for maternal BMI and metabolically favourable adiposity on other birth anthropometric outcomes, adjusted for offspring genotype. The x-axis shows the change in outcomes in SDs per 1 SD increase in maternal body fat percentage (6.5%) and BMI (4 kg/m2)
Higher maternal metabolically favourable adiposity showed suggestive evidence of causing lower cord-blood leptin levels and, in contrast, there was strong evidence of higher maternal BMI resulting in higher cord-blood leptin levels (ESM Fig. 10). There was no detectable effect of maternal metabolically favourable adiposity or BMI on cord-blood insulin, C-peptide or adiponectin levels, though the small sample sizes made these results imprecise (ESM Fig. 10).
Validity of the genetic instrumental variables
The BMI SNPs were positively and consistently associated with pregnancy BMI across all of the cohorts used for our secondary analyses (ESM Fig. 11).
There was no evidence that collider bias influenced the results of maternal metabolically favourable adiposity on outcomes (ESM Table 8 and ESM Fig. 12), and the effect was directionally consistent irrespective of which adiposity exposure weights were used (ESM Fig. 13, ESM Fig. 14).
The metabolically favourable adiposity genetic score was not associated with maternal smoking or maternal education level in UKB, ALSPAC, EFSOCH or BiB but it was associated with lower female educational level in HAPO (ESM Table 9). The BMI genetic score was associated with a higher prevalence of current smoking status in women in UKB and pregnancy smoking status in ALSPAC and with lower female educational level in UKB (ESM Table 9). Given these findings, we undertook further analyses using multivariable MR to see whether maternal education and smoking status confounded the association. Results from the multivariable adjusted MR analyses were consistent with those from the unadjusted MR analyses, for both smoking and years of education, adjusted for individually and when both were included in the multivariable model (ESM Table 10).
Mediation analysis
In multivariable MR analyses, there was evidence that the effect of BMI on birthweight is mediated by its effect on fasting glucose, with the effect of BMI in the main inverse variance weighted (IVW) analyses (35 g [95% CI 6, 63] difference in mean birthweight per 1 SD increase in BMI, p = 0.02), attenuating with adjustment for fasting glucose (14 g [95% CI −18, 46] difference in mean birthweight per 1 SD increase in BMI, p = 0.39; ESM Table 11).
Discussion
We have found evidence that higher maternal favourable adiposity lowers offspring birthweight and that higher general adiposity (BMI) increases birthweight. Our secondary analyses provided some evidence that higher maternal favourable adiposity causes lower fasting glucose in contrast to the glucose-increasing effect of higher BMI. Additional analyses of effects of maternal favourable vs general adiposity on neonatal anthropometric measures, including head circumference, were consistent in direction with those of birthweight but larger sample sizes will be needed to determine whether effect sizes varied between measures that capture fat mass (e.g. skinfold thickness) vs lean mass (e.g. head circumference). There was evidence to suggest that cord-blood leptin levels are lowered by maternal favourable adiposity and raised by higher maternal BMI. There was insufficient power to detect precise effects on cord-blood markers of fetal insulin response, as the studies used different measures of this (insulin, C-peptide or adiponectin).
Metabolically favourable adiposity is a composite of individual traits, some of which have been found to be causally associated with birthweight. In particular, higher maternal fasting glucose (often resulting from insulin resistance, a component of the metabolically unfavourable adiposity trait) has consistently been found to be causally associated with higher offspring birthweight in MR studies [2, 3]. We have shown higher metabolically favourable adiposity to be tentatively associated with lower fasting glucose, suggesting that the effect of higher maternal metabolically favourable adiposity on lower offspring birthweight may be mediated by its effect on fasting glucose levels. Though we have focused on fasting glucose, metabolically favourable adiposity is also associated with other exposures, in particular lower triacylglycerol levels [9]. However, MR studies have failed to find any evidence of an association between maternal circulating triacylglycerol levels and offspring birthweight [3, 28]. Furthermore, glucose crosses the placenta by facilitated diffusion [29] and directly influences fetal growth; for this reason, our focus in this study was on maternal fasting glucose. Our multivariable MR analysis provides some evidence that higher maternal fasting glucose has a mediating role in the effect of maternal BMI on offspring birthweight. However, it is not possible to explore this mediation with metabolically favourable adiposity because of the composite nature of this exposure and because metabolically favourable adiposity SNPs were selected based on their indirect association with glucose traits (given insulin resistance is one of the defining features of this composite trait), therefore making the results difficult to interpret (see ESM Methods: Multivariable MR analyses for glucose mediation for more details).
The metabolically favourable adiposity phenotype used here is based on the adipose ‘tissue expandability’ hypothesis. Other metabolically favourable adiposity phenotypes have been developed that focus on BMI [30], BMI and WHR [31], or BMI, WHR and body fat percentage independently [32] rather than body fat percentage alone. Unlike the metabolically favourable adiposity phenotype used in this study, those studies used correlation rather than clustering analyses. Of the two studies that reported the genetic variants they identified, the phenotype reported by Winkler et al. 2018 [31] overlapped with four of the 14 loci used in this study (GRB14/COBLL1, VEGFA, CCDC92/DNAH10 and FAM13A) while Huang et al. 2021 [32] captured all but two of the loci used in this study (CITED2 and TRIB1/LRATD2). Neither of these have been used in MR studies of birthweight. The Winkler et al. 2018 [31] phenotype might not be comparable with our phenotype and could produce different results compared with this study, though the broader Huang et al. 2021 [32] phenotype might produce a similar result. Nonetheless, further studies are needed in future to test other genetic instruments for metabolically favourable adiposity phenotypes.
Study strengths and limitations
To the best of our knowledge, this is the first study to use MR to investigate the effect of maternal metabolically favourable adiposity on offspring birthweight. We used data from a large GWAS of birthweight and for the first time examined potential effects on maternal glucose traits as well as additional newborn anthropometric and cord-blood measurements in exploratory analyses. We explored the validity of our genetic instrumental variables using multiple sensitivity analyses, including the recently developed Radial MR method [26], and found that overall results from these sensitivity analyses were consistent with our main findings. Our BMI genetic variants explained 2.7% of the variance in BMI in the original GWAS cohort [11]. As metabolically favourable adiposity is not a directly measured single trait, it is not possible to measure how much ‘variance’ is explained by the genetic variants. However, for the primary study with birthweight the MR estimates were precise.
Only 5% of those invited participated in the UKB and offspring birthweight was reported several years later. However, the similarity of the UKB results with those from the four birth cohorts with response rates of at least 70% suggests that the UKB results are unlikely to be importantly biased. Self-report of own and first child’s birthweight and the rounding to the nearest one pound (~0.454 kg, first child’s birthweight only) may have introduced error in the birthweight measure in UKB, but this would be random with respect to genotype and would not be expected to substantially bias results. In UKB there is a relatively lower reported birthweight than in most of the other cohorts used in this study, likely reflecting secular trends of increasing birth size over time.
In UKB, genetically instrumented BMI was found to be associated with both educational attainment and smoking status. However, results of the multivariable IVW analyses of maternal BMI on offspring birthweight adjusted for smoking and years of education were not substantially different from the main result, suggesting the association was not heavily confounded (ESM Table 10).
While there was evidence of between-SNP heterogeneity of both metabolically favourable adiposity and BMI, results were directionally consistent across the main estimate, leave-one-out analyses, MR-Egger and weighted-median analyses. The Radial MR analyses for both maternal metabolically favourable adiposity and maternal BMI found evidence of SNPs with outlier effects (four for maternal metabolically favourable adiposity, 18 for maternal BMI). However, in both cases, removal of the outlier SNPs from the analyses resulted in estimates consistent with the main estimates, suggesting horizontal pleiotropy is unlikely to be a major source of bias for our analyses. In our secondary studies we had insufficient power to undertake these sensitivity analyses.
In conclusion, our results suggest that maternal metabolically favourable adiposity has the opposite effect on offspring birthweight to that of maternal BMI. This means that higher adiposity in mothers does not necessarily lead to higher offspring birthweight and may result in lower offspring birthweight if accompanied by a favourable metabolic profile. In the future, methods to stratify overweight and obese pregnant women by their metabolically favourable adiposity status could allow for targeted interventions to achieve healthy birthweight.
Data availability
Our study uses two-sample MR. We used both published summary results (i.e. taking results from published research papers and websites) and individual participant cohort data: journal-published and website summary data were used for sample one of the two-sample MR (published GWAS of BMI and body fat percentage). The references to the published data sources are provided in the main paper. The data for the GWAS of BMI are available at https://portals.broadinstitute.org/collaboration/giant/index.php/GIANT_consortium_data_files. The data for the GWAS of body fat percentage are available at https://walker05.u.hpc.mssm.edu. We used individual participant data for the second MR sample and for undertaking sensitivity analyses from the UKB, ALSPAC, BiB, EFSOCH and HAPO cohorts. The data in UKB, ALSPAC and BiB are fully available, via managed systems, to any researchers. The managed system for both studies is a requirement of the study funders but access is not restricted on the basis of overlap with other applications to use the data or on the basis of peer review of the proposed science. Researchers have to pay for a dataset to be prepared for them. Full information on how to access UKB data can be found at www.ukbiobank.ac.uk/using-the-resource/. The ALSPAC data management plan (www.bristol.ac.uk/alspac/researchers/data-access/documents/alspac-data-management-plan.pdf) describes, in detail, the policy regarding data sharing, which is through a system of managed open access. The following steps highlight how to apply for access to the data included in this paper and all other ALSPAC data: (1) please read the ALSPAC access policy (PDF, 627 kB), which describes the process of accessing the data and samples in detail, and outlines the costs associated with doing so; (2) you may also find it useful to browse the fully searchable ALSPAC research proposals database, which lists all research projects that have been approved since April 2011; (3) please submit your research proposal for consideration by the ALSPAC Executive Committee and you will receive a response within 10 working days to advise you whether your proposal has been approved. If you have any questions about accessing data, please e-mail alspac-data@bristol.ac.uk. Full information on how to access BiB data can be found at https://borninbradford.nhs.uk/research/how-to-access-data/. Requests for access to the original EFSOCH dataset should be made in writing in the first instance to the EFSOCH data team via the Exeter Clinical Research Facility (crf@exeter.ac.uk).
Abbreviations
- ALSPAC:
-
Avon Longitudinal Study of Parents and Children
- BiB:
-
Born in Bradford
- d.f:
-
Degrees of freedom
- EFSOCH:
-
Exeter Family Study of Childhood Health
- EGG:
-
Early Growth Genetics
- GWAS:
-
Genome-wide association studies
- HAPO:
-
Hyperglycemia and Adverse Pregnancy Outcomes
- IVW:
-
Inverse variance weighted
- MAGIC:
-
Meta-Analysis of Glucose and Insulin-related traits Consortium
- MR:
-
Mendelian randomisation
- SEM:
-
Structural equation modelling
- UKB:
-
UK Biobank
- WLM:
-
Weighted linear model
References
McDonald SD, Han Z, Mulla S, Beyene J (2010) Overweight and obesity in mothers and risk of preterm birth and low birth weight infants: systematic review and meta-analyses. BMJ 341:c3428. https://doi.org/10.1136/bmj.c3428
Warrington NM, Beaumont RN, Horikoshi M et al (2019) Maternal and fetal genetic effects on birth weight and their relevance to cardio-metabolic risk factors. Nat Genet 51(5):804–814. https://doi.org/10.1038/s41588-019-0403-1
Tyrrell J, Richmond RC, Palmer TM et al (2016) Genetic evidence for causal relationships between maternal obesity-related traits and birth weight. JAMA 315(11):1129–1140. https://doi.org/10.1001/jama.2016.1975
Rossi AC, Mullin P, Prefumo F (2013) Prevention, management, and outcomes of macrosomia: a systematic review of literature and meta-analysis. Obstet Gynecol Surv 68(10):702–709. https://doi.org/10.1097/01.ogx.0000435370.74455.a8
Lawlor DA, Relton C, Sattar N, Nelson SM (2012) Maternal adiposity--a determinant of perinatal and offspring outcomes? Nat Rev Endocrinol 8(11):679–688. https://doi.org/10.1038/nrendo.2012.176
The HAPO Study Cooperative Research Group (2009) Hyperglycemia and Adverse Pregnancy Outcome (HAPO) study: associations with neonatal anthropometrics. Diabetes 58(2):453–459. https://doi.org/10.2337/db08-1112
Yaghootkar H, Scott RA, White CC et al (2014) Genetic evidence for a normal-weight “metabolically obese” phenotype linking insulin resistance, hypertension, coronary artery disease, and type 2 diabetes. Diabetes 63(12):4369. https://doi.org/10.2337/db14-0318
Yaghootkar H, Lotta LA, Tyrrell J et al (2016) Genetic evidence for a link between favorable adiposity and lower risk of type 2 diabetes, hypertension, and heart disease. Diabetes 65(8):2448. https://doi.org/10.2337/db15-1671
Ji Y, Yiorkas AM, Frau F et al (2019) Genome-wide and abdominal MRI data provide evidence that a genetically determined favorable adiposity phenotype is characterized by lower ectopic liver fat and lower risk of type 2 diabetes, heart disease, and hypertension. Diabetes 68(1):207. https://doi.org/10.2337/db18-0708
Lu Y, Day FR, Gustafsson S et al (2016) New loci for body fat percentage reveal link between adiposity and cardiometabolic disease risk. Nat Commun 7:10495. https://doi.org/10.1038/ncomms10495
Locke AE, Kahali B, Berndt SI et al (2015) Genetic studies of body mass index yield new insights for obesity biology. Nature 518:197. https://doi.org/10.1038/nature14177
Pulit SL, Stoneman C, Morris AP et al (2018) Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum Mol Genet 28(1):166–174. https://doi.org/10.1093/hmg/ddy327
Burgess S, Davies NM, Thompson SG (2016) Bias due to participant overlap in two-sample Mendelian randomization. Genet Epidemiol 40(7):597–608. https://doi.org/10.1002/gepi.21998
Horikoshi M, Yaghootkar H, Mook-Kanamori DO et al (2013) New loci associated with birth weight identify genetic links between intrauterine growth and adult height and metabolism. Nat Genet 45(1):76–82. https://doi.org/10.1038/ng.2477
Boyd A, Golding J, Macleod J et al (2013) Cohort profile: the ‘children of the 90s’—the index offspring of the Avon longitudinal study of parents and children. Int J Epidemiol 42(1):111–127. https://doi.org/10.1093/ije/dys064
Fraser A, Macdonald-Wallis C, Tilling K et al (2013) Cohort profile: the Avon longitudinal study of parents and children: ALSPAC mothers cohort. Int J Epidemiol 42(1):97–110. https://doi.org/10.1093/ije/dys066
Wright J, Small N, Raynor P et al (2012) Cohort profile: the Born in Bradford multi-ethnic family cohort study. Int J Epidemiol 42(4):978–991. https://doi.org/10.1093/ije/dys112
Knight B, Shields BM, Hattersley AT (2006) The Exeter Family Study of Childhood Health (EFSOCH): study protocol and methodology. Paediatr Perinat Epidemiol 20(2):172–179. https://doi.org/10.1111/j.1365-3016.2006.00701.x
The HAPO Study Cooperative Research (2008) Hyperglycemia and adverse pregnancy outcomes. N Engl J Med 358(19):1991–2002. https://doi.org/10.1056/NEJMoa0707943
Dupuis J, Langenberg C, Prokopenko I et al (2010) New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet 42(2):105–116. https://doi.org/10.1038/ng.520
Warrington NM, Freathy RM, Neale MC, Evans DM (2018) Using structural equation modelling to jointly estimate maternal and fetal effects on birthweight in the UK Biobank. Int J Epidemiol 47(4):1229–1241. https://doi.org/10.1093/ije/dyy015
Stephen B, Dylan SS, Simon GT (2015) A review of instrumental variable estimators for Mendelian randomization. Stat Methods Med Res 26(5):2333–2355. https://doi.org/10.1177/0962280215597579
Steichen T (2001) METANINF: Stata module to evaluate influence of a single study in meta-analysis estimation. Boston College Department of Economics, Boston, MA
Bowden J, Davey Smith G, Burgess S (2015) Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 44(2):512–525. https://doi.org/10.1093/ije/dyv080
Bowden J, Davey Smith G, Haycock PC, Burgess S (2016) Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol 40(4):304–314. https://doi.org/10.1002/gepi.21965
Davey Smith G, Spiller W, Bowden J et al (2018) Improving the visualization, interpretation and analysis of two-sample summary data Mendelian randomization via the radial plot and radial regression. Int J Epidemiol 47(4):1264–1278. https://doi.org/10.1093/ije/dyy101
Sanderson E, Davey Smith G, Windmeijer F, Bowden J (2018) An examination of multivariable Mendelian randomization in the single-sample and two-sample summary data settings. Int J Epidemiol 48(3):713–727. https://doi.org/10.1093/ije/dyy262
Hwang L-D, Lawlor DA, Freathy RM, Evans DM, Warrington NM (2019) Using a two-sample Mendelian randomization design to investigate a possible causal effect of maternal lipid concentrations on offspring birth weight. Int J Epidemiol 48(5):1457–1467. https://doi.org/10.1093/ije/dyz160
Hay WW Jr (2006) Placental-fetal glucose exchange and fetal glucose metabolism. Trans Am Clin Climatol Assoc 117:321–340
Pigeyre M, Sjaarda J, Mao S et al (2019) Identification of novel causal blood biomarkers linking metabolically favorable adiposity with type 2 diabetes risk. Diabetes Care 42(9):1800. https://doi.org/10.2337/dc18-2444
Winkler TW, Günther F, Höllerer S et al (2018) A joint view on genetic variants for adiposity differentiates subtypes with distinct metabolic implications. Nat Commun 9(1):1946–1946. https://doi.org/10.1038/s41467-018-04124-9
Huang LO, Rauch A, Mazzaferro E et al (2021) Genome-wide discovery of genetic loci that uncouple excess adiposity from its comorbidities. Nat Metab 3(2):228–243. https://doi.org/10.1038/s42255-021-00346-2
Beaumont RN, Warrington NM, Cavadino A et al (2018) Genome-wide association study of offspring birth weight in 86 577 women identifies five novel loci and highlights maternal genetic effects that are independent of fetal genetics. Hum Mol Genet 27(4):742–756. https://doi.org/10.1093/hmg/ddx429
Hartwig FP, Davies NM, Hemani G, Davey Smith G (2016) Two-sample Mendelian randomization: avoiding the downsides of a powerful, widely applicable but potentially fallible technique. Int J Epidemiol 45(6):1717–1726. https://doi.org/10.1093/ije/dyx028
Acknowledgements
This research has been conducted using the UKB Resource under application number 7036. We would like to thank the participants and researchers from the UKB who contributed or collected data. We are extremely grateful to all of the families who took part in ALSPAC, the midwives for their help in recruiting them, and the whole ALSPAC team, which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists and nurses. BiB is only possible because of the enthusiasm and commitment of the children and parents. We are grateful to all the participants, practitioners and researchers who have made BiB happen. We are also grateful to the families that took part in EFSOCH and the researchers that collected data. We are grateful to the Genetics of Complex traits team at the University of Exeter (Exeter, UK), for their assistance in learning the methods and navigating the study data; in particular, we are grateful to F. Casanova who helped with the data extraction for BiB and J. Bowden who helped in calculating the F statistic for the BMI genetic instrument. The authors would like to acknowledge the use of the University of Exeter High-Performance Computing (HPC) facility in carrying out this work.
Authors’ relationships and activities
DAL has received support from Medtronic Ltd. and Roche Diagnostics for biomarker research that is not related to the study presented in this paper. TMF has received support from GSK, Sanofi and Boehringer Ingelheim that is not related to the study presented in this paper. All other authors declare that there are no relationships or activities that might bias, or be perceived to bias, their work.
Funding
This study was supported by the US National Institute of Health (R01 DK10324), the European Research Council under the European Union’s Seventh Framework Programme (FP7/2007-2013) / ERC grant agreement no 669545, the British Heart Foundation (CS/16/4/32482 and AA/18/7/34219) and the NIHR Biomedical Centre at the University Hospitals Bristol NHS Foundation Trust and the University of Bristol. Core funding for ALSPAC is provided by the UK Medical Research Council and Wellcome (217065/Z/19/Z) and the University of Bristol. Genotyping of the ALSPAC maternal samples was funded by the Wellcome Trust (WT088806) and the offspring samples were genotyped by Sample Logistics and Genotyping Facilities at the Wellcome Trust Sanger Institute and LabCorp (Laboratory Corporation of America) using support from 23andMe. A comprehensive list of grants funding is available on the ALSPAC website (http://www.bristol.ac.uk/alspac/external/documents/grant-acknowledgements.pdf). BiB data used in this research were funded by the Wellcome Trust (WT101597MA), a joint grant from the UK Medical Research Council (MRC) and UK Economic and Social Science Research Council (ESRC) (MR/N024397/1) and the National Institute for Health Research (NIHR) under its Collaboration for Applied Health Research and Care (CLAHRC) for Yorkshire and Humber and the Clinical Research Network (CRN). The Exeter Family Study of Childhood Health (EFSOCH) was supported by South West NHS Research and Development, Exeter NHS Research and Development, the Darlington Trust and the Peninsula National Institute of Health Research (NIHR) Clinical Research Facility at the University of Exeter. Genotyping of the EFSOCH study samples was funded by Wellcome Trust and Royal Society grant 104150/Z/14/Z. WDT is supported by the GW4 BIOMED DTP awarded to the Universities of Bath, Bristol, Cardiff and Exeter from the UK Medical Research Council (MRC). MCB was supported by a UK MRC Skills Development Fellowship (MR/P014054/1). RMF and RNB were funded by a Wellcome Trust and Royal Society Sir Henry Dale Fellowship (104150/Z/14/Z). MCB, RMF and DAL work in / are affiliated with a unit that is supported by the University of Bristol and UK Medical Research Council (MC_UU_00011/6). DAL is an NIHR Senior Investigator (NF-0616-10102). ATH is supported by a NIHR Senior Investigator award and also a Wellcome Trust Senior Investigator award (098395/Z/12/Z). The funders had no role in the design of the study, the collection, analysis or interpretation of the data; the writing of the manuscript, or the decision to submit the manuscript for publication. The views expressed in this paper are those of the authors and not necessarily those of any funder.
Author information
Authors and Affiliations
Contributions
DAL and RMF designed this study, with WDT and MCB further developing the design; all four made substantial contributions to the conception and design. The following authors made substantial contributions to data acquisition: DAL (ALSPAC); ATH and BAK (EFSOCH); DMS and WLL (HAPO); GS, RA, DM and DAL (BiB); and ARW, RNB and JT (preparing imputed genotype and phenotype data for the UKB sample of European ancestry participants). The following authors made substantial contributions to analyses: AK and DMS (analyses of HAPO data); and WDT, with support from JT, MCB, RNB, RMF and DAL (all other analyses, including pooling all results analysed by others). The following authors made substantial contributions to the interpretation of data: TMF, HY and YJ (multivariable GWAS to discover favourable adiposity genetic variants); and NMW and DME (WLM GWAS using the UKB + EGG genetic and birthweight data). WDT wrote the first draft of the paper with support from MCB, RMF and DAL; all other authors read and made critical revisions to the paper. All authors approved the final submission of the paper. WDT, RMF and DAL act as guarantors for the paper’s integrity.
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM
(PDF 1.73 kb)
ESM Tables
(XLSX 144 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Thompson, W.D., Beaumont, R.N., Kuang, A. et al. Higher maternal adiposity reduces offspring birthweight if associated with a metabolically favourable profile. Diabetologia 64, 2790–2802 (2021). https://doi.org/10.1007/s00125-021-05570-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-021-05570-9